

Abstract
Crosstalk between G-protein signaling and glutamatergic transmission within the brain reward circuits is critical for long-term emotional effects (depression and anxiety), cravings, and negative withdrawal symptoms associated with opioid addiction. A previous study showed that Regulator of G-protein signaling 4 (RGS4) may be implicated in opiate action in the nucleus accumbens (NAc). However, the mechanism of the NAc-specific RGS4 actions that induce the behavioral responses to opiates remains largely unknown. The present study used a short hairpin RNA (shRNA)-mediated knock-down of RGS4 in the NAc of the mouse brain to investigate the relationship between the activation of ionotropic glutamate receptors and RGS4 in the NAc during morphine reward. Additionally, the shRNA-mediated RGS4 knock-down was implemented in NAc/striatal primary-cultured neurons to investigate the role that striatal neurons have in the morphine-induced activation of ionotropic glutamate receptors. The results of this study show that the NAc-specific knockdown of RGS4 significantly increased the behaviors associated with morphine and did so by phosphorylation of the GluR1 (Ser831) and NR2A (Tyr1325) glutamate receptors in the NAc. Furthermore, the knock-down of RGS4 enhanced the phosphorylation of the GluR1 and NR2A glutamate receptors in the primary NAc/striatal neurons during spontaneous morphine withdrawal. These findings show a novel molecular mechanism of RGS4 in glutamatergic transmission that underlies the negative symptoms associated with morphine administration.
Keywords: addiction, glutamatergic transmission, morphine, nucleus accumbensm, regulator of G-protein signaling 4
INTRODUCTION
Opioids, specifically μ-opioid receptor (MOR) agonists, are the gold standard for controlling severe pain; however, the cessation of chronic opioid treatment induces withdrawal syndromes characterized by physical symptoms and drug-seeking behaviors (Chartoff et al., 2014). The nucleus accumbens (NAc) consisting of approximately 95% γ-aminobutyric acid (GABA)ergic medium spiny neurons expressing dopamine D1 and D2 receptors receives extensive glutamatergic and dopaminergic inputs from several brain regions (Kim et al., 2016). The NAc may have a modulatory role in opiate reward and withdrawal (Garcia-Perez et al., 2016). For example, intra-NAc injections of opioids have induced drug-seeking behaviors in animal models (Goeders et al., 1984; Liang et al., 2006), whereas intra-NAc injections of opioid receptor antagonists have induced physical withdrawal symptoms in morphine-dependent animals (Jaw et al., 1994). Taken together, these findings suggest that both the reward and withdrawal states may be mediated by the action of morphine in the NAc.
Morphine-activated MORs are distributed throughout the brain reward circuitry including the NAc and ventral tegmental area (VTA) and are G-protein-coupled receptors (GPCRs) linked to Gαi. While MORs inhibit cyclic adenosine monophosphate (cAMP) production during acute morphine treatment, prolonged morphine exposure causes neuronal adaptations that ultimately result in a compensatory upregulation of adenylyl cyclase function so that the cAMP levels reach approximately normal which is a phenomenon known as cAMP superactivation (Nestler, 2004). Precipitated morphine withdrawal due to administration of naloxone, an opioid receptor antagonist, induces dramatic increases in cAMP levels in a morphine-dependent state (Copeland et al., 1989). Thus, the upregulation of cAMP signaling is believed to contribute to the addictive features of morphine and results in a high rate of relapse due to withdrawal symptoms that are manifested by the continuance of drug-taking behaviors (Nestler, 2004). Additionally, chronic morphine treatment increases intracellular Ca2+ levels by the activation of MORs; these, in turn, activate diverse proteins, such as calcium-calmodulin kinase II (CaMKII) and cAMP response element-binding protein (CREB) which are thought to have important roles as molecular switches that modulate opioid reward and withdrawal (Liu et al., 2012; Valverde et al., 2004). Thus, targeting these signaling pathways might be a reliable therapeutic approach that can ameliorate the negative consequences associated with chronic morphine treatment (Liu et al., 2012).
An increasing amount of evidence has shown that repeated psychostimulant exposure influences glutamatergic transmission in the brain reward circuitry (Popik and Kolasiewicz, 1999; Popik and Wrobel, 2002). Under conditions of psychostimulant addiction, accumbal glutamate is thought to underlie the psychostimulant-induced neuronal plasticity that ultimately results in the manifestation of progressive negative behaviors including cravings and withdrawal symptoms (Dobi et al., 2011). However, few studies have investigated opioid (e.g., morphine and heroin)-induced changes in glutamatergic transmission in the NAc. Because the cellular and behavioral consequences of opioids often differ from those of psychostimulants, it is important to elucidate the changes in glutamatergic transmission under the states of opioid reward and withdrawal.
The regulators of the G-protein signaling (RGS) family have important roles in the modulation of GPCR-mediated signaling pathways through the interaction of G protein subunits with various effectors (Bilodeau and Schwendt, 2016; Levitt et al., 2006). RGS4 is widely distributed throughout brain regions such as the prefrontal cortex, hippocampus, locus coeruleus, and striatum (Grillet et al., 2005). In particular, several studies have shown the modulatory function of RGS4 on various GPCRs including the muscarinic and dopamine receptors (Ding et al., 2006; Taymans et al., 2003) as well as on changes in accumbal RGS4 expression following morphine treatment (Han et al., 2010). Furthermore, the involvement of RGS4 in morphine reward and tolerance has been implicated in conditioned place preference (CPP) tests and hot plate assays (Han et al., 2010). However, there is little evidence of the possible influence of the molecular mechanisms of RGS4 on morphine-induced behavioral consequences. Thus, the present study used a NAc-specific knock-down model to examine the role of RGS4 in chronic morphine-induced reward. Additionally, in vivo and in vitro experiments were done with the RGS4 knock-down model to determine whether ionotropic glutamatergic transmission in the NAc is involved in morphine reward and withdrawal.
MATERIALS AND METHODS
Animals
For the behavioral tests and molecular assays, 7-week old male C57BL/6J mice (21–23 g) were purchased from the Korea Institute of Science and Technology Animal Facility (KiSAF; Korea). For the in vitro experiments, pregnant female C57BL/6J mice were obtained on day 12 of gestation from a specific-pathogen-free colony at the Damul Experimental Animal Center (Korea). All animals were housed in a room maintained at 23 C ± 2°C with a relative humidity of 50 ± 5% and artificial lighting from 08:00 to 20:00 and air changes every hour. The animals were provided tap water and commercial rodent chow (Samyang Feed; Korea) ad libitum. The Institutional Animal Care and Use Committee of the KIST approved all protocols used in this study (approval no. 2016-081).
Lentivirus-mediated RGS4 knock-down
psi-LVRU6GP RGS4 shRNA, which targets mouse RGS4 mRNA (GenBank accession number NM_009062.3), and psi-LVRU6GP control shRNA, which does not target any specific mRNA, were purchased from GeneCopoeia (USA); these characteristics were established and confirmed by Genecopoeia. The lentivirus particles were produced by co-transfection into HEK-293T cells with the Lenti-Pac HIV Packaging Mix (GeneCopoeia). Following transfection, the medium was collected and subjected to ultracentrifugation at 28,000 g for 90 min. with a SW28 rotor (Optima L-90K, Beckman Coulter; USA). The virus pellet was re-suspended with phosphate-buffered saline (PBS) and immediately stored at −80°C. The concentrated virus titer was determined by infecting HEK-293T cells after a serial dilution with each lentivirus, and the enhanced green fluorescent protein (eGFP) expression of the infected cells was measured with flow cytometry 72 h after the injection; the titer was approximately 1 × 109 infectious units (IFU) per ml.
Stereotaxic injections
The stereotactic surgical procedures were performed while the animals were under ketamine (120 mg kg−1; Yuhan Co., Korea) and xylazine (0.6 mg kg−1; Bayer AG, Leverkusen, Germany) anesthesia. A total of 2 μl (1 × 109 IFU/ml) of the lentiviral vectors were bilaterally injected into the NAc (anteroposterior +1.3, mediolateral ±0.9, and dorsoventral −3.7 from bregma) at a rate of 0.1 μl/min. using a micro-infusion pump (New Era Pump Systems, Inc.; USA). At least 4 weeks of recovery were allowed prior to the behavioral and biochemical assays. Following the behavioral procedures, the injection sites were confirmed using the expression of eGFP in coronal brain sections. Data from mice with mistargeted injections were excluded from the analyses.
Reagents and antibodies
Morphine hydrochloride was purchased from Myungmoon Pharm. Co., LTD (Korea). Rabbit polyclonal anti-RGS4 antibody (cat. No. PA5-22332) was purchased from ThermoFisher Scientific Inc. (USA). Rabbit polyclonal anti-phospho-NR2A (Tyr1325; cat. No. ab16646) and polyclonal anti-NR2A antibody (cat. No. ab118587) were purchased from Abcam (USA). Rabbit monoclonal anti-phospho-GluR1 (Ser831; cat. No. 04-823) and rabbit polyclonal anti-GluR1 (cat. No. ABN241) were purchased from Millipore (USA). Rabbit monoclonal anti-phospho-CaMKII (Thr286; cat. No. 12716) and rabbit polyclonal anti-CaMKII antibody (cat. No. 3362), rabbit polyclonal anti-phospho-PKA-C (Thr197) antibody (cat. No. 4781) and rabbit polyclonal anti-PKA-C antibody (cat. No. 4782) were purchased from Cell Signaling Technology (USA). Monoclonal mouse anti-beta-actin was purchased from Sigma-Aldrich (USA). For the immunoblot procedures, horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgG were obtained from Thermo Fisher Scientific, Inc. (USA).
CPP test
The standard CPP protocol was modified for the present study (Moron et al., 2010). Four weeks after the stereotaxic injection surgery, the mice were placed in the central chamber (gray) of the CPP apparatus and allowed to explore three distinct chambers for 20 min. Mice showing a > 75% place preference for either of the two conditioning chambers (black and white) were excluded from the procedure. On the subsequent 5 days, the mice received intraperitoneal (i.p.) injections of morphine twice per day with a 6-h interval. On the first day of the conditioning session, the mice were injected with 15 mg/kg of morphine and confined to a randomly assigned chamber (morphine-paired) for 30 min. After 6 h, the mice received a second injection of saline and were confined to the other chamber (saline-paired) for 30 min. The injection order for the morphine and saline were reversed daily; however, the assigned chambers did not change during the conditioning session. The saline control groups were injected with saline twice per day and confined to a randomly assigned chamber for 30 min. during the conditioning sessions. On the test day, the mice were placed in the central gray chamber and allowed to freely explore the apparatus for 20 min. The time spent in each chamber was recorded for each mouse using EthoVision XT (Noldus; USA), and the difference between the time spent in the morphine-paired chamber and the time spent in the saline-paired chamber on the test day was calculated. The mice were immediately sacrificed using cervical dislocation after the CPP test, and the brains were frozen at −80°C until the biochemical assays.
Primary NAc/striatal culture and treatment
The primary cell culture method used in in this study was performed as previously described (Kim et al., 2014). Briefly, striatal tissue samples were dissected from the pups of C57BL/6 mice at gestational days 17–18 and then prepared for culturing. Following dissection, the tissues were chopped and digested with 10 units/ml of papain (Worthington; USA) and 100 units/ml of DNase I (Roche; Switzerland) in dissociation buffer at 37°C for 30 min, and the digested tissue was triturated with Neurobasal A medium (Invitrogen; USA). The cells were seeded at a density of 4 × 105 cells/well on poly-D-lysine hydrobromide (150 μg/ml; Sigma-Aldrich)-coated 24-well plates (NUNC™; Thermo Fisher Scientific); 1 h after plating, the Neurobasal A was replaced with a growth medium consisting of Neurobasal A, 1× B27 supplement (Invitrogen), 100 units/ml of penicillin, 0.1 mg/ml of streptomycin, and 0.5 mM of glutamine (Invitrogen). All cultures were kept at 37°C and 5% CO2, and the cultured neurons were transduced with Lv-shRNA-eGFP and Lv-eGFP at a multiplicity of infection of 5 on 3 day in vitro (3 DIV). To evaluate the chronic effects of morphine, the neurons (9 DIV) were incubated with 10 μM of morphine for 3 days.
Western blot analysis
Tissue sections (100-μm thick) were sliced from the frozen samples at −30°C with a CM1950 cryostat (Leica Biosystems; Germany), and the NAc tissue was collected from the prepared slices with a 16-gauge tissue punch. The samples were then separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a poly-vinylidene difluoride membrane; the membranes were blocked using a combination of 1% normal goat serum and 1% bovine serum albumin (BSA). Next, the samples were incubated with the following primary antibodies in PBS with Tween-20 overnight at 4°C: rabbit anti-RGS4 (1:500 dilution), rabbit anti phospho-NR2A (1:1,000 dilution), rabbit anti phospho-GluR1, rabbit anti phospho-CaMKII (1: 1,000 dilution), rabbit anti-phospho-PKA-C (1:1,000 dilution), rabbit anti-NR2A (1:1,000 dilution), rabbit anti-GluR1 (1:1,000 dilution), rabbit anti-CaMKII (1:1,000 dilution) and rabbit anti-PKA-C (1:1,000 dilution). After extensive washing and incubation with an HRP-conjugated anti-rabbit antibody (1:5,000 dilution; Thermo Fisher Scientific, Inc.), the signals were visualized using a chemiluminescence kit (SuperSignal® West Pico or Femto; Thermo Fisher Scientific, Inc.) and read on a C-DiGit®Blot Scanner (LI-COR; USA). To normalize the signals, the membranes were reprobed with an antibody against β-actin (1: 5,000 dilution). The bands were quantified using the ImageJ software (National Institutes of Health [NIH]; USA).
Real-time polymerase chain reaction
RNA extraction and real-time PCR procedures were performed as previously described by our research group (Kim et al., 2014). Total RNA was isolated from NAc tissue using the RNAeasy® Lipid Tissue Mini kit (Qiagen; USA) according to the manufacturer’s instructions, and the concentrations of the RNA samples were determined by measuring the optical density with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc). Next, first-strand complementary DNA (cDNA) was prepared using random primers (Takara Bio; Tokyo, Japan) with Superscript™ II reverse transcriptase (Invitrogen) according to the manufacturer’s instructions, and the cDNA was diluted to 8 ng/μl with RNase-free water. The primer sequences for RGS4 (NM_009062.3) were as follows: forward sequence (5′-3′), ATGAAACATCGG CTGGGGTT and reverse sequence (5′-3′), TTGAAAGCTGCC AGTCCACA. Real-time PCR amplifications were performed using TOPreal™ qPCR 2× PreMIX (Enzynomics; Daejeon, Korea) with a Stratagene MX3000P (Agilent Technologies; USA) according to the manufacturer’s instructions. The thermal cycling profile consisted of a pre-incubation step at 94°C for 10 min, followed by 45 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and elongation at 72°C for 20 s. Additionally, a melting program was used to verify that only one product was amplified. The amplification curves from each real-time PCR reaction were generated within the software, and the threshold cycle values were determined; β-actin was used as a housekeeping gene for normalization to an internal control for each sample. All results are expressed as a mean fold change using the 2−ΔΔ CT method (Livak and Schmittgen, 2001).
Statistical analysis
All data are reported as the means ± standard error of the mean (SEM) and were analyzed with unpaired t-tests and one-way or two-way analysis of variance (ANOVA) tests. Next, Fisher’s least significant difference (LSD) post hoc tests for multiple comparisons were performed, and p values < 0.05 were considered to indicate statistical significance.
RESULTS
RGS4 knock-down in the NAc increased morphine-induced behaviors
To examine the influence of RGS4 on the rewarding effects of morphine in the NAc, lentivirus-expressing shRNA tagged with eGFP (Lv-shRNA-eGFP) targeting RGS4 and Lv-eGFP were injected into the bilateral NAc (Figs. 1A and 1B); the infected area was confirmed 4 weeks after the injection based on the expression of eGFP (Fig. 1C). To assess the efficacy of the knock-down of RGS4 in the NAc, NAc tissues were collected, and protein levels were measured with Western blot analyses. The level of RGS4 expression in the NAc of mice injected with Lv-shRNA-eGFP was reduced by > 60% compared to mice injected with Lv-eGFP (Fig. 1D).
Fig. 1. RGS4 controlled morphine-induced reward in the NAc.

(A) Schematic representation of the shRNA-targeting RGS4 construct. (B) Injection target site in the mouse brain (NAc; red circle). (C) Representative photographs of eGFP expression in the NAc of mice injected with Lv-eGFP and Lv-shRNA-eGFP. (D) Representative immunoblots of RGS4 expression in the NAc of mice injected with Lv-eGFP and Lv-shRNA-eGFP. All data are expressed as means ± SEM; n = 6 for each group. (E) Schematic diagram of the schedule for the morphine CPP test. (F) Mice infected with Lv-shRNA-eGFP in the NAc showed an increased response to morphine (15 mg/kg, i.p.) in the CPP paradigm. All data are expressed as means ± SEM (n = 6 for each group). Data were analyzed with unpaired t-tests. *p < 0.05 vs. the Lv-eGFP infected group.
To assess whether the knock-down of RGS4 influenced morphine reward, the CPP paradigm was used (Fig. 1E). In the CPP test, mice injected with Lv-shRNA-eGFP exhibited an increased sensitivity to the rewarding effects of morphine relative to the mice injected with Lv-eGFP (Fig. 1F). Additionally, to examine the effects of the lentivirus infection in the NAc on locomotor changes that can affect behavior in the CPP paradigm, open field tests were performed. No significant changes in locomotion were detected in the mice injected with the Lv-shRNA-eGFP compared to the mice injected with the Lv-eGFP (Supplementary Fig. S1). Taken together, these findings suggest that RGS4 in the NAc is involved in the behaviors related to morphine reward.
Morphine-induced activation of ionotropic glutamate receptors was enhanced by the knock-down of RGS4 in the NAc
Several studies have shown that changes in ionotropic glu-tamatergic transmission occur during prolonged MOR activation and that these changes are involved in the rewarding effects of morphine (Gonzalez et al., 1997; Martin et al., 1999; Murray et al., 2007). Because there are questions regarding the changes in the time course of neuronal activity in the NAc after chronic morphine treatment, this study examined the expressions of c-fos (immediate early gene) in the NAc at 30 min., 2 h, and 24 h after 5 consecutive days of i.p. morphine injections. Nestler (2001) described that c-fos expression in the NAc reached the maximum point at 2 h after the injection of acute psychotic drugs, and that c-fos expression showed a delayed and gradually increasing pattern which is correlated to negative behavioral symptoms such as dependence, tolerance, and withdrawal symptoms after treatment with chronic psychotic drugs. The present results show that the mRNA expressions of c-fos increased by approximately six-fold at 30 min. after the last morphine injection and that c-fos expression was maintained by approximately four-fold at 2 and 24 h after the last morphine injection (Supplementary Fig. S2).
It was also hypothesized that the temporal changes in neuronal activity due to chronic morphine exposure might be strongly related to changes in ionotropic glutamatergic transmission. Because there is some evidence that c-fos is regulated by ionotropic glutamate receptors (Das et al., 1997; Lerea et al., 1993), we examined the phosphorylation levels of ionotropic glutamate receptors GluR1 and NR2A as well as PKA and CaMKII, which are considered to be involved with the signaling pathways of inotropic glutamate receptors, using Western blot analysis (Fig. 2).
Fig. 2. RGS4 regulated the activation of glutamate receptors in the NAc during the morphine-conditioned place preference test.
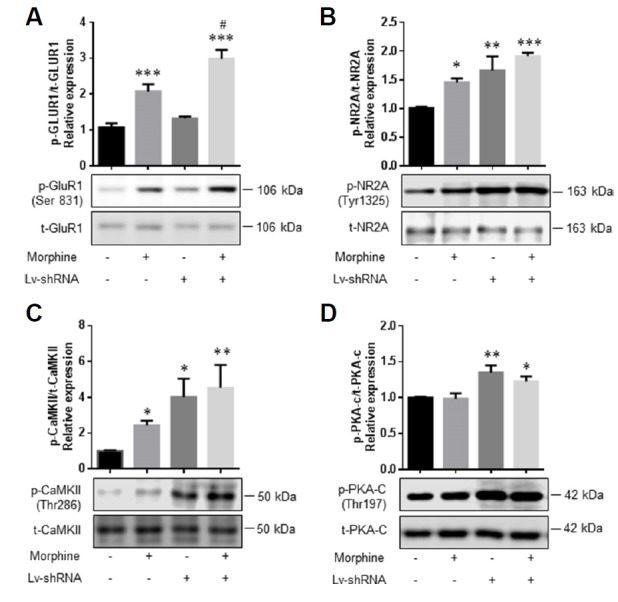
For the CPP paradigm, mice were bilaterally injected with Lv-shRNA-eGFP and Lv-eGFP and then morphine. Subsequently, the mice were sacrificed, and the levels of glutamate receptors, CaMKII and PKA-C phosphorylations were examined. (A–D) Representative images and bar graphs of immunoblots for phospho-GluR1 (Ser831), total-GluR1, phospho-NR2A (Tyr1325), total-NR2A, phospho-CaMKII (Thr286), total-CaMKII, phospho-PKA-C (Thr197), and total-PKA-C in the NAc (left to right: control, morphine, Lv-shRNA-eGFP, and Lv-shRNA-eGFP + morphine-treated groups). All data are expressed as means ± SEM; n = 6 for each group and were analyzed with a two-way ANOVA followed by a Fisher’s LSD post hoc test. *p < 0.05 vs. vehicle-treated control Lv-eGFP-infected group, **p < 0.01 vs. vehicle-treated control Lv-eGFP-infected group, and ***p < 0.001 vs. vehicle-treated control Lv-eGFP-infected group. #p < 0.05 vs. vehicle-treated control Lv-shRNA-eGFP-infected group.
AMPA and NMDA glutamate receptors are regulated by the protein kinase C (PKC)/CaMKII signaling pathway (Boehm et al., 2006; Dias et al., 2012), and the phosphorylation levels of GluR1 at Ser831 and NR2A at Tyr1325 are known to be regulated by PKC and CaMKII, which increase the AMPA receptor function by changes in channel conductance (Derkach et al., 1999; Ohnishi et al., 2011; Snyder et al., 2000). Thus, the activation of CaMKII in the NAc of mice injected with Lv-shRNA-eGFP and Lv-eGFP was assessed after the morphine CPP post-test (Fig. 2). The present results show that chronic treatment with morphine during the CPP paradigm significantly increased the phosphorylation levels of GluR1 (Fig. 2A, F (1, 20) = 60.21, P < 0.0001), NR2A (Fig. 2B, F (1, 20) = 17.32, P = 0.0005), and CaMKII (Fig. 2C, F (1, 20) = 9.413, P = 0.0061) in the NAc compared to the vehicle-treated controls.
cAMP-dependent protein kinase A (PKA) signaling is considered to be one of the most important μ-opioid dependent signaling pathways (Ai et al., 1999; Shen et al., 2000). Therefore, we also tested whether chronic morphine treatment induces phosphorylation of PKA-C in the NAc. However, the phosphorylation of PKA-C was not changed after the morphine treatment (Fig. 2D).
Additionally, the expression level of p-GluR1 in the Lv-shRNA-eGFP + morphine group was significantly increased compared to those of the Lv-eGFP-infected control and Lv-shRNA-eGFP-infected groups (Fig. 2A, F (1, 20) = 11.47, P = 0.0029). The expression of p-NR2A (Fig. 2B, F (1, 20) = 6.879, P = 0.0163) and p-CaMKII (Fig. 2C, F (1, 20) = 5.697, P = 0.0270) significantly increased in both the vehicle-treated control group and the morphine-treated group infected by Lv-shRNA-eGFP compared to the Lv-eGFP-infected control group.
Although we found an increase of p-PKA-C (Thr197) in the Lv-shRNA-eGFP and Lv-shRNA-eGFP + morphine group, there was no change in the p-PKA-C in the NAc of mice injected with Lv-shRNA-eGFP compared to that in the NAc of mice injected with Lv-eGFP after chronic morphine treatment during the CPP (Fig. 2D, F (1, 20) = 0.161, P = 0.6924).
Chronic morphine treatment increased the activation of ionotropic glutamate receptors in primary NAc/striatal neurons
To clarify the role of neuronal involvement in the activation of glutamate receptors after chronic morphine treatment, Western blot analyses were done to assess the protein expressions of RGS4, p-GluR1 (Ser831), GluR1, p-NR2A (Tyr1325), NR2A, p-PKA-C (Thr197), PKA-C, p-CaMKII (Thr286), and CaMKII in primary NAc/striatal neurons after 3 days of treatment with morphine (10 μM). The expression level of RGS4 protein was significantly decreased after the morphine treatment (Figs. 3A and 3B, saline control group: 1.00 ± 0.06, morphine treated group: 0.50 ± 0.04, fold change, Unpaired t-test, p < 0.01 vs. saline control group). The morphine treatment significantly increased the phosphorylation levels of GluR1 (Figs. 3A and 3C, saline control group: 1.00 ± 0.06, morphine treated group: 1.57 ± 0.14, fold change, Unpaired t-test, p < 0.05 vs. saline control group), NR2A (Figs. 3A and 3D, saline control group: 1.00 ± 0.07, morphine treated group: 2.75 ± 0.33, fold change, Unpaired t-test, p < 0.01 vs. saline control group), and CaMKII (Figs. 3A and 3E, saline control group: 1.00 ± 0.05, morphine treated group: 2.55 ± 0.35, fold change, Unpaired t-test, p < 0.05 vs. saline control group). However, the phosphorylation level of PKA was significantly decreased (Figs. 3A and 3F, saline control group: 1.00 ± 0.06, morphine treated group: 0.60 ± 0.02, fold change, Unpaired t-test, p < 0.05 vs. saline control group).
Fig. 3. Morphine induced the activation of ionotropic glutamate receptors in primary NAc/striatal neurons.

To examine the effects of morphine on glutamate receptors in NAc/striatal neurons, the neurons (9 DIV) were incubated with morphine (10 μM) for 3 days. (A) Representative photographs of immunoblots for RGS4, phospho-GluR1 (Ser831), total-GluR1, phospho-NR2A (Tyr1325), total-NR2A, phospho-CaMKII (Thr286), total-CaMKII, phospho-PKA-C (Thr197), and total-PKA-C. (B–F) Bar graphs showing semi-quantitative analyses of the phosphorylation levels of GluR1, NR2A, CaMKII, and PKA-C in NAc/striatal neurons after vehicle and morphine treatment. All data are expressed as means ± SEM; n = 6 for each group and were analyzed with unpaired t-tests. p < 0.05 vs. vehicle-treated control and **p < 0.01 vs. vehicle-treated control.
RGS4 knock-down enhanced ionotropic glutamate receptor activation during chronic morphine treatment and withdrawal
To investigate whether RGS4 might have a modulatory role during the activation of ionotropic glutamate receptors in primary NAc/striatal neurons, 3 DIV primary NAc/striatal neurons were transduced with Lv-shRNA-eGFP and Lv-eGFP. At 12 DIV, the neurons were assessed for transduction and knock-down efficiency; > 95% of the neurons expressed eGFP fluorescence, and there were significant decreases in the RGS4 protein and mRNA levels (71.12 ± 1.18% and 68.97 ± 5.54%, respectively) after Lv-shRNA-eGFP infection of primary striatal/NAc neurons (Figs. 4A–4C).
Fig. 4. RGS4 regulated glutamate receptors in primary NAc/striatal neurons.

At 3 DIV, neurons were infected with Lv-eGFP and Lv-shRNA-eGFP, and the cells were collected on 12 DIV. (A) Representative photographs of Lv-eGFP (left panel) and Lv-shRNA-eGFP (right panel) expression levels in NAc/striatal neurons. (B and C) Bar graphs showing semi-quantitative analyses of the protein and mRNA expression levels of RGS4 in Lv-eGFP- and Lv-shRNA-eGFP-infected NAc/striatal neurons. (D) Representative photographs of immunob-lots for phospho-GluR1 (Ser831), total-GluR1, phospho-NR2A (Tyr1325), total-NR2A, phospho-CaMKII (Thr286), total-CaMKII, and phospho-PKA-C (Thr197), and total-PKA-C. (E–G) Bar graphs showing semi-quantitative analyses of the phosphorylation levels of GluR1, NR2A, CaMKII, and PKA-C in the Lv-eGFP- and Lv-shRNA-eGFP-infected groups. All data are expressed as means ± SEM; n = 6 for each group, and were analyzed with unpaired t-tests. **p < 0.01 vs. Lv-eGFP-infected group and ***p < 0.001 vs. Lv-eGFP-infected group.
Next, the expression levels of p-GluR1 (Ser831), GluR1, p-NR2A (Tyr1325), NR2A, p-PKA-C (Thr197), p-PKA-C, p-CaMKII (Thr286) and CaMKII were assessed. There were significant increases in the phosphorylation levels of p-GluR1, p-NR-2A, and p-CaMKII in primary NAc/striatal neurons infected with Lv-shRNA-eGFP relative to the levels in neurons infected with Lv-eGFP (Figs. 4D–4G). However, there was no change in the p-PKA-C (Thr197) expression between the Lv-shRNA-eGFP and Lv-eGFP infected primary striatal/NAc neurons (Fig. 4D and 4H).
To better understand the effects of RGS4 on ionotropic glutamate receptors during chronic morphine treatment and withdrawal, Lv-shRNA-eGFP- and Lv-eGFP-infected primary NAc/striatal neurons were incubated with 10 μm of morphine for 3 days beginning on 9 DIV followed by 1, 3, and 6 h of spontaneous withdrawal. Next, the expression patterns of p-GluR1 (Ser831), GluR1, p-NR2A (Tyr1325), NR2A, p-PKA-C (Thr197), PKA-C, p-CaMKII (Thr286) and CaMKII were examined at 0, 1, 3, and 6 h after morphine cessation. There were significant increases in the phosphorylation levels of GluR1 (F (1, 40) = 212.3, P < 0.0001), NR2A (F (1, 40) = 159, P < 0.0001), and CaMKII (F (1, 40) = 93.55, P < 0.0001) throughout the duration of the morphine withdrawal in the Lv-shRNA-eGFP-infected group compared to the Lv-eGFP-infected group (Figs. 5A–5D). Expression of p-PKA-C (The197) did not change (F (1, 40) = 0.2546, P = 0.6166) between the Lv-shRNA-eGFP and Lv-eGFP infected neurons (Figs. 5A and 5E).
Fig. 5. RGS4 knock-down enhanced glutamate receptor activation after morphine withdrawal in primary NAc/striatal neurons.

At 3 DIV, the neurons were infected with Lv-eGFP and Lv-shRNA-eGFP for 9 days. To test the effects of RGS4 on glutamate receptor activation during morphine withdrawal, the neurons were incubated with morphine (10 μM) for 3 days at 12 DIV, washed three times with growth medium, and cultured for the indicated time to induce spontaneous withdrawal. (A) Representative images of immunoblots for phospho-GluR1 (Ser831), total-GluR1, phospho-NR2A (Tyr1325), total-NR2A, phospho-CaMKII (Thr286), total-CaMKII, phospho-PKA-C (Thr197), and total-PKA-C. (B–E) Bar graphs showing semi-quantitative analyses of the phosphorylation levels of GluR1, NR2A, CaMKII, and PKA after spontaneous morphine withdrawal (0, 1, 3, and 6 h) in the Lv-eGFP- and Lv-shRNA-eGFP-infected groups. All data are expressed as means ± SEM; n = 6 for each group and were analyzed with a two-way ANOVA followed by a Fisher’s LSD post hoc test. *p < 0.05 vs. Lv-eGFP-infected group, **p < 0.001 vs. Lv-eGFP-infected group at the same time points and ***p < 0.001 vs. Lv-eGFP-infected group at the same time points. ##p < 0.01 and ###p < 0.001 vs. 0 h after morphine treatment in Lv-shRNA-infected group. &&&p < 0.001 vs. 0 h after morphine treatment in Lv-eGFP-infected group.
DISCUSSION
The findings of this study revealed the NAc/striatal neuron-specific roles of RGS4 in morphine reward. Because previous evidence indicates that the RGS family is associated with a variety of psychiatric conditions including depression, schizophrenia, and drug addiction, it is important to clarify the function and cellular mechanisms of RGS proteins in specific signaling pathways and cell types (Lomazzi et al., 2008; Terzi et al., 2014). Many studies have reported on the modulatory role of RGS4 in response to psychostimulant treatment in the NAc and the dorsal striatal regions through GPCRs including metabotropic glutamate receptor (mGluR5) and dopamine D1 and D2 receptors (Schwendt et al., 2006; 2007; 2012). However, the influence of RGS4 on the regulation of ionotropic glutamatergic transmission through GPCRs during psychostimulant addiction has yet to be considered. Thus, this study used a NAc/striatal-selective knockdown model to elucidate the influence of striatal RGS4 on ionotropic glutamate receptors during morphine reward. The findings of the behavioral tests in this study were supported by in vivo and in vitro biochemical assays which indicated that RGS4 modulates the morphine-induced activation of glutamate receptors.
The present results also indicate that RGS4 can modulate morphine reward. The NAc-specific RGS4 knock-down enhanced morphine CPP after 5 consecutive days of i.p. morphine injections (15 mg/kg). These findings are similar to those of previous studies showing that the conditional deletion of RGS4 with the Cre-lox system increases morphine CPP after a low-dose (3 mg/kg) morphine treatment (Han et al., 2010) and that other RGS genes, such as RGS9-2, increase sensitivity to morphine reward (Gaspari et al., 2014). Interestingly, these two independent studies failed to identify the common molecular mechanisms underlying the identical behavioral consequences of chronic morphine treatment, which suggests the involvement of different signaling pathways for each RGS family. RGS4 is also involved in the response to psychostimulants including morphine in a variety of brain regions (Bishop et al., 2002; Lomazzi et al., 2008; Schwendt et al., 2007; 2012). However, the cellular mechanism of accumbal RGS4 contributing to the rewarding effects of morphine is not clear. Therefore, in this study, we investigated the possible cellular mechanisms of accumbal RGS4 in the morphine rewarding effect.
The present findings indicate that one of the plausible mechanisms underlying the development of morphine reward may be caused by glutamatergic transmission in the NAc. Even though numerous studies have examined changes in glutamatergic transmission in the NAc after psychostimulant administration (Jedynak et al., 2016; Parsegian and See, 2014), little is known about the effects of chronic morphine exposure on ionotropic glutamatergic transmission in the NAc. Thus, this study investigated whether morphine would change the action of ionotropic glutamate receptors in the NAc following the morphine CPP procedure and in primary NAc/striatal neurons. Using the CPP paradigm, this study found significant increases in the phosphorylation levels of the AMPA receptor subunit GluR1 (Ser831) and NMDAR receptor subunit NR2A (Tyr1325) in the NAc after chronic morphine treatment. Additionally, the in vitro results confirmed the increases in the phosphorylation levels of GluR1 and NR2A after chronic morphine treatment. Taken together, these data suggest that chronic morphine treatment increases glutamatergic transmission in the NAc and supports previous findings showing that morphine reward is altered by glutamate receptor inhibition (Gonzalez et al., 1997; Trujillo and Akil, 1991). Moreover, Robbe et al. (2002) showed that morphine-dependent mice exhibit decreases in long-term depression (LTD) through (mGluR2/3) at glutama-tergic synapses in the NAc supporting our data which show the involvement of glutamatergic transmission in the NAc in morphine addiction.
This study also identified an upregulation in the phosphorylation of CaMKII after chronic morphine treatment was done both in vivo and in vitro, which suggests that the morphine-induced activation of ionotropic glutamate receptors in the NAc may be closely linked with CaMKII signaling. Consistent with these findings, previous evidence has shown that the inhibition of CaMKII in the NAc attenuates the reinstatement of morphine reward (Liu et al., 2012). However, further research is necessary to elucidate the effects of the crosstalk between RGS4 and CaMKII signaling within brain reward circuits and to determine the critical factors that control RGS4 and inotropic glutamate receptors. Furthermore, the in vivo results show an unaltered expression of p-PKA-C, an active form of PKA, during the CPP paradigm, and the in vitro results show a decrease in the p-PKA-C expression after chronic morphine treatment, which suggests that the chronic morphine-induced activation of glutamate receptors might not be dependent on PKA signaling, and a future study is needed for the complete understanding of the involvement of PKA signaling in the RGS4 action in morphine reward.
Previous studies have shown that glutamatergic transmission is associated with morphine withdrawal symptoms (Inoue et al., 2003; Manzoni and Williams, 1999). For example, NR2A null knockout mice exhibit attenuated morphine withdrawal symptoms (Inoue et al., 2003). In this study, primary NAc/striatal neurons infected with Lv-shRNA targeting RGS4 showed a significant increase in the phosphorylation levels of glutamate receptors and CaMKII signaling during spontaneous morphine withdrawal compared to the Lv-eGFP-infected group. Thus, the in vitro findings of this study suggest that RGS4 modulates morphine-induced withdrawal through ionotropic glutamate receptors and CaMKII signaling. Interestingly, in the present study, the NAc of mice infected with Lv-shRNA showed increased phosphorylation of p-PKA-C in both the saline and morphine CPP groups. Conversely, the in vitro results showed that chronic morphine treatment and spontaneous morphine withdrawal significantly decreased the p-PKA-C expression, and the knock-down of RGS4 failed to alter the p-PKA-C expression of primary NAc/striatal cultured neurons in response to morphine treatment. This discrepancy can be due to a lack of interaction with other brain regions for regulating the striatal PKA activity in a single primary culture system. In a future study, co-culture experiments with the hippocampus or prefrontal cortex are needed to elucidate whether the prefrontal cortex or hippocampus might be involved in the striatal PKA activity in response to chronic morphine treatment.
In conclusion, the present findings confirmed that the NAc-specific RGS4 levels regulate morphine reward in mice. Furthermore, the molecular and biochemical approaches used in this study indicate that chronic morphine treatment and spontaneous withdrawal induced the activation of ionotropic glutamate receptors in the NAc and that this process was modulated by RGS4. These findings provide new insights into the molecular mechanisms underlying negative morphine-induced symptoms.
Supplementary data
ACKNOWLEDGMENTS
This research was funded by a National Research Council of Science & Technology (NST) grant by the Korean government (MSIP) (No. CRC-15-04-KIST). This research was also supported by the National Research Foundation of Korea under the following grant: NRF-2017R1A2B2003993 (“Mid-career Researcher Program”). In addition, this work was supported by the Education and Research Encouragement Fund of Seoul National University Hospital (2017).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ai W., Gong J., Yu L. MAP kinase activation by mu opioid receptor involves phosphatidylinositol 3-kinase but not the cAMP/PKA pathway. FEBS Lett. 1999;456:196–200. doi: 10.1016/s0014-5793(99)00949-7. [DOI] [PubMed] [Google Scholar]
- Bilodeau J., Schwendt M. Post-cocaine changes in regulator of G-protein signaling (RGS) proteins in the dorsal striatum: Relevance for cocaine-seeking and protein kinase C-mediated phosphorylation. Synapse. 2016;70:432–440. doi: 10.1002/syn.21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop G., Cullinan W., Curran E., Gutstein H. Abused drugs modulate RGS4 mRNA levels in rat brain: comparison between acute drug treatment and a drug challenge after chronic treatment. Neurobiol Dis. 2002;10:334–343. doi: 10.1006/nbdi.2002.0518. [DOI] [PubMed] [Google Scholar]
- Boehm J., Kang M.G., Johnson R.C., Esteban J., Huganir R.L., Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Chartoff E.H., Connery H.S. It’s MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system. Front Pharmacol. 2014;5:116. doi: 10.3389/fphar.2014.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R.L., Jr, Pradhan S.N., Dillon-Carter O., Chuang D.M. Rebound increase of basal cAMP level in NG108-15 cells during chronic morphine treatment: effects of naloxone and chloramphenicol. Life Sci. 1989;44:1107–1116. doi: 10.1016/0024-3205(89)90338-x. [DOI] [PubMed] [Google Scholar]
- Das S., Grunert M., Williams L., Vincent S.R. NMDA and D1 receptors regulate the phosphorylation of CREB and the induction of c-fos in striatal neurons in primary culture. Synapse. 1997;25:227–233. doi: 10.1002/(SICI)1098-2396(199703)25:3<227::AID-SYN1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Derkach V., Barria A., Soderling T.R. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias R.B., Ribeiro J.A., Sebastiao A.M. Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. Hippocampus. 2012;22:276–291. doi: 10.1002/hipo.20894. [DOI] [PubMed] [Google Scholar]
- Ding J., Guzman J.N., Tkatch T., Chen S., Goldberg J.A., Ebert P.J., Levitt P., Wilson C.J., Hamm H.E., Surmeier D.J. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- Dobi A., Seabold G.K., Christensen C.H., Bock R., Alvarez V.A. Cocaine-induced plasticity in the nucleus accumbens is cell specific and develops without prolonged withdrawal. J Neurosci. 2011;31:1895–1904. doi: 10.1523/JNEUROSCI.5375-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez D., Nunez C., Laorden M.L., Milanes M.V. Regulation of dopaminergic markers expression in response to acute and chronic morphine and to morphine withdrawal. Addict Biol. 2016;21:374–386. doi: 10.1111/adb.12209. [DOI] [PubMed] [Google Scholar]
- Gaspari S., Papachatzaki M.M., Koo J.W., Carr F.B., Tsimpanouli M.E., Stergiou E., Bagot R.C., Ferguson D., Mouzon E., Chakravarty S., Deisseroth K., Lobo M.K., Zachariou V. Nucleus accumbens-specific interventions in RGS9-2 activity modulate responses to morphine. Neuropsychopharmacology. 2014;39:1968–1977. doi: 10.1038/npp.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeders N.E., Lane J.D., Smith J.E. Self-administration of methionine enkephalin into the nucleus accumbens. Pharmacol Biochem Behav. 1984;20:451–455. doi: 10.1016/0091-3057(84)90284-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez P., Cabello P., Germany A., Norris B., Contreras E. Decrease of tolerance to, and physical dependence on morphine by, glutamate receptor antagonists. Eur J Pharmacol. 1997;332:257–262. doi: 10.1016/s0014-2999(97)01099-6. [DOI] [PubMed] [Google Scholar]
- Grillet N., Pattyn A., Contet C., Kieffer B.L., Goridis C., Brunet J.F. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25:4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.H., Renthal W., Ring R.H., Rahman Z., Psifogeorgou K., Howland D., Birnbaum S., Young K., Neve R., Nestler E.J., et al. Brain region specific actions of regulator of G protein signaling 4 oppose morphine reward and dependence but promote analgesia. Biol Psychiatry. 2010;67:761–769. doi: 10.1016/j.biopsych.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M., Mishina M., Ueda H. Locus-specific rescue of GluRepsilon1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci. 2003;23:6529–6536. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaw S.P., Makimura M., Oh K.W., Hoskins B., Ho I.K. Involvement of kappa-opioid receptors in opioid dependence/withdrawal: studies using butorphanol. Eur J Pharmacol. 1994;257:153–160. doi: 10.1016/0014-2999(94)90707-2. [DOI] [PubMed] [Google Scholar]
- Jedynak J., Hearing M., Ingebretson A., Ebner S.R., Kelly M., Fischer R.A., Kourrich S., Thomas M.J. Cocaine and amphetamine induce overlapping but distinct patterns of AMPAR plasticity in nucleus accumbens medium spiny neurons. Neuropsychopharmacology. 2016;41:464–476. doi: 10.1038/npp.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Yang M., Son Y., Jang H., Kim D., Kim J.C., Kim S.H., Kang M.J., Im H.I., Shin T., et al. Glial activation with concurrent up-regulation of inflammatory mediators in trimethyltin-induced neurotoxicity in mice. Acta Histochem. 2014;116:1490–1500. doi: 10.1016/j.acthis.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Kim J., Ham S., Hong H., Moon C., Im H.I. Brain reward circuits in morphine addiction. Mol Cells. 2016;39:645–653. doi: 10.14348/molcells.2016.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerea L.S., McNamara J.O. Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signaling pathways. Neuron. 1993;10:31–41. doi: 10.1016/0896-6273(93)90239-n. [DOI] [PubMed] [Google Scholar]
- Levitt P., Ebert P., Mirnics K., Nimgaonkar V.L., Lewis D.A. Making the case for a candidate vulnerability gene in schizophrenia: Convergent evidence for regulator of G-protein signaling 4 (RGS4) Biol Psychiatry. 2006;60:534–537. doi: 10.1016/j.biopsych.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Liang J., Li Y., Ping X., Yu P., Zuo Y., Wu L., Han J.S., Cui C. The possible involvement of endogenous ligands for mu-, delta- and kappa-opioid receptors in modulating morphine-induced CPP expression in rats. Peptides. 2006;27:3307–3314. doi: 10.1016/j.peptides.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Liu Z., Zhang J.J., Liu X.D., Yu L.C. Inhibition of CaMKII activity in the nucleus accumbens shell blocks the reinstatement of morphine-seeking behavior in rats. Neurosci Lett. 2012;518:167–171. doi: 10.1016/j.neulet.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lomazzi M., Slesinger P.A., Luscher C. Addictive drugs modulate GIRK-channel signaling by regulating RGS proteins. Trends Pharmacol Sci. 2008;29:544–549. doi: 10.1016/j.tips.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni O.J., Williams J.T. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G., Ahmed S.H., Blank T., Spiess J., Koob G.F., Siggins G.R. Chronic morphine treatment alters NMDA receptor-mediated synaptic transmission in the nucleus accumbens. J Neurosci. 1999;19:9081–9089. doi: 10.1523/JNEUROSCI.19-20-09081.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moron J.A., Gullapalli S., Taylor C., Gupta A., Gomes I., Devi L.A. Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology. 2010;35:955–966. doi: 10.1038/npp.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray F., Harrison N.J., Grimwood S., Bristow L.J., Hutson P.H. Nucleus accumbens NMDA receptor subunit expression and function is enhanced in morphine-dependent rats. Eur J Pharmacol. 2007;562:191–197. doi: 10.1016/j.ejphar.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Nestler E.J. Molecular basis of long-term plasticity underlying addiction. Nature reviews neuroscience. 2001;2:119. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Nestler E.J. Historical review: Molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Ohnishi H., Murata Y., Okazawa H., Matozaki T. Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011;34:629–637. doi: 10.1016/j.tins.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Parsegian A., See R.E. Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology. 2014;39:811–822. doi: 10.1038/npp.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik P., Kolasiewicz W. Mesolimbic NMDA receptors are implicated in the expression of conditioned morphine reward. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:288–294. doi: 10.1007/pl00005354. [DOI] [PubMed] [Google Scholar]
- Popik P., Wrobel M. Morphine conditioned reward is inhibited by MPEP, the mGluR5 antagonist. Neuropharmacology. 2002;43:1210–1217. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Robbe D., Bockaert J., Manzoni O.J. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–2235. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- Schwendt M., McGinty J.F. Regulator of G-protein signaling 4 interacts with metabotropic glutamate receptor subtype 5 in rat striatum: relevance to amphetamine behavioral sensitization. J Pharmacol Exp Ther. 2007;323:650–657. doi: 10.1124/jpet.107.128561. [DOI] [PubMed] [Google Scholar]
- Schwendt M., Gold S.J., McGinty J.F. Acute amphetamine down-regulates RGS4 mRNA and protein expression in rat forebrain: distinct roles of D1 and D2 dopamine receptors. J Neurochem. 2006;96:1606–1615. doi: 10.1111/j.1471-4159.2006.03669.x. [DOI] [PubMed] [Google Scholar]
- Schwendt M., Hearing M.C., See R.E., McGinty J.F. Chronic cocaine reduces RGS4 mRNA in rat prefrontal cortex and dorsal striatum. Neuroreport. 2007;18:1261–1265. doi: 10.1097/WNR.0b013e328240507a. [DOI] [PubMed] [Google Scholar]
- Schwendt M., Sigmon S.A., McGinty J.F. RGS4 overexpression in the rat dorsal striatum modulates mGluR5- and amphetamine-mediated behavior and signaling. Psychopharmacology. 2012;221:621–635. doi: 10.1007/s00213-011-2606-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Benedict Gomes A., Gallagher A., Stafford K., Yoburn B.C. Role of cAMP-dependent protein kinase (PKA) in opioid agonist-induced mu-opioid receptor downregulation and tolerance in mice. Synapse. 2000;38:322–327. doi: 10.1002/1098-2396(20001201)38:3<322::AID-SYN11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Snyder G.L., Allen P.B., Fienberg A.A., Valle C.G., Huganir R.L., Nairn A.C., Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans J.M., Leysen J.E., Langlois X. Striatal gene expression of RGS2 and RGS4 is specifically mediated by dopamine D1 and D2 receptors: clues for RGS2 and RGS4 functions. J Neurochem. 2003;84:1118–1127. doi: 10.1046/j.1471-4159.2003.01610.x. [DOI] [PubMed] [Google Scholar]
- Terzi D., Gaspari S., Manouras L., Descalzi G., Mitsi V., Zachariou V. RGS9-2 modulates sensory and mood related symptoms of neuropathic pain. Neurobiol Learn Mem. 2014;115:43–48. doi: 10.1016/j.nlm.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo K.A., Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Valverde O., Mantamadiotis T., Torrecilla M., Ugedo L., Pineda J., Bleckmann S., Gass P., Kretz O., Mitchell J.M., Schütz G., et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology. 2004;29:1122–1133. doi: 10.1038/sj.npp.1300416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.