

Abstract
Peroxisome proliferators induce hepatic peroxisome proliferation and hepatocellular carcinomas in rodents. These chemicals increase the expression of the peroxisomal β-oxidation pathway and the cytochrome P-450 4A family, which metabolizes lipids, including fatty acids. Mice lacking fatty acyl-CoA oxidase (AOX−/−), the first enzyme of the peroxisomal β-oxidation system, exhibit extensive microvesicular steatohepatitis, leading to hepatocellular regeneration and massive peroxisome proliferation. To investigate proteins involved in peroxisome proliferation, we adopted a novel surface-enhanced laser desorption/ionization (SELDI) ProteinChip technology to compare the protein profiles of control (wild-type), AOX−/−, and wild-type mice treated with peroxisome proliferator, Wy-14,643. The results indicated that the protein profiles of AOX−/− mice were similar to the wild-type mice treated with Wy14,643, but significantly different from the nontreated wild-type mice. Using four different ProteinChip Arrays, a total of 40 protein peaks showed more than twofold changes. Among these differentially expressed peaks, a downregulated peak was identified as the major urinary protein in both AOX−/− and Wy14,643-treated mice by SELDI. The identification of MUP was further confirmed by two-dimensional electro-phoresis and liquid chromatography coupled tandem mass spectrometry (LC-MS-MS). This SELDI method offers several technical advantages for detection of differentially expressed proteins, including ease and speed of screening, no need for chromatographic processing, and small sample size.
Keywords: ProteinChip Array, SELDI, Mass spectrometry, Peroxisome proliferator, Peroxisome proliferator-activated receptor, Acyl-CoA oxidase, Major urinary protein
PEROXISOMES in liver parenchymal cells proliferate in response to structurally diverse nonmutagenic compounds designated as peroxisome proliferators (PP) (20–22). The induction of peroxisome proliferation is mediated by PP-activated receptor alpha (PPARα), a member of a group of transcription factors that regulate the expression of genes involved in lipid metabolism and adipocyte differentiation (21). In association with peroxisome proliferation, peroxisomal β-oxidation system enzymes, which consist of three enzymes, namely H2O2-generating fatty acyl-CoA oxidase (AOX), enoyl-CoA hydratase/l-3-hydroxyacyl-CoA dehydrogenase (l-bifunctional enzyme, l-PBE), and 3-ketoacyl-CoA thiolase (THL), are dramatically increased (13,21). Mice deficient in AOX (AOX−/−), the first and rate-limiting enzyme of this inducible peroxisomal β-oxidation pathway, initially exhibit extensive microvesicular fatty metamorphosis of liver parenchymal cells and inflammatory reaction, and further develop regenerative hepatocytes that display massive spontaneous peroxisome proliferation. Subsequently, liver tumors develop in AOX-deficient mice by 15 months of age, a result similar to sustained induction of PPARα by exogenous peroxisome proliferator (7,19). These results imply that the deletion of AOX leads to accumulation of endogenous ligands responsible for the transcriptional activation of PPARα in vivo. The magnitude of peroxisome proliferation occurring spontaneously in the liver of adult AOX−/− mice as a result of activation of PPARα by unmetabolized AOX substrates is comparable with that induced in the liver of wild-type mice exposed to exogenous peroxisome proliferators (4,21). However, due to the inactivation of AOX gene, the lipid metabolism pathway and associated biochemical events in AOX−/− mice are obviously different from wild-type mice exposed to peroxisome proliferators. Identifying those differences will be helpful to the understanding of the molecular base of this peroxisome proliferation-associated hepatocarcinogenesis.
Several gene expression-based methods, such as PCR-based gene expression screen, differential display, and subtraction hybridization, have been used to study the mechanism of peroxisome proliferation-associated immediate and delayed pleiotropic responses (1,5). More recently, cDNA microarray technology has been used to study the full spectrum of target genes that become activated in response to PPARα ligands in liver (4). These approaches are primarily focused on changes occurring at the mRNA level. Recent studies on yeast have revealed that it is necessary to determine the protein expression level directly as mRNA levels may or may not correlate with the protein level (9,12). A proteomics approach may provide information that could not be obtained at the RNA level, due to poor correlation between mRNA and protein levels or due to posttranslational modifications that may result in several isoforms generated from one mRNA (19). As part of our effort to elucidate the molecular mechanism of peroxisome proliferator-induced hepatocarcinogenesis, we used proteomics approaches to study global changes of protein expression and to identify protein targets involved in peroxisome proliferation.
In this study we employed the surface-enhanced laser desorption/ionization (SELDI) ProteinChip technology to determine the differential protein expression profile between AOX−/− mice and wild-type mice treated with Wy14,643, a potent peroxisome proliferator. This recently developed ProteinChip technology (Ciphergen Biosystems, Inc., Fremont, CA) couples the sensitive analytical capabilities of mass spectrometry with novel surface chemistry (8,14,16). SELDI allows for the accurate mass detection of hundreds of proteins simultaneously down to a level of a few femtomoles of protein (16). The key component of this technology, in addition to the mass spectrometer, is the ProteinChip Arrays, which contain 2-mm-diameter adsorptive sample spots. Each of these sample spots represents either chemically or biochemically based surfaces enhanced for affinity capture of a population of proteins of defined chemical nature. After removal of unbound proteins and interfering substances, the molecular masses of the proteins retained on the ProteinChip Array are determined by laser desorption time-of-flight mass spectrometry analysis, resulting in a “protein profile” that is specific to the sample condition. Differences in protein composition between different status are further analyzed by ProteinChip Software (Ciphergen Biosystems) (16). The SELDI ProteinChip Biology System has been used successfully in discovery of biomarkers, including bladder carcinoma (28), Alzheimer’s disease (2,6), and prostate cancer (30,31), etc. In this study, we applied SELDI ProteinChip technology in profiling mouse liver proteins and further identified major urinary protein (MUP) as a biomarker of peroxisome proliferation. By comparison with one-dimensional and two-dimensional gel electrophoresis approaches, the advantages and disadvantages of SELDI ProteinChip technology are also addressed.
MATERIALS AND METHODS
Animals
Wild-type [C57BL/6J0 and AOX-null (AOX−/−] (7) male mice, 3–4 months of age, were housed in a controlled environment with a 12-h light/dark cycle with free access to water and standard laboratory chow. For positive control, peroxisome proliferator Wy-14,643 (0.125%, W/W) was administered to wild-type mice in powdered diet ad libitum for 2 weeks. Control (wild-type) mice received 0.15 ml of dimethyl sulfoxide by gavage, which was used as the solvent for Wy-14,643. All animals were sacrificed by terminal anesthesia, and the liver tissue was kept at −80°C before preparation of protein samples by homogenization. All animal procedures used in this study were reviewed and approved by the Institutional Review Board for Animal Research of Northwestern University.
Protein Sample Preparation and Electrophoresis
Individual mouse livers were homogenized separately in 5 ml of homogenization buffer (10 mM Tris-HCl, pH 7.4, 0.25 M sucrose, 1 mM EDTA, 1 mM PMSF, 2.5 mg/ml Aprotinin, antipain, and leupeptin) on ice using a potter-type homogenizer with 15 strokes. The homogenates were pooled by treatment group and centrifuged at 25,000 × g for 15 min and the supernatants were collected. Protein concentrations of each group were determined using BCA protein assay (Pierce, Rockford, IL), and the final concentration was adjusted to 20 mg/ml using the homogenization buffer. The protein samples were aliquoted to 100 μl each and frozen at −80°C before use. For standard (1D) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, 75 μg of protein was loaded onto each lane of gels prepared by standard techniques or purchased from Invitrogen (Carlsbad, CA). For 2D gel analysis, the pooled samples were labeled by fluorescent dyes as described elsewhere (27). Briefly, 300 μg of protein from each pooled sample was diluted to a final volume of 200 (μl of labeling buffer (7 M urea, 2 M thiourea, 4% CHAPS in 10 mM HCl, pH 9.0). Three cyanine dyes (Cy2, Cy3, and Cy5, from Amersham Biosciences, Piscataway, NJ) were used to label three groups of the liver protein (Wt, AOX−/−, and Wy14,643 treated), respectively, in a ratio of 50 μg protein/200 pmol dye for 30 min. The reaction was terminated by 10 mM lysine and the labeled proteins were mixed together with the addition of Pharmalyte (pH 3–10) (Amersham Biosciences) to 0.5% and dithiothreitol to 10 mg/ml. The mixture was applied to Immobiline DryStrips (24 cm, pH 4–7 and pH 3–10, Amersham Biosciences) with a total of 120 KVH isoelectric focusing. The second dimension was carried out with 10% SDS-PAGE gels, and gel images were generated using 2920-2D Master Imager (Amersham Biosciences).
Protein Identification by MS
Bands from 1D gels and spots from 2D gels were processed for mass spectrometric analysis following a modified procedure originally developed by Shevchenko et. al. (26). Briefly, gel pieces from 1D and 2D gels stained with Coomassie or SyproRuby, respectively, were destained first by using two changes of equal volumes of 25 mM ammonium bicarbonate and 50% acetonitrile. Destained gel pieces were dried, then rehydrated in 25 mM ammonium bicarbonate buffer containing 12.5 μg/ml trypsin (Promega, Madison, WI) and incubated overnight at 37°C to generate tryptic fragments. Resulting tryptic peptides were extracted from the gels by one change of 25 mM ammonium bicarbonate and 50% acetonitrile and two changes of 5% formic acid and 50% acetonitrile. The extracts were then analyzed by micro liquid chromatography electrospray ionization tandem mass spectrometry (μ LC-ESI-MS/MS) as described by Yates et al. (10,32). Briefly, samples containing tryptic peptides were loaded onto a fritless 365 × 100 μm fused silica capillary (FSC) column, packed with 5 μm Zorbax XDB-C18 packing material (Agilent Technologies, Palo Alto, CA) at a length of 7 cm. During MS data collection, the flow rate at the tip was maintained at about 300 μl/min using a precolumn restriction column. The tryptic peptides from Coomassie-stained 1D gels were separated by a 30-min linear gradient of 0–60% solvent B (80% acetonitrile/0.02% heptafluorobutyric acid) whereas the peptides from SyproRuby-stained 2D gels were separated by a 60-min linear gradient of 0-60% solvent B. Separated peptides were electrosprayed and entered a LCQ Deca ion-trap mass spectrometer (ThermoFinnigan, San Jose, CA). Tandem mass spectra were automatically collected in data-dependent mode with the top three most abundant ions from a full MS scan. Resultant MS/MS spectra were used directly to search a mammalian subset of the NCBI nonredundant database with SpectrumMill (Millennium Pharmaceuticals, Inc. Cambridge, MA) database search engine.
SELDI Protein Analysis
Three types of ProteinChip Arrays, strong anion exchange (SAX2), weak cation exchange (WCX2), and immobilized metal affinity capture (IMAC3), all from Ciphergen Biosystems, were used for liver protein profiling according to the manufacture’s protocols. The IMAC3 arrays were chelated with nickel (IMAC3-Ni) or copper (IMAC3-Cu), respectively, for capturing metal-binding proteins. Mouse liver protein samples (20 μg/μl) were diluted to a final concentration of 1 μg/μl with respective array binding buffers (SAX2: 50 mM Tris-HCl, 0.05% Triton-100, pH 8.0; WCX2: 50 mM NaOAc, pH 4.5; IMAC3: 250 mM NaCl, 100 mM sodium phosphate, 0.05% Triton-100, pH 7.2). After equilibration with respective binding buffer, each array spot was loaded with 5 μl of the diluted sample and incubated at room temperature in a humidified environment for 30 min. The unbound sample was removed and the array was washed three times in a 15-ml Corning tube with 8 ml of the same binding buffer to remove nonspecific binding. The ProteinChip Array was quickly rinsed with 8 ml of 1 mM HEPES (pH 8) before air drying and each spot was loaded twice with 0.5 μl of saturated sinapinic acid solution (SPA) in 50% acetonitrile (v/v) containing 0.5% trifluoroacetic acid (v/v). The ProteinChip Arrays were then read by a Protein-Chip Reader (Ciphergen Biosystems).
The reader was calibrated with the “All-in-1” peptide standard (Ciphergen) and each spot was scanned for both low mass region (high mass to 30000 Da, optimized from 1000 to 20000 Da, starting laser intensity 255) and high mass region (high mass to 200000 Da, optimized from 25000 to 65000 Da, starting laser intensity 300). The TOF mass spectra were collected and analyzed using Ciphergen’s Protein-Chip Software 2.1b. Peaks with signal/noise ratio of at least 3 were considered meaningful. Each sample was duplicated in two spots in the same array and the experiments were repeated three times.
Identification of Major Urinary Protein (MUP) by SELDI ProteinChip System
The MUP was enriched from the original mouse liver lysate and semipurified using micro spin columns. To prepare micro spin columns, 100 μl of QAE Sephadex A-50 (Amersham Biosciences) pre-equilibrated with binding buffer (50 mM Na2HPO4, pH 7) was packed in Micro Bio-Spin columns (BIO-RAD, Hercules, CA). For each treatment group, 1.6 mg of the original liver lysate was diluted into 200 μl of binding buffer and loaded on a micro spin column. The binding was carried out for 15 min in a cold room with gentle shaking. Unbound proteins were washed from the column with the same binding buffer (3 washes, 200 μl each wash) to minimize contamination. The bound proteins were eluted with 200 μl of elution buffer (50 mM NaOAc) at pH 5, 4, and 3, respectively. For analysis, 1 μl of each fraction was loaded directly on a normal phase array (NP1, Ciphergen) spot and air dried. After addition of 0.5 μl SPA, the array was read in the ProteinChip Reader. Fractions eluted at pH 4 and pH 3 were further separated by 4–20% SDS-PAGE gels and an 18.7 kDa wild-type mouse-specific band was excised for in-gel tryptic digestion according to a protocol suggested by Ciphergen. The resultant tryptic peptides were resolved by the ProteinChip Reader and analyzed by the ProteinChip Software. The protein ID was obtained by searching the NCBI database using ProFound search engine (33).
RESULTS
Analysis of Mouse Liver Protein by SDS-PAGE Coupled With MS/MS
Acyl-CoA oxidase (AOX), along with enoyl-CoA hydratase/l-3-hydroxyacyl-CoA dehydrogenase (l-bifunctional enzyme, l-PBE) and 3-ketoacyl-CoA thiolase, form the peroxisomal β-oxidation system, which is responsible for oxidation of long chain and very long chain fatty acids (13). Mice lacking AOX (AOX−/−) display massive spontaneous peroxisome proliferation and further develop hepatocellular carcinomas (7). To investigate changes of protein expression involved in peroxisome proliferation, total liver protein of AOX-deficient mice was first compared with control (wild-type) and peroxisome proliferator Wy-14,643-treated mice by SDS-PAGE analysis. As shown in Figure 1, four major protein bands (b1–b4), showed reduced expression in both AOX-null mice and Wy14,643-treated mice when compared with control mice. On the other hand, the intensity of three abundant protein bands (b5, b6, and b7), with approximate molecular weight of 78, 45, and 26 kDa, respectively, were clearly increased in both AOX-null mice and Wy14,643-treated mice. LC-MS-MS analysis of these bands revealed protein IDs as shown in Table 1. Due to the nature of SDS-PAGE, b2, b3, and b7 were shown to contain multiple protein IDs. Alternation of protein expression in these three bands may be due to one of these identified proteins or a combination. Among those identified proteins, glutathione S-transferase (GST), peroxisomal bifunctional enzyme (PBE), and peroxisomal 3-ketoacyl-CoA thiolase have been reported previously to be related to peroxisome proliferation (11,15,17,21–25,29). Although the downregulation of carbamyl-phosphate synthetase 1 in peroxisome proliferator-treated mice was not reported before, this observation was consistent with the results from 2D gel-coupled LC-MS-MS analysis (data not shown). The SDS-PAGE coupled with the MS/MS approach is simple and straightforward. Yet, due to the limited resolution of SDS-PAGE separation, a different approach has to be considered for the analysis of complex samples.
Figure 1.
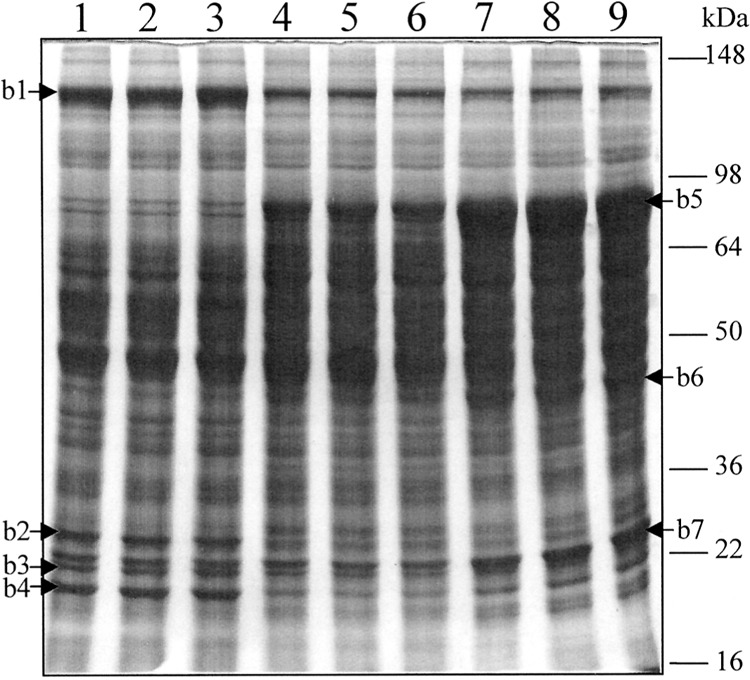
SDS-PAGE analysis of mouse liver proteins. Mouse liver homogenates (75 μg) were loaded to each lane of a 10% homogenous gel and stained with Coomassie Brilliant Blue R-250. Lanes 1–3: control (wild type); lanes 4–6: AOX−/−; lanes 7–9, wild-type treated with Wy14,643. Arrows indicate visible differentially expressed protein bands: b1–4, downregulated; b5–7: upregulated.
TABLE 1.
IDENTIFICATION OF DIFFERENTIALLY EXPRESSED PROTEINS BY LC-MS-MS FROM ONE-DIMENSIONAL GEL ELECTROPHORESIS OF MOUSE LIVER PROTEINS
| Band | MS/MS (# of Matched Peptides) | Database (NCBI) Accession # | MW (Da) | PI | Protein Candidate |
|---|---|---|---|---|---|
| 1 | 12 | 8393186 | 164580.8 | 6.33 | carbamoyl-phosphate synthase mitochondrial precursor (CPSASE I) |
| 2 | 6 | 10717134 | 29366.4 | 6.89 | carbonic anhydrase 3 |
| 2 | 7106389 | 27855.0 | 8.59 | proteasome subunit alpha type 7 (proteasome subunit RC6-1) | |
| 3 | 3 | 11120720 | 24664.8 | 4.35 | membrane associated progesterone receptor component (acidic 25-kDa protein) (25-DX) |
| 2 | 13879286 | 22197.4 | 6.49 | similar to biliverdin reductase B (flavin reductase (NADPH)) | |
| 4 | 8 | 10092608 | 23609.3 | 7.69 | glutathione S-transferase P 1 (GST Class-PI) |
| 5 | 23 | 12836375 | 78243 | 9.26 | peroxisomal bifunctional emzyme (PBE) includes: enoyl-CoA hydratase; 3,2-trans-enoyl-CoA iosomerase; 3-hydroxyacyl-CoA dehydrogenase |
| 6 | 7 | 6978429 | 44839.6 | 8.33 | peroxisomal 3-ketoacyl-CoA thiolase |
| 7 | 14 | 6754084 | 25970.2 | 7.72 | glutathione S-transferase GT8.7 (GST 1-1) (GST Class-MU) |
| 6 | 68423 | 26712.8 | 6.90 | triosephosphate iosomerase (TIM) | |
| 5 | 3219774 | 24870.8 | 5.71 | antioxidant protein2 |
Profiling of Mouse Liver Protein by SELDI ProteinChip Biology System
The SELDI ProteinChip Biology System combines protein capture on arrayed surfaces with direct detection by mass spectrometry. SELDI ProteinChip System has been used for analysis of various types of biological samples (14,16). To explore the suitability of this system for mouse liver protein profiling and further identify protein targets involved in peroxisome proliferation, liver sample from control mice was first used to establish experimental conditions. Liver lysates of control mice containing 1, 2.5, 5, and 10 μg of protein were loaded on SAX2, WCX2, IMAC3-Ni, and IMAC3-Cu array spots, respectively, with duplication. After stringent washes to eliminate nonspecifically bound proteins, retained proteins were analyzed by the ProteinChip Reader. Under the tested condition, 5 μg was the optimal protein load, which produced the best sensitivity and peak resolution. Figure 2 displays liver protein profiles of control mice captured by SAX2, WCX2, IMAC3-Ni, and IMAC3-Cu arrays in low mass range. With 5 μg of protein load, prominent peaks occurred between 5 and 20 kDa in low mass range and between 25 and 100 kDa in high mass range (data not shown). While the strong anion exchange (SAX2) array captured more peaks than the weak cation exchange (WCX2) array, the IMAC3-Ni affinity array captured more metal-binding proteins than IMAC3-Cu. A ∼15 kDa protein cluster was captured by IMAC3-Cu and IMAC3-Ni arrays as the dominant peak and appeared as a doubly charged peak with molecular mass of 7507. These results revealed distinct profiles of proteins captured by four different ProteinChip Arrays, indicating that different ProteinChip Array chemical surfaces allow differential capture of proteins according to their unique biochemical properties.
Figure 2.
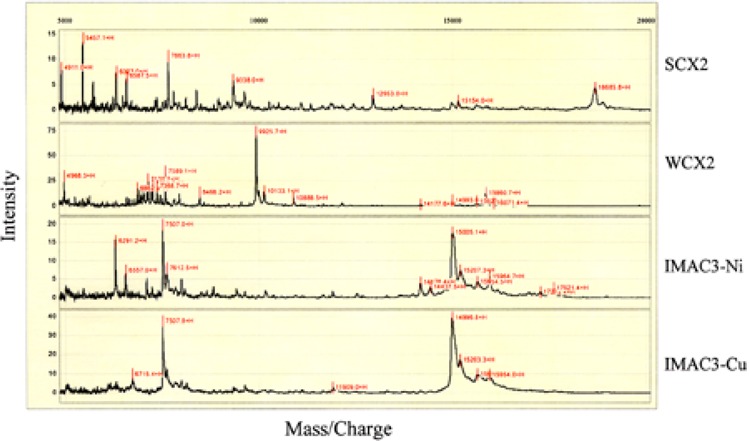
Surface-enhanced laser desorption/ionization (SELDI) time-of-flight (TOF) mass spectra of wild-type mouse liver proteins captured by different ProteinChip Array surfaces. SAX2: strong anion exchange; WCX2: weak cation exchange; IMAC3-Cu: immobilized metal affinity capture-nickel chaleted; IMAC3-Cu: immobilized metal affinity capture-copper chaleted.
SELDI-TOF Profiles of AOX-Null Mice Are Similar to Those of Wy14,643-Treated Mice
Similar to activation of PPARα by peroxisome proliferators, in AOX-null mice liver, some enzymes, such as peroxisomal β-oxidation enzymes, are markedly upregulated and certain others are down-regulated (4). Comparison of SELDI profiles of AOX-null mouse liver proteins with the profiles of control and Wy14,643-treated mice revealed a similarity between AOX−/− and Wy14,643-treated mice, but a significant difference from the control mice. Figure 3 displays SELDI-TOF profiles of SAX2 array and IMAC3-Cu array in selected mass ranges. Peaks, as indicated by arrows at the molecular mass of 18685, 59434, 15924, and 78352 Da, respectively, were found to be substantially different. The 18685-Da peak of the control mice captured by the SAX2 array was not detectable in AOX−/− and Wy14,643-treated mice, representing one of the downregulated proteins. In contrast, the 15924- and 78352-Da peaks detected in IMAC3-Cu array, along with the 59434-Da peak found in SAX2 array, showed increased peak intensities in AOX−/− and Wy14,643-treated mice, indicating that these were the upregulated proteins. Notably, the 78352-Da upregulated protein captured by IMAC3-Cu array showed an elevated expression level in Wy14,643-treated mice compared with AOX-null mice. To further quantitatively analyze these SELDI profiles, the Biomarker Wizard feature of the ProteinChip Software was used to compare these captured protein peaks. As summarized in Table 2, a total of 40 peaks in the mass range between 4500 and 100,000 Da exhibited more than twofold difference among control, AOX−/−, and Wy14,643-treated mice. These 40 peaks represent a subset of proteins whose expression levels were altered in AOX−/− and/or Wy14,643-treated mice in comparison to the control mice. Among these 40 peaks, 15 of them were downregulated whereas 25 of them were upregulated. Although the trend of changes between AOX−/− and Wy14,643-treated mice was similar, the extent of repression or induction was not always the same. For example, the IMAC3-Cu h16 cluster, which was later identified as the peroxisomal bifunctional enzyme (data not shown), showed three times higher induction in Wy14,643-treated mice than in AOX-null mice. On the other hand, a few peaks, such as WCX11 and IMAC3-Cu17, showed roughly twice higher peak intensity in AOX−/− mice than in Wy14,643-treated mice. Nevertheless, these results revealed similarities of protein profiles between AOX-null and Wy14,643-treated mice, which were significantly different from the control mice. Of these differences found, majorities were in the 5000–20000-Da mass ranges.
Figure 3.
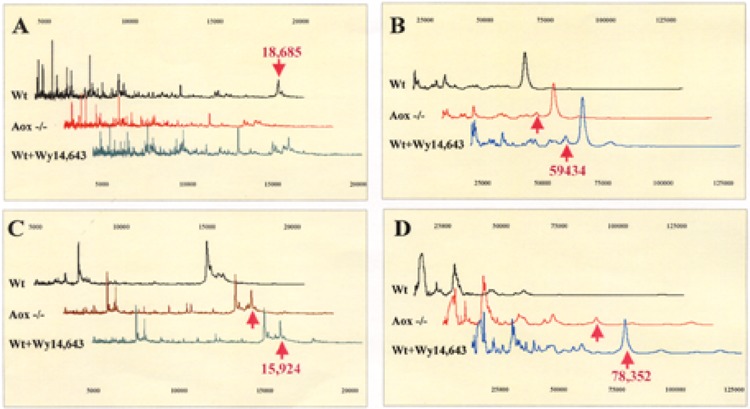
SELDI protein profiles of mouse liver proteins. (A) SAX2 array in low mass range; (B) SAX2 array in high mass range; (C) IMAC3-Cu array in low mass range; (D) IMAC3-Cu array in high mass range. Vertical direction: mass intensity; horizontal direction: mass charge.
TABLE 2.
DIFFERENTIALLY EXPRESSED PROTEIN CLUSTERS IDENTIFIED BY SELDI FROM SAX, WAX, IMAC-Cu, AND IMAC-Ni ARRAYS
| Cluster ID* | M/Z | Peak Intensity | Trend | ||
|---|---|---|---|---|---|
| wt | AOX−/− | wt + Wy | |||
| SAX1 | 4577 | 5.2 | — | – | down |
| SAX2 | 4651 | 9.4 | 1.3 | 1.9 | down |
| SAX10 | 9338 | 5.9 | 2 | 2 | down |
| SAX11 | 9358 | 4.7 | 0.7 | 0.6 | down |
| SAX13 | 9749 | 1.3 | 3 | 6.5 | up |
| SAX15 | 12954 | 3.1 | 3.4 | 7 | up |
| SAX16 | 14986 | 1.5 | 1.8 | 3.4 | up |
| SAX19 | 15920 | 0.7 | 1.5 | 4.4 | up |
| SAX20 | 18685 | 4.5 | – | – | down |
| SAXh10† | 45105 | 3.0 | 5.1 | 5 | up |
| SAXh14† | 59434 | 2.5 | 4.3 | 4.8 | up |
| SAXh16† | 84911 | 0.4 | – | \– | down |
| WCX2 | 3648 | 15.2 | 48.3 | 39.8 | up |
| WCX5 | 5451 | 7.7 | 31.9 | 28.1 | up |
| WCX8 | 6588 | 8.9 | 33.2 | 29.1 | up |
| WCX9 | 6645 | 8.1 | 33 | 31.3 | up |
| WCX11 | 6991 | 11.2 | 49 | 22.7 | up |
| WCX14 | 7589 | 34 | 20.4 | 16.5 | down |
| WCX21 | 9542 | 4.3 | – | – | down |
| WCX24 | 10134 | 14.8 | 22 | 46.3 | up |
| WCX25 | 10889 | 7.9 | 14.9 | 19.7 | up |
| WCX35 | 19948 | 4 | 8.7 | 14.4 | up |
| WCXh6 | 55822 | – | 0.8 | 0.4 | up |
| WCXh9 | 76601 | 0.5 | 1 | 0.7 | up |
| IMAC-Cu2 | 5816 | 3.3 | 1.3 | 0.8 | down |
| IMAC-Cu5 | 6716 | 9.8 | 7.6 | 4.6 | down |
| IMAC-Cu9 | 7993 | 6.5 | 24.9 | 22 | up |
| IMAC-Cu14 | 11117 | 1.5 | 7.4 | 4.8 | up |
| IMAC-Cu17 | 12378 | 0.4 | 9.2 | 4.7 | up |
| IMAC-Cu18 | 13777 | 0.7 | 5.2 | 4.5 | up |
| IMAC-Cu22 | 15924 | 9.5 | 22 | 20.3 | up |
| IMAC-Cu24 | 17812 | 0.6 | 2 | 4.1 | up |
| IMAC-Cuh16† | 78352 | 0.5 | 8.3 | 40 | up |
| IMAC-Cuh17† | 93529 | – | 1.8 | 4 | up |
| IMAC-Ni1 | 6293 | 15.2 | 11.6 | 2.4 | down |
| IMAC-Ni2 | 6558 | 7.4 | 4.1 | 1 | down |
| IMAC-Ni3 | 7097 | 4.8 | 2.4 | 1 | down |
| IMAC-Ni11 | 11912 | 1.6 | 1.5 | 3.4 | up |
| IMAC-Ni13 | 14179 | 4.1 | 2.1 | 0.4 | down |
| IMAC-Ni19 | 17626 | 2.5 | 1.6 | – | down |
| IMAC-Nih8† | 78367 | — | 0.9 | 10.2 | up |
The ID was assigned based on mass orders from low to high on each ProteinChip Array; peaks with greater than twofold changes are listed.
These peaks are from high mass range.
Comparison of SELDI Protein Profiles With SDS-PAGE Gel
To validate the SELDI profile, we converted mass spectrum data into a virtual gel format (gel view) by using the ProteinChip Software and then compared the gel view with SDS-PAGE results. Figure 4 shows the gel view of SAX2 and IMAC3-Cu profiles in comparison with a 4–20% gradient SDS-PAGE gel profile using the same samples. As arrows indicated, the 18.7-kDa downregulated and the 78-kDa upregu-lated proteins were captured by SAX2 and IMAC3-Cu arrays, respectively. Both proteins appeared in the same SDS-PAGE gel with expression patterns similar to those found in the ProteinChip Arrays. In contrast to the 78-kDa band, which was a prominent band in Wy14,643-treated mice samples, the 18.7-kDa band appeared as a faint band in the control mice samples. These results revealed a correlation between SELDI and SDS-PAGE gel profiles. The difference is, SDS-PAGE gel separates total proteins whereas the ProteinChip Array only captures a subset of proteins. Thus, if the SELDI ProteinChip Biology System is used to compare total protein profiles, combinations of different array surfaces with all possible binding and washing conditions will be necessary.
Figure 4.

Comparison of SELDI protein profiles with SDS-PAGE protein profile. (A) Virtue gel format (gel view) of SELDI protein profile in low mass range (15000–30000 Da) on SAX2 array. (B) Virtue gel format (gel view) of SELDI-TOF protein profile in high mass range (23000–100000 Da) on IMAC-Cu array. (C) 4–20% gradient SDS-PAGE gel loaded with 75 μg mouse liver protein. Lanes 1 and 2: wild-type; lanes 3 and 4: AOX−/−; lanes 5 and 6: wild-type treated with Wy14,643. Arrows indicate the differentially expressed proteins of 18.7 and 78 kDa identified by SELDI ProteinChip System and SDS-PAGE, respectively.
Identification of MPU by SELDI
In the SELDI-TOF profiling experiment, a total of 40 peaks exhibited more than twofold changes among control, AOX−/−, and Wy14,643 mice. In an attempt to assign biological significance to some of these differentially expressed proteins, the control (wild-type) mouse-specific 18.7-kDa protein captured on strong anion exchange (SAX2) array was chosen for protein identification. The fact that it binds to a strong anion exchange (SAX2) array surface indicates that the 18.7-kDa protein is negatively charged and can be purified by an anion exchange column. Hence, QAE Sephadex A-50 (Amersham Biosciences) resin was used to make micro spin columns. After preequilibration, mouse liver lysates from each treatment group were loaded on the micro spin columns and unbound proteins were washed away. Bound proteins were then eluted stepwise with buffers at descending pH. Direct SELDI-TOF analysis of the elutions at pH 3 from control, AOX-null, and Wy14,643-treated mice samples on NP1 array (Ciphergen) revealed a 18689-Da peak only presented in the control sample (Fig. 5A). Accordingly, SDS-PAGE analysis of the same elution revealed a control mouse-specific 18.7-kDa protein band (Fig. 5B). Although there were other proteins found in the same elution, clearly, the 18.7-kDa protein was enriched and other components were appreciably reduced compared with the total mouse liver proteins separated by the same gradient SDS-PAGE gels (Fig. 4C).
Figure 5.
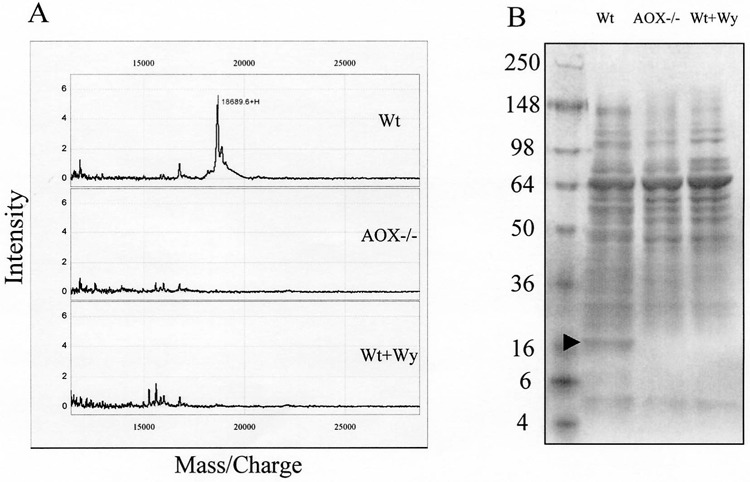
Partial purification and characterization of the PP-downregulated 18.7-kDa protein. Mouse liver homogenates of wild-type, AOX−/−, and wild-type treated with Wy14,643 were loaded to micro spin columns filled with QAE Sephadex A-50 resin. After washing to remove nonspecific banding, the bound proteins were eluted and subjected to SELDI-TOF protein profiling on NP1 array (A) or SDS-PAGE analysis (B). Note that the 18.7-kDa protein was found only in the wild-type mouse liver.
For protein identification, the enriched 18.7-kDa protein band was excised and half of the gel slice was subjected to in-gel digestion and protein identification using SELDI ProteinChip Biology System. According to Ciphergen’s User Guide, the resultant tryptic peptides were resolved by the ProteinChip Reader and analyzed by the ProteinChip Software. Using ProFound search engine to search the NCBI protein database, eight peptides from the tryptic digestion matched a 166-aa mouse major urinary protein (MUP) (3) with 54% coverage (Table 3). To confirm the SELDI result, the other half of the gel slice was subjected to LC-MS-MS analysis and the same protein identification was obtained (data not shown). In addition, the downregulation of mouse MUP in AOX-null and peroxisome proliferator-treated mice was also previously detected at the mRNA level (1,5). This evidence supports a conclusion that the detection and identification of MPU as a downregulated protein in AOX-null and Wy14,643-treated mice by SELDI ProteinChip Biology System are reliable.
TABLE 3.
PEPTIDE FRAGMENT FROM THE TRYPSIN DIGESTION OF THE PARTIALLY PURIFIED SAX20 PEAK MATCHED WITH MUP (NCBI ACCESSION #494384, SEARCHED BY PROFOUND)
| Measured Mass (M) | Avg/Mono | Computed Mass | Error (Da) | Residues | Missed Cut | Peptide Sequence | |
|---|---|---|---|---|---|---|---|
| Start | To | ||||||
| 1752.672 | A | 1752.991 | −0.319 | 19 | 33 | 1 | INGEWHTIILASDKR |
| 1894.572 | A | 1895.273 | −0.701 | 44 | 59 | 0 | LFLEQIHVLENSLVLK |
| 2009.362 | A | 2010.187 | −0.825 | 81 | 98 | 0 | AGEYSVTYDGFNTFTIPK |
| 2031.492 | A | 2030.250 | 1.243 | 150 | 166 | 2 | ENIIDLSNANRCLQARE |
| 2047.892 | A | 2049.209 | −1.317 | 1 | 18 | 1 | CVHAEEASSTGRNFNVEK |
| 2470.912 | A | 2471.727 | −0.815 | 114 | 135 | 1 | DGETFQLMGLYGREPDLSSDIK |
| 2486.562 | A | 2484.798 | 1.764 | 13 | 33 | 2 | NFNVEKINGEWHTIILASDKR |
| 3626.942 | A | 3627.997 | −1.055 | 1 | 32 | 2 | CVHAEEASSTGRNFNVEK INGEWHTIILASDK |
Characterization of MUP by 2D gel and LC-MS-MS
To further characterize MUP, mouse liver lysates from control, AOX-null, and Wy14,643-treated were analyzed by 2D gel electrophoresis. Figure 6 displays the results of which the first dimension was isoelectrically focused by pH 4–7 IPG strips and the second dimension was separated by 10% SDS-PAGE gels. Arrows indicate three protein spots named P1, P2, and P3, which were only seen in the control mice. The molecular weights of these three spots were the same, approximately 19 kDa. However, their pIs were slightly different, at about 4.8, 4.9, and 5.0, respectively. The expression pattern of these three spots along with their molecular weight and pI nature led to a suspicion that these three spots might be MUPs. Therefore, P1, P2, and P3 were excised from the 2D gel and analyzed by LC-MS-MS. Database searches indicated that these three spots, indeed, were different forms of MUP, as shown in Table 4. Interestingly, all three spots that were very well separated by 2D gels at pI ranging from 4 to 7 matched the same gene sequences of MUP1 in mouse and alpha-2u-globulin I precursor in rat with an exception that only P2 matched MUP2. There are three MUP mRNAs species (I, II, and III) found in mouse liver as a result of polymorphic multigene family (3). Although our LC-MS-MS identification was based on amino acid sequences, we were not able to obtain complete amino acid sequence information from these three MUP spots. It is not clear if the difference in pIs is due to different amino acid sequences or caused by posttranslational modifications. However, our results indicate that they are all downregulated in AXO −/− and Wy14,643-treated mice, suggesting these three MUP isoforms are regulated by the same mechanism.
Figure 6.
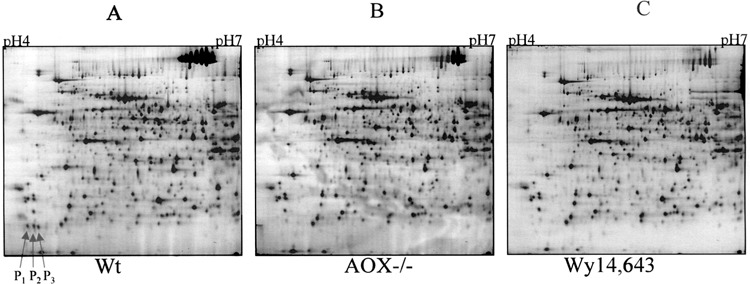
2D gel electrophoresis of mouse liver proteins. Mouse liver homogenates of wild-type (A), AOX−/− (B), and wild-type treated with Wy-14,643 (C) were first separated by isoelectric focusing (horizontal axis, pH 4–7) and further separated by 10% SDS-PAGE (vertical axis), which stretches from approximately 15 kDa (bottom) to about 120 kDa (top). Arrows indicate the PP-downregulated 18.7-kDa triple spots.
TABLE 4.
IDENTIFICATION OF THE “TRIPLE” SPOTS FROM 2D GEL BY LC-MS-MS
| Spot # | MS/MS (# of Matched Peptides) | MW (Da) | pI | Database (NCBI) Accession # | Protein Identity |
|---|---|---|---|---|---|
| P1 | 4 | 20649.5 | 4.89 | 72073 | alpha-2u-globulin I precursor or |
| 20648.6 | 5.04 | 127527 | major urinary protein 1 precursor (MUP 1) | ||
| P2 | 8 | 20663.6 | 5.04 | 127527 | MUP 2 or |
| 20648.6 | 5.03 | 13654245 | MUP 1 or | ||
| 20649.5 | 4.89 | 72073 | alpha-2u-globulin I precursor | ||
| P3 | 6 | 20648.6 | 5.03 | 13654245 | MUP 1 or |
| 20649.5 | 4.89 | 72073 | alpha-2u-globulin I precursor |
DISCUSSION
This study applied SELDI ProteinChip technology to examine liver protein profiles of wild-type, AOX−/−, and Wy14,643-treated mice. As an initial screen, SELDI-based analysis offers clear advantages over more conventional SDS-PAGE methodologies in that it is capable of easily and rapidly screening large numbers of biological samples, no initial processing (chromatographic and/or electrophoretic) is required and only a small sample volume is needed for each analysis.
Using SELDI technology and a standard panel of four different chemically based ProteinChip Arrays, 40 differentially expressed protein peaks were observed from liver extracts of wild-type and AOX−/− or Wy14,643-treated mice. The molecular masses of these differentially regulated species ranged from 4577 to 93529 Da, with the majority of them (80%) being less than 20000 Da. In contrast, using a 1D SDS-PAGE approach, only seven bands could be identified as differentially expressed proteins from the same samples. This difference may be explained in that SDS-PAGE is typically insensitive to proteins in the low mass range, the mass region where SELDI excels.
Using SELDI and SDS-PAGE as complementary technologies, the 18.7-kDa protein that was found to be downregulated in AOX−/− and Wy14,643-treated mice by SELDI analysis was enriched and semipurified by a predictive spin column approach. Based on the retention characteristics learned from SAX2 ProteinChip Arrays, the binding and elution characteristics were predicted in a preparative scale anionic exchange column. This provided an 18.7-kDa preparation that was simplified enough to allow for final purification on a 1D SDS-PAGE followed by identification by mass fingerprinting using the ProteinChip System. This protein was identified as MUP and further confirmed by subsequent LC-MS-MS analysis.
When comparing the SELDI ProteinChip Biology System and 2D SDS-PAGE coupled with LC-MS-MS, it becomes clear that the two technologies are complementary. Both technologies were able to distinguish the 18.7-kDa protein species as differentially regulated in wild-type vs. AOX−/− or Wy14,643-treated mice. Although SELDI analysis provided for rapid discovery, validation, and preparative purification leading to final identification, 2D-SDS-PAGE data further revealed that three different isoforms of MUP were present. Additionally, SELDI analysis provides exceptional screening potential in the low mass range, an area traditionally difficult by SDS-PAGE analysis.
In mouse urine, up to 15 MUP forms with pI ranging from 4.6 to 5.3 have been observed. Although different isoforms can be attributed to polymorphism of the MUP genes, the contribution of posttranslational modifications cannot be ruled out (3). It is not clear, at present, how these three forms of liver MUP correlate to the different isoforms of urine MUP. Once a correlation is established, MUP may serve as a prime biomarker of peroxisome proliferation in toxicology studies.
In conclusion, the data presented here demonstrate the applicability of SELDI for differential profiling of mouse liver proteins and the potential of this technique for the identification of marker proteins to aid in the prediction of toxicological effects during procedures such as drug development. As a proof of principle, we have identified one of the peaks in the SELDI protein profiles (i.e., that at 18.7 kDa) as MUP. Application of the SELDI ProteinChip technology to studies of peroxisome proliferation has the potential to advance this field, making SELDI ProteinChip technology an important predictive and diagnostic tool.
ACKNOLEDGMENTS
This work was partially supported by the National Institutes of Health Grant GM 23750 (to J.K.R.) and the Veterans Affairs Merit Review Grant (to A.V.Y.). We thank Dick Rubin and Lee Lomas for critical discussion.
REFERENCES
- 1. Alvares K.; Subbarao V.; Rao M. S.; Reddy J. K. Ciprofibrate represses alpha 2u-globulin expression in liver and inhibits d-limonene nephrotoxicity. Carcinogenesis 17:311–316; 1996. [DOI] [PubMed] [Google Scholar]
- 2. Austen B. M.; Frears E. R.; Davies H. The use of seldi proteinchip arrays to monitor production of Alzheimer’s betaamyloid in transfected cells. J. Pept. Sci. 6:459–469; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Cavaggioni A.; Mucignat-Caretta C. Major urinary proteins, alpha(2U)-globulins and aphrodisin. Biochim. Biophys. Acta 1482:218–228; 2000. [DOI] [PubMed] [Google Scholar]
- 4. Cherkaoui-Malki M.; Meyer K.; Cao W.; Latruffe N.; Yeldandi A. V.; Rao M. S.; Bradfield C.; Reddy J. K. Identification of novel peroxisome proliferator-activated receptor α (PPARα) target genes in mouse liver using cDNA microarray analysis. Gene Expr. 9:291–304; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dadras S. S.; Cook W. S.; Yeldandi A.V.; Cao W.; Rao M. S.; Wang Z.; Reddy J. K. Peroxisome proliferator-activated receptor α-dependent induction of cell surface antigen: Ly-6D gene in the mouse liver. Gene Expr. 9:173–181; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies H.; Lomas L.; Austen B. Profiling of amyloid beta peptide variants using SELDI ProteinChip arrays. Biotechniques 27:1258–1261; 1999. [PubMed] [Google Scholar]
- 7. Fan C. Y.; Pan J.; Usuda N.; Yeldandi A. V.; Rao M. S.; Reddy J. K. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J. Biol. Chem. 273:15639–15645; 1998. [DOI] [PubMed] [Google Scholar]
- 8. Fung E. T.; Wright G. L. Jr.; Dalmasso E. A. Proteomic strategies for biomarker identification: Progress and challenges. Curr. Opin. Mol. Ther. 2:643–650; 2000. [PubMed] [Google Scholar]
- 9. Futcher B.; Latter G. I.; Monardo P.; McLaughlin C. S.; Garrels J. I. A sampling of the yeast proteome. Mol. Cell. Biol. 19:7357–7368; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gatlin C. L.; Kleemann G. R.; Hays L. G.; Link A. J.; Yates J. R. Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal. Biochem. 263:93–101; 1998. [DOI] [PubMed] [Google Scholar]
- 11. Grasl-Kraupp B.; Waldhor T.; Huber W.; Schulte-Hermann R. Glutathione S-transferase isoenzyme patterns in different subtypes of enzyme-altered rat liver foci treated with the peroxisome proliferator nafenopin or with phenobarbital. Carcinogenesis 14:2407–2412; 1993. [DOI] [PubMed] [Google Scholar]
- 12. Gygi S. P.; Rochon Y.; Franza B. R.; Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19:1720–1730; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hashimoto T.; Fujita T.; Usuda N.; Cook W.; Qi C.; Peters J. M.; Gonzalez F. J.; Yeldandi A. V.; Rao M. S.; Reddy J. K. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase: Genotype correlation with fatty liver phenotype. J. Biol. Chem. 274:19228–19236; 1999. [DOI] [PubMed] [Google Scholar]
- 14. Johnston-Wilson N. L.; Bouton C. M.; Pevsner J.; Breen J. J.; Torrey E. F.; Yolken R. H. Emerging technologies for large-scale screening of human tissues and fluids in the study of severe psychiatric disease. Int. J. Neuropsychopharmacol. 4:83–92; 2001. [DOI] [PubMed] [Google Scholar]
- 15. Mehrotra K.; Morgenstern R.; Ahlberg M. B.; Georgellis A. Hypophysectomy and/or peroxisome proliferators strongly influence the levels of phase II xenobiotic metabolizing enzymes in rat testis. Chem. Biol. Interact. 122:73–87; 1999. [DOI] [PubMed] [Google Scholar]
- 16. Merchant M.; Weinberger S. R. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis 21:1164–1177; 2000. [DOI] [PubMed] [Google Scholar]
- 17. O’Brien M. L.; Cunningham M. L.; Spear B. T.; Glauert H. P. Effects of peroxisome proliferators on glutathione and glutathione-related enzymes in rats and hamsters. Toxicol. Appl. Pharmacol. 171:27–37; 2001. [DOI] [PubMed] [Google Scholar]
- 18. Pandey A.; Mann M. Proteomics to study genes and genomes. Nature 405:837–846; 2000. [DOI] [PubMed] [Google Scholar]
- 19. Qi C.; Zhu Y.; Pan J.; Usuda N.; Maeda N.; Yeldandi A. V.; Rao M. S.; Hashimoto T.; Reddy J. K. Absence of spontaneous peroxisome proliferation in enoyl-CoA hydratase/L-3-hydroxyacyl-CoA dehydrogenase-deficient mouse liver: Further support for the role of fatty acyl CoA oxidase in PPARalpha ligand metabolism. J. Biol. Chem. 274:15775–15780; 1999. [DOI] [PubMed] [Google Scholar]
- 20. Rao M. S.; Reddy J. K. Hepatocarcinogenesis of peroxisome proliferators. Ann. NY Acad. Sci. 804:573–587; 1996. [DOI] [PubMed] [Google Scholar]
- 21. Reddy J. K.; Chu R. Peroxisome proliferator-induced pleiotropic responses: Pursuit of a phenomenon. Ann. NY Acad. Sci. 804:176–201; 1996. [DOI] [PubMed] [Google Scholar]
- 22. Ren B.; Thelen A. P.; Peters J. M.; Gonzalez F. J.; Jump D. B. Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 272:26827–26832; 1997. [DOI] [PubMed] [Google Scholar]
- 23. Santagostino A.; Colleoni M.; Arias E.; Pacces Zaffaroni N.; Zavanella T. Changes in catalase, glutathione peroxidase and glutathione-S-transferase activities in the liver of newts exposed to 2-methyl-4-chlorophenoxyacetic acid (MCPA). Pharmacol. Toxicol. 65:136–139; 1989. [DOI] [PubMed] [Google Scholar]
- 24. Sato K.; Satoh K.; Tsuchida S.; Hatayama I.; Shen H.; Yokoyama Y.; Yamada Y.; Tamai K. Specific expression of glutathione S-transferase Pi forms in (pre) neoplastic tissues: Their properties and functions. Tohoku J. Exp. Med. 168:97–103; 1992. [DOI] [PubMed] [Google Scholar]
- 25. Schramm H.; Friedberg T.; Robertson L. W.; Oesch F.; Kissel W. Perfluorodecanoic acid decreases the enzyme activity and the amount of glutathione S-transferases proteins and mRNAs in vivo. Chem. Biol. Interact. 70:127–143; 1989. [DOI] [PubMed] [Google Scholar]
- 26. Shevchenko A.; Wilm M.; Vorm O.; Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858; 1996. [DOI] [PubMed] [Google Scholar]
- 27. Tonge R.; Shaw J.; Middleton B.; Rowlinson R.; Rayner S.; Young J.; Pognan F.; Hawkins E.; Currie I.; Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 1:377–396; 2001. [DOI] [PubMed] [Google Scholar]
- 28. Vlahou A.; Schellhammer P. F.; Medrinos S.; Kondylis F. I.; Gong L.; Nasim S.; Wright G. L. Jr. Development of a novel proteomic approach for the detection of transitional cell carcinoma of the bladder in urine. Am. J. Pathol. 158:1491–1501; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voskoboinik I.; Drew R.; Ahokas J. T. Differential effect of peroxisome proliferators on rat glutathione S-transferase isoenzymes. Toxicol. Lett. 87:147–155; 1996. [DOI] [PubMed] [Google Scholar]
- 30. Wang S.; Diamond D. L.; Hass G. M.; Sokoloff R.; Vessella R. L. Identification of prostate-specific membrane antigen (PMSA) as the target of monoclonal antibody 107-1A4 by ProteinChip array surface-enhanced laser desorption/ionization (SELDI) technology. Int. J. Cancer 92:871–876; 2001. [DOI] [PubMed] [Google Scholar]
- 31. Xiao Z.; Adam B. L.; Casares L. H.; Clements M. A.; Davis J. W.; Schhammer P. F.; Dalmasso E. A.; Wright G. L. Jr. Quantitation of serum prostate-specific membrane antigen by a novel protein biochip immunoassay discriminates benign from malignant prostate disease. Cancer Res. 61:6029–6033; 2001. [PubMed] [Google Scholar]
- 32. Yates J. R.; Carmack E.; Hays L.; Link A. J.; Eng J. K. Automated protein identification using microcolumn liquid chromatography-tandem mass spectrometry. Methods Mol. Biol. 112:553–569; 1999. [DOI] [PubMed] [Google Scholar]
- 33. Zhang W.; Chait B. T. ProFound: An expert system for protein identification using mass spectrometric peptide mapping information. Anal. Chem. 72:2482–2489; 2000. [DOI] [PubMed] [Google Scholar]