

Abstract
Voltage‐gated sodium (NaV) channel gating is a complex phenomenon which involves a distinct contribution of four integral voltage‐sensing domains (VSDI, VSDII, VSDIII and VSDIV). Utilizing accrued pharmacological and structural insights, we build on an established chimera approach to introduce animal toxin sensitivity in each VSD of an acceptor channel by transferring in portable S3b–S4 motifs from the four VSDs of a toxin‐susceptible donor channel (NaV1.2). By doing so, we observe that in NaV1.8, a relatively unexplored channel subtype with distinctly slow gating kinetics, VSDI–III participate in channel opening whereas VSDIV can regulate opening as well as fast inactivation. These results illustrate the effectiveness of a pharmacological approach to investigate the mechanism underlying gating of a mammalian NaV channel complex.
Keywords: scorpion toxin, spider toxin, NaV1.8, S3b‐S4 motif, voltage‐sensing domain, gating
Introduction
Within every phylum of the animal kingdom, voltage‐gated Na+ (NaV) channels are nature's answer to the need for intra‐organism communication and coordination, particularly when speed is a biological necessity (Hille, 2001). Utilizing the Na+ gradient across the cell membrane, these proteins generate electrical signals that telegraph messages throughout the organism (Ahern et al. 2016). As such, NaV channels support a myriad of critical physiological processes such as sensory perception, heart and brain function and muscle movement (George, 2005; Cannon, 2006; Catterall, 2012; Waxman et al. 2014). Structurally, eukaryotic NaV channels are large 24‐pass transmembrane proteins composed of four homologous domains (DI, II, III and IV) which form a pseudo‐fourfold symmetric channel encompassing a central Na+‐selective pore (segments 5–6; S5–S6) surrounded by four voltage sensors (segments 1–4; S1–S4), one from each domain (VSDI–IV) (Hille, 2001; Bezanilla, 2008; Ahern et al. 2016; Clairfeuille et al. 2016; Shen et al. 2017; Yan et al. 2017). Accruing data gleaned from a subset of the nine mammalian NaV channel subtypes (NaV1.1–NaV11.9) suggest that channel gate opening is driven by VSDI–III activation whereas the subsequent movement of VSDIV initiates fast and/or slow inactivation (Chen et al. 1996; Kontis & Goldin, 1997; Cha et al. 1999; Kuhn & Greeff, 1999; Sheets et al. 1999; Jurkat‐Rott et al. 2000; Mitrovic et al. 2000; Chanda & Bezanilla, 2002; Bosmans et al. 2008; Capes et al. 2013; Silva & Goldstein, 2013; Osteen et al. 2017). Under physiological conditions, channel opening is associated with membrane depolarization and action potential initiation whereas fast inactivation prevents channels from reopening for a short period of time, thereby allowing the unidirectional propagation of action potentials (Kandel et al. 2012).
The vital physiological role of NaV channels makes them a prime target for toxins produced by venomous animals which use these short peptides as a potent hunting tool or predator deterrent (Gilchrist et al. 2014). Researchers have greatly benefitted from toxins by taking advantage of their exquisite target specificity to elucidate structural and functional aspects of voltage‐gated ion channels or to explore their contribution to cellular excitability (Dutertre & Lewis, 2010; Kalia et al. 2015). In general, toxins can bind to the pore region to impede Na+ flux or they can interact with one or more VSDs to (1) inhibit channel opening; (2) induce channel opening at more negative voltages; or (3) delay fast inactivation to produce a persistent current (Bosmans & Swartz, 2010). Typically, gating‐modifier toxins bind to a specific region within VSDs, the S3b–S4 loop, a helix‐turn‐helix (paddle) motif that flexes in response to changes in membrane potential and makes few contacts with the rest of the channel protein (Li‐Smerin & Swartz, 2000; Jiang et al. 2003; Long et al. 2007; Chakrapani et al. 2008; Bosmans & Swartz, 2010; Xu et al. 2010; Payandeh et al. 2011; Martin‐Eauclaire et al. 2015; Ahern et al. 2016; Shen et al. 2017; Yan et al. 2017). Consequently, this loop can be transplanted into corresponding voltage‐gated K+ (KV) channel regions without disrupting the voltage‐sensing process (Alabi et al. 2007; Bosmans et al. 2008, 2011). Moreover, the resulting chimeric KV channels gain sensitivity to an array of NaV channel toxins, a powerful tool that can be used to discover novel ligands that target specific VSDs (Bende et al. 2014; Klint et al. 2015).
Here, we extend this chimera approach by swapping S3b–S4 loops between NaV channel subtypes (Fig. 1 A) and treating the S3b–S4 loop–toxin pair as a transferrable module to introduce toxin sensitivity. We employ this method to establish the role of individual VSDs in the gating process of NaV1.8, a slow‐inactivating NaV channel subtype involved in nociception (Waxman et al. 2014).
Figure 1. NaV channel S3b–S4 motifs and NaV1.8 current rundown.
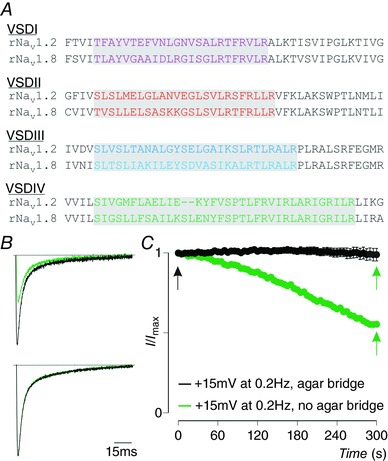
A, partial sequence alignment of VSDI–IV within rNaV1.2 and rNaV1.8 (used in this work) organized per domain. Transplantable regions are indicated in colour against a grey background. B, example of ∼50% NaV1.8 current rundown at +15 mV at a depolarizing pulse frequency of 0.2 Hz from a holding potential of −90 mV (top trace; black is first recording, green is current remaining after 5 min). Bottom trace shows no current rundown upon replacing the Ag+ electrodes with agar bridges (3 m NaCl). C, progression of NaV1.8 current rundown over a 5 min timeframe without (green) and with (black) agar bridges. Arrows indicate time points at which example traces shown in B were recorded. Error bars represent SEM, with n = 3.
Methods
Toxins and chemicals
ProTx‐II from the tarantula Thrixopelma pruriens and ATX‐II from the sea anemone Anemonia sulcata were acquired from Peptides International (Louisville, KY, USA) and Alomone Labs (Jerusalem, Israel), respectively. AaHII from the Androctonus australis hector scorpion and TsVII (or Ts1 or Tsγ) from the Tityus serrulatus scorpion were a gift from Marie‐France Martin‐Eauclaire and Pierre Bougis (University of Marseille, France). HaTx from the Grammostola rosea tarantula was a gift from Kenton Swartz (NIH/NINDS, USA). Purified toxins were kept at −20°C and aliquots were dissolved in appropriate solutions containing 0.1% (m V−1) BSA. Chemicals used were from Sigma‐Aldrich (USA) unless otherwise noted.
Two‐electrode voltage‐clamp recordings from Xenopus laevis oocytes
The cDNA sequence of rat (r)NaV1.2a, rNaV1.8, rβ1 (Origene, Rockville, MD, USA), rKV2.1, NaV1.x–KV2.1 VSD chimeras and mutant NaV channels was confirmed by automated DNA sequencing and cRNA was synthesized using T7 or SP6 polymerase (mMessage mMachine kit, Life Technologies, USA) after linearizing the DNA with applicable restriction enzymes. NaV channels were expressed with β1 (1:5 molar ratio) in Xenopus laevis oocytes (toads obtained from Xenopus one, Dexter, MI, USA) and studied following 2–4 days incubation after cRNA injection (incubated at 17°C in 96 mm NaCl, 2 mm KCl, 5 mm Hepes, 1 mm MgCl2 and 1.8 mm CaCl2, 50 μg ml−1 gentamycin, pH 7.6 with NaOH) using the two‐electrode voltage‐clamp recording technique (OC‐725C, Warner Instruments, Hamden, CT, USA) with a 150 μl recording chamber. Data were filtered at 4 kHz and digitized at 20 kHz using pClamp 10 software (Molecular Devices, Sunnyvale, CA, USA). Microelectrode resistances were 0.5–1.5 MΩ when filled with 3 m KCl. For NaV channel experiments, the external recording solution contained (in mm): 100 NaCl, 5 Hepes, 1 MgCl2 and 1.8 CaCl2, pH 7.6 with NaOH. For NaV1.x/KV2.1 chimera channel experiments, the external recording solution was (in mm): 50 KCl, 50 NaCl, 5 Hepes, 1 MgCl2 and 0.3 CaCl2, pH 7.6 with NaOH. All experiments were performed at ∼20°C. Leak and background conductances, identified by blocking the channel with agitoxin‐2 (gift from Kenton Swartz (NIH/NINDS, USA)), were subtracted for KV channel currents. The use of animals was in compliance with US NIH guidelines and was approved by the Johns Hopkins University Animal Care and Use Committee.
After addition of a toxin to the recording chamber, equilibration between channel and toxin was monitored using depolarizations at 5 s intervals. Voltage–activation curves were obtained by measuring steady‐state currents and calculating conductance for NaV channels or tail currents for KV channels, and a single Boltzmann function was fitted to the data according to I/I max = [1 + exp(−zF(V − V 1/2)/RT)]−1, in which I/I max is the normalized tail‐current amplitude, z is the equivalent charge, V 1/2 is the half‐activation voltage, F is Faraday's constant, R is the gas constant and T is temperature in kelvin. P values mentioned in the text result from a statistical analysis using the paired Student's t test, typically comparing control to toxin application. Data are presented as means ± SEM Off‐line data analysis was performed using Clampfit 10 (Molecular Devices, Sunnyvale, CA, USA), Origin 8.0 (Originlab, Northampton, MA, USA) and Microsoft Excel (Microsoft, Redmond, WA, USA).
Results
NaV1.8 as a candidate acceptor for NaV1.2 S3b–S4 regions
We focused on NaV1.8 as the recipient of previously defined NaV1.2 S3b–S4 loops because its gating properties are relatively unexplored compared to other channel subtypes; yet, macroscopic current kinetics are substantially slower than those from neuronal or muscle NaV channel isoforms (NaV1.1–NaV1.7) (Akopian et al. 1996; Zhang et al. 2017). Moreover, NaV1.8 is insensitive to most toxins available to us, thereby providing a suitable background for generating gain‐of‐function chimeras. Aside from low expression levels, NaV1.8‐mediated currents in oocytes suffer from an apparent rundown upon repeated electrical stimulation (Choi & Soderlund, 2004). Since this phenomenon typically does not occur when NaV1.8 currents are recorded from mammalian cells, we hypothesized that removing the Ag+ electrodes from the recording chamber by using agar bridges would resolve this issue. Indeed, NaV1.8 current reduction tends to stabilize at ∼50% after 5 min of depolarizing the membrane potential to +15 mV at 0.2 Hz from a holding potential of −90 mV (Fig. 1 B and C). In contrast, no rundown is observed with the same pulse protocol when using agar bridges, suggesting a possible partial channel block upon interaction of Ag+ with one of the species‐ and subtype‐specific cysteines in the S5–S6 pore‐forming regions (i.e. Cys814/832/1333). In subsequent NaV1.8 experiments, we therefore used agar bridges when recording from wild‐type (WT) and chimeric channels.
VSDI is involved in NaV1.8 opening
To determine the role of VSDI in NaV1.8 gating, we exploited high‐affinity ProTx‐II binding to NaV1.2 VSDI (Bosmans et al. 2008) by replacing this S3b–S4 loop sequence in NaV1.8 with that of NaV1.2 (Fig. 1 A) and measure changes in ProTx‐II sensitivity. (Chimeras are symbolized as follows; 8:8888 or 8:2888 where the number before the colon indicates the channel subtype (i.e. NaV1.8) and after the colon each number represents the VSD identity of origin (i.e. 8:2888; VSDI from NaV1.2 and VSDI–IV from NaV1.8).) The conductance (G)–voltage (V) and steady‐state inactivation (SSI) relationships of the 8:2888 chimera are comparable to that of the WT channel (8:8888) (Fig. 2). After treatment with 100 nm ProTx‐II, 8:2888 currents are dramatically reduced in a voltage‐dependent manner and replicate the toxin‐induced effect observed with WT NaV1.2 (2:2222) (Fig. 3). Thus, this experiment illustrates the role of VSDI in NaV1.8 opening.
Figure 2. Gating characteristics of NaV1.8 and NaV1.2–NaV1.8 S3b–S4 loop chimeras.
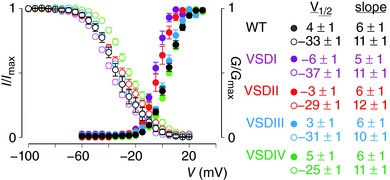
Shown are the G–V (G/G max) and SSI (steady‐state inactivation; I/I max) relationships of WT NaV1.8 (black) and the four S3b–S4 NaV1.2–NaV1.8 chimeras (VSDI–IV in magenta, red, blue and green, respectively). See Fig. 1 for sequence alignment. V 1/2 and slope values were obtained from a Boltzmann fit of the data. Error bars are SEM, with n = 4.
Figure 3. Effect of ProTx‐II on NaV1.2, NaV1.8 and the 8:2888 chimera.
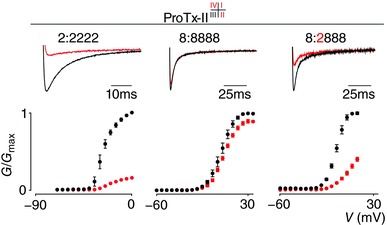
ProTx‐II interacts with VSDI, II and IV (see superscript summary after ‘ProTx‐II’) in NaV1.2 to inhibit channel opening (see 2:2222 column). Top shows a current trace whereas bottom depicts a G–V relationship before (black) and after (red) application of 100 nm ProTx‐II. A small inhibitory effect was observed on WT NaV1.8 (8:8888 column). In contrast, 100 nm ProTx‐II strongly inhibits opening of the 8:2888 chimera. Current traces shown were recorded at voltages near the foot of the G–V curve for each construct (holding potential was −90 mV with 5 s between depolarizing pulses). V 1/2 = −27 ± 1 mV (2:2222 black), −8 ± 1 mV (2:2222 red), 4 ± 1 mV (8:8888 black), 6 ± 1 mV (8:8888 red), −6 ± 1 mV (8:2888 black), 9 ± 1 mV (8:2888 red). Error bars represent SEM, with n = 3.
VSDII is coupled to NaV1.8 opening
In most NaV channel subtypes, VSDII is the prime target for a large number of animal toxins characterized thus far (Gilchrist et al. 2014). At 100 nm, ProTx‐II also interacts with the S3b–S4 loop in NaV1.2 VSDII to stabilize the voltage sensor in the closed state (Bosmans et al. 2008; Xiao et al. 2014). When transferring only this loop into the corresponding location in NaV1.8 (Figs 1 A and 2), 100 nm ProTx‐II impedes 8:8288 activation, thereby suggesting coupling between VSDII and channel opening (Fig. 4 A). In contrast to ProTx‐II, the β‐scorpion toxin TsVII is thought to stabilize VSDII of NaV1.2, but not NaV1.8, in an activated state, thus allowing the channel to open at more hyperpolarized voltages (Marcotte et al. 1997; Campos et al. 2007; Bosmans et al. 2008). Applying 100 nm TsVII to the 8:8288 chimera (Fig. 4 B) indeed recapitulates the effects seen on NaV1.2 (Bosmans et al. 2008; Gilchrist et al. 2013), an observation that further supports the role of NaV1.8 VSDII in channel opening.
Figure 4. Effect of ProTx‐II and TsVII on NaV1.2, NaV1.8 and the 8:8288 chimera.
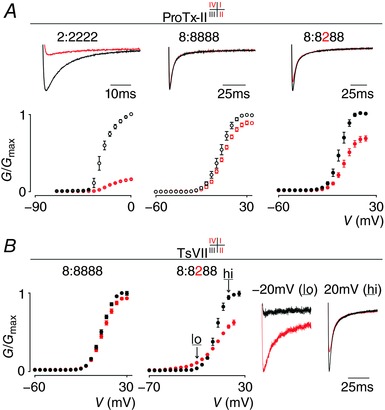
A, ProTx‐II interacts with VSDI, II and IV in NaV1.2 to inhibit channel opening. Top shows a current trace whereas bottom depicts a G–V relationship before (black) and after (red) application of 100 nm ProTx‐II. Left (2:2222) and middle (8:8888) panel were taken from Fig. 3 for comparison (indicated with open circles). Right panel shows that 100 nm ProTx‐II inhibits opening of the 8:8288 chimera. Current traces shown were recorded at voltages near the foot of the G–V curve (holding potential was −90 mV with 5 s between depolarizing pulses). V 1/2 = −4 ± 1 mV (8:8288 black), 0 ± 1 mV (8:8288 red). Error bars represent SEM, with n = 3. B, TsVII interacts with VSDII, III and IV in NaV1.2 to influence channel opening (Bosmans et al. 2008; Gilchrist et al. 2013) but does not affect NaV1.8 when 1 μm is applied (left panel). In contrast, 100 nm TsVII clearly influences the 8:8288 chimera (middle panel) by promoting channel opening at hyperpolarized voltages (‘lo’ at −20 mV) and inhibiting currents at depolarized voltages (‘hi’ at 20 mV). These effects are exemplified by current traces in the right panel (holding potential was −90 mV). V 1/2 = 5 ± 1 mV (8:8888 black), 6 ± 1 mV (8:8888 red), −1 ± 1 mV (8:8288 black), −7 ± 1 mV (8:8288 red). Error bars represent SEM, with n = 5.
VSDIII plays a role in NaV1.8 opening
To our knowledge, NaV channel VSDIII‐specific toxins have yet to be identified. One hypothesis for the lack of such peptides may be that nature found it more impactful to target VSDI, II, or IV to alter NaV channel function. Alternatively, toxin access to the VSDIII S3b–S4 region may be hampered by local lipid environment constraints (Lee & MacKinnon, 2004; Swartz, 2008; Milescu et al. 2009; Mihailescu et al. 2014; Gupta et al. 2015). To circumvent this resource gap, we employed TsVII which binds to VSDII, III and IV in NaV1.2 to hyperpolarize channel activation‐voltage threshold while inhibiting current at depolarized voltages (Bosmans et al. 2008). After substituting the endogenous VSDIII S3b–S4 loop in NaV1.8 with that of NaV1.2 (Fig. 1 A), we measured changes in susceptibility to 1 μm TsVII. The 8:8828 chimera is functional and the G–V and SSI relationship resemble that of 8:8888 (Figs 2 and 5). Upon application of 1 μm TsVII, 8:8888 function is not altered; however, 8:8828 opening is affected similarly to NaV1.2 and the 8:8288 chimera (Fig. 4 B). In contrast to the 8:8288 chimera, which is sensitive to 100 nm TsVII, the 8:8828 chimera requires 1 μm TsVII to trigger an effect, suggesting that NaV1.2 VSDIII is less susceptible to toxin binding.
Figure 5. Effect of TsVII on NaV1.8 and the 8:8828 chimera.
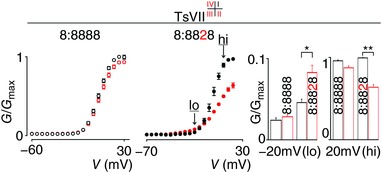
TsVII interacts with NaV1.2 VSDI–IV to alter channel opening (Bosmans et al. 2008; Gilchrist et al. 2013) but has no effect on NaV1.8 when 1 μm is applied (left panel, taken from Fig. 4 B, indicated with open circles). In contrast, 1 μm TsVII influences the 8:8828 chimera (middle panel) by mildly promoting channel opening at hyperpolarized voltages (‘lo’ at −20 mV) and inhibiting currents at depolarized voltages (‘hi’ at 20 mV). These effects are statistically significant and are quantified in the right panel (‘lo’ is 0.02 ± 0.01 (8:8888 black) and 0.03 ± 0.01 (8:8888 red), 0.05 ± 0.01 (8:8828 black) and 0.08 ± 0.01 (8:8828 red); ‘hi’ is 0.96 ± 0.01 (8:8888 black) and 0.87 ± 0.2 (8:8888 red), 0.99 ± 0.01 (8:8828 black) and 0.64 ± 0.03 (8:8828 red). V 1/2 = 5 ± 1 mV (8:8888 black), 6 ± 1 mV (8:8888 red), −1 ± 1 mV (8:8828 black), 4 ± 1 mV (8:8828 red). Error bars represent SEM, with n = 3; * P < 0.01, ** P < 0.001.
VSDIV regulates NaV1.8 inactivation and opening
Among the four NaV channel VSDs, VSDIV is unique because transferring its S3b–S4 loop into KV channels consistently slows channel kinetics when compared to constructs containing S3b–S4 motifs from VSDI–III (Bosmans et al. 2008, 2011; Bende et al. 2014). This observation fits the view that VSDIV plays a distinct role in inactivating the channel after it has opened (Sheets et al. 1999; Chanda & Bezanilla, 2002; Capes et al. 2013; Ahern et al. 2016). As a result, animal toxins that primarily target the VSDIV S3b–S4 region commonly impede fast inactivation (Gilchrist et al. 2014). To delineate the role of VSDIV in NaV1.8 gating, we substituted the endogenous S3b–S4 loop sequence in this VSD with that of NaV1.2 (Fig. 1 A) and measured changes in susceptibility to ATX‐II and AaHII, a sea anemone and α‐scorpion toxin that interact with NaV1.2 VSDIV (Rogers et al. 1996; Bosmans et al. 2008). The 8:8882 chimera is functional and the G–V and SSI relationship is similar to that of the WT channel (8:8888) (Figs 6 A and B and 2). Upon application of 100 nm ATX‐II or AaHII, 8:8888 is not affected; however, 8:8882 fast inactivation slows down substantially and a persistent current appears at the end of a 50 ms depolarizing test pulse. These effects are similar to those seen with NaV1.2 when applying 100 nm ATX‐II or AaHII (Fig. 6 A and B).
Figure 6. Effect of ATX‐II and AaHII on NaV1.2, NaV1.8 and the 8:8882 chimera.
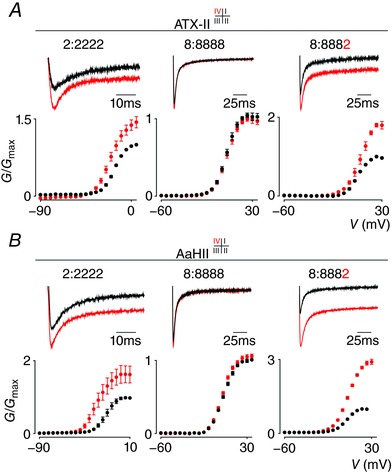
A, ATX‐II interacts exclusively with VSDIV in NaV1.2 to inhibit channel fast inactivation and increase currents over a wide voltage range (see 2:2222 column). Top shows a current trace whereas bottom depicts a G–V relationship before (black) and after (red) application of 100 nm ATX‐II. No effect was observed on WT NaV1.8 (8:8888 column). In contrast, 100 nm ATX‐II does inhibit fast inactivation of the 8:8882 chimera. V 1/2 = −16 ± 1 mV (2:2222 black), −21 ± 1 mV (2:2222 red), 4 ± 1 mV (8:8888 black), 4 ± 1 mV (8:8888 red), 8 ± 1 mV (8:8882 black), 5 ± 1 mV (8:8882 red). Current traces shown were recorded using a 50 ms voltage step near the foot of the G–V curve for each construct (holding potential was −90 mV with 5 s between depolarizing pulses). Error bars represent SEM from n = 3–5 measurements. B, AaHII (100 nm) also interacts with VSDIV in NaV1.2 (2:2222), but not NaV1.8 (8:8888), in a manner similar to ATX‐II. V 1/2 = −16 ± 1 mV (2:2222 black), −26 ± 1 mV (2:2222 red), 5 ± 1 mV (8:8888 black), 4 ± 1 mV (8:8888 red), 4 ± 1 mV (8:8882 black), 6 ± 1 mV (8:8882 red). Panel organization is identical to A with error bars representing SEM, with n = 4.
To further substantiate the critical role of VSDIV in NaV1.8 fast inactivation, we employed HaTx (100 nm) which does not affect WT NaV1.8 gating but primarily targets VSDI and VSDII in NaV1.2 to inhibit channel opening (Bosmans et al. 2008). At this concentration, HaTx also binds to VSDIV but since channel opening typically occurs before inactivation (i.e. open‐state inactivation), the principal effect on NaV1.2 is to stabilize the closed state. If NaV1.8 VSDIV is indeed coupled to fast inactivation, we expect HaTx to slow down this gating parameter upon transferring only the S3b–S4 region of NaV1.2 VSDIV into NaV1.8. Indeed, the 8:8882 chimera is sensitive to 100 nm HaTx and fast inactivation is strongly inhibited (Fig. 7).
Figure 7. Effect of HaTx on NaV1.2, NaV1.8 and the 8:8882 chimera.
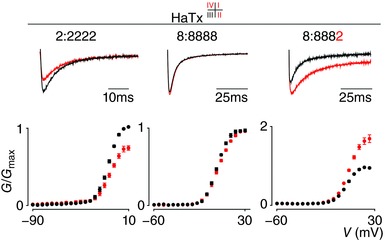
HaTx interacts with VSDI, II and IV in NaV1.2 to inhibit channel opening (see 2:2222 column). Top shows a current trace whereas bottom depicts a G–V relationship before (black) and after (red) application of 100 nm HaTx. No effect was observed on WT NaV1.8 (8:8888 column). In contrast, 100 nm HaTx inhibits fast inactivation of the 8:8882 chimera. Current traces shown were recorded using a 50 ms voltage step near the foot of the G‐‐V curve for each construct (holding potential was −90 mV with 5 s between depolarizing pulses). V 1/2 = −11 ± 1 mV (2:2222 black), −9 ± 1 mV (2:2222 red), 6 ± 1 mV (8:8888 black), 7 ± 1 mV (8:8888 red), 8 ± 1 mV (8:8882 black), 10 ± 1 mV (8:8882 red). Error bars represent SEM from n = 4 measurements.
Generally, VSDIV‐targeting toxins as well as (disease) mutations in this region hamper fast inactivation without noticeably disrupting channel opening (Ji et al. 1996; Rogers et al. 1996; Campos et al. 2008; Gilchrist et al. 2014). However, the inactivation gate may also close before the channel reaches a conducting state (i.e. closed‐state inactivation). These phenomena inspired the formulation of a model in which VSDIV movement occurs in two consecutive stages: (1) partial VSDIV activation associated with channel opening after either VSDI–II or VSDIII activates, and (2) full activation of VSDIV after which the inactivation particle is free to prevent further conduction (Bean, 1981; Aldrich & Stevens, 1983; Cha et al. 1999; Horn et al. 2000; Chanda & Bezanilla, 2002; Chanda et al. 2004; Armstrong, 2006). Similar to HaTx, prior work with chimeric KV2.1 channels revealed that ProTx‐II targets VSDI, VSDII and VSDIV of NaV1.2 (Bosmans et al. 2008; Xiao et al. 2014). As a result, 100 nm ProTx‐II strongly inhibits NaV1.2 opening whereas WT NaV1.8 is much less affected (Fig. 8 A). (Correspondingly, 100 nm ProTx‐II does not inhibit the four S3b–S4 NaV1.8–KV2.1 chimeras (Fig. 8 B).) When applying ProTx‐II to the 8:8882 chimera, we observe that the toxin binds to the channel to impede Na+ influx over a wide voltage range (Fig. 8). This result suggests that ProTx‐II may affect the first, partial VSDIV movement to decrease channel conductance whereas HaTx, AaHII and ATX‐II prevent full VSDIV activation resulting in fast inactivation inhibition.
Figure 8. Effect of ProTx‐II on NaV1.2, NaV1.8 and the 8:8882 chimera.
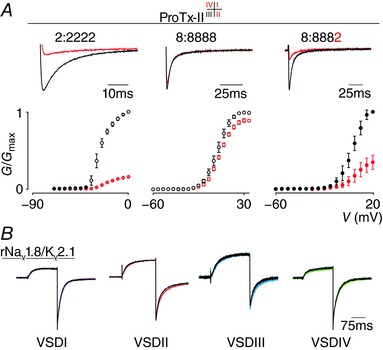
A, ProTx‐II interacts with VSDI, II and IV in NaV1.2 to hamper channel opening (see 2:2222 column). Top shows a current trace whereas bottom depicts a G–V relationship before (black) and after (red) application of 100 nm ProTx‐II. A minor inhibitory effect was observed on WT NaV1.8 (8:8888 column). In contrast, 100 nm ProTx‐II strongly inhibits opening of the 8:8882 chimera. Current traces shown were recorded at voltages near the foot of the G‐‐V curve for each construct (holding potential was −90 mV with 5 s between depolarizing pulses). Error bars represent SEM, with n = 3. B, shown is a representative example of the effect of 100 nm ProTx‐II on the four S3b–S4 NaV1.8–KV2.1 chimeras (VSDI–IV in magenta, red, blue and green, respectively). No significant inhibitory effect was observed.
Discussion
NaV channel gating is a multifaceted process in which VSDI–III and partial VSDIV activation is thought to contribute to channel opening whereas a subsequent VSDIV movement initiates inactivation (Armstrong, 2006; Ahern et al. 2016), resulting in a complex biophysical landscape. Here, we expand a previously established chimera approach to anatomize the role of individual VSDs in NaV channel gating (Alabi et al. 2007; Bosmans et al. 2008; Milescu et al. 2009; Bende et al. 2014; Klint et al. 2015; Osteen et al. 2016). Our pharmacological method comprises a binary module consisting of an animal toxin and its S3b–S4 loop target that can be transferred between NaV channel subtypes (Fig. 1) to activate or inhibit particular VSDs that may be coupled to channel opening or inactivation. A key asset of this approach is that swapping an S3b–S4 helix‐turn‐helix loop has little impact on expression or function of the chimeric channel because structural constraints are minimal (Bosmans et al. 2008; Shen et al. 2017; Yan et al. 2017). Moreover, animal toxins that target this region are highly specific and commonly bind with low nanomolar affinities (Gilchrist et al. 2014).
We used this approach to investigate the role of the four VSDs in the gating of NaV1.8, a relatively understudied NaV channel subtype involved in nociception, with slow kinetics and low expression levels in oocytes. By using agar bridges to record rat NaV1.8‐mediated currents from oocytes, we avoided the apparent rundown as a response to repeated electrical stimulation (Fig. 1). Upon transplanting previously defined S3b–S4 motifs from NaV1.2 into NaV1.8 (Bosmans et al. 2008, 2011), we were able to introduce sensitivity to a range of toxins from spider, scorpion and sea anemone venom and demonstrate that VSDI, II and III participate in channel opening (Figs 2, 3, 4, 5, 6, 7). In contrast, toxin‐mediated slowing of fast inactivation and ProTx‐II‐mediated inhibition of activation of the 8:8882 chimera suggest that VSDIV can mediate fast inactivation as well as opening of the channel, possibly because VSDIV movement occurs in two consecutive stages (Armstrong, 2006). Altogether, our results illustrate that transferring the S3b–S4 loop–toxin module to introduce toxin sensitivity can help elucidate the role of individual VSDs in ion channel gating, studies that have typically been conducted using a combination of electrophysiology and sophisticated fluorescence measurements (Chanda & Bezanilla, 2002; Pless et al. 2014; Varga et al. 2015). Propelled by emerging ion channel structural insights and the ongoing search for new VSD‐targeting toxins, there is reason to think that this chimera method can be refined further (e.g. VSDI‐ or VSDIII‐specific toxins) and that it could be used to probe the gating mechanisms of other voltage‐gated ion channel families that share similar features (Payandeh et al. 2011; Wu et al. 2015, 2016; Salari et al. 2016; Shen et al. 2017).
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
John Gilchrist and Frank Bosmans conceived the study, and designed and performed molecular biology and electrophysiological experiments. Both authors were involved in writing of the manuscript and approved the final version and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by a Ruth Kirschstein NIH predoctoral fellowship (F31NS084646 to J.G.), the Human Frontier Science Program Grant RGY0064/2013, a Synergy Award of the Johns Hopkins Medicine Discovery Fund, and the National Institutes of Health (R01NS091352 to F.B.).
Author's present address
J. Gilchrist: UCSF, 1550 4th St RH 482, San Francisco, CA 94158, USA.
Acknowledgements
We thank Al Goldin (UCIrvine, USA) for sharing rNaV1.2a, John Wood (UCL, UK) for rNaV1.8 and Kenton J. Swartz (NIH, USA) for sharing rKV2.1.
Biographies
John Michael Gilchrist graduated from Johns Hopkins University School of Medicine in 2017, receiving a PhD in physiology. His thesis work took place in the lab of Dr Frank Bosmans, where he used animal toxins to explore the interaction between voltage‐gated sodium channels and beta‐subunits. John is currently a postdoc in the laboratory of Lily Jan at UCSF.

Frank Bosmans received his Pharmacist degree and PhD in pharmaceutical sciences at the University of Leuven, Belgium. He continued working on voltage‐gated ion channel function and toxin pharmacology during his post‐doctoral studies with Dr Kenton Swartz at the National Institutes of Health. He is currently an Associate Professor in the Department of Physiology and Solomon H. Snyder Department of Neuroscience at Johns Hopkins University School of Medicine.
Edited by: Ole Petersen & Yasushi Okamura
This review was presented at the symposium ‘Shared and unique aspects of the gating mechanisms of ligand‐ and voltage‐gated ion channels’ which took place at IUPS 38th World Congress, Rio de Janeiro, Brazil, 1–5 August 2017.
Contributor Information
John Gilchrist, Email: johnmichael.gilchrist@ucsf.edu.
Frank Bosmans, Email: frankbosmans@jhmi.edu.
References
- Ahern CA, Payandeh J, Bosmans F & Chanda B (2016). The hitchhiker's guide to the voltage‐gated sodium channel galaxy. J Gen Physiol 147, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian AN, Sivilotti L & Wood JN (1996). A tetrodotoxin‐resistant voltage‐gated sodium channel expressed by sensory neurons. Nature 379, 257–262. [DOI] [PubMed] [Google Scholar]
- Alabi AA, Bahamonde MI, Jung HJ, Kim JI & Swartz KJ (2007). Portability of paddle motif function and pharmacology in voltage sensors. Nature 450, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW & Stevens CF (1983). Inactivation of open and closed sodium‐channels determined separately. Cold Spring Harb Sym 48, 147–153. [DOI] [PubMed] [Google Scholar]
- Armstrong CM (2006). Na channel inactivation from open and closed states. Proc Natl Acad Sci USA 103, 17991–17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (1981). Sodium‐channel inactivation in the crayfish giant‐axon ‐ must channels open before inactivating. Biophys J 35, 595–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bende NS, Dziemborowicz S, Mobli M, Herzig V, Gilchrist J, Wagner J, Nicholson GM, King GF & Bosmans F (2014). A distinct sodium channel voltage‐sensor locus determines insect selectivity of the spider toxin Dc1a. Nat Commun 5, 4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F (2008). How membrane proteins sense voltage. Nat Rev Mol Cell Biol 9, 323–332. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Martin‐Eauclaire MF & Swartz KJ (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F, Puopolo M, Martin‐Eauclaire MF, Bean BP & Swartz KJ (2011). Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J Gen Physiol 138, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F & Swartz KJ (2010). Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol Sci 31, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos FV, Chanda B, Beirao PS & Bezanilla F (2007). β‐Scorpion toxin modifies gating transitions in all four voltage sensors of the sodium channel. J Gen Physiol 130, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos FV, Chanda B, Beirao PSL & Bezanilla F (2008). Alpha‐scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J Gen Physiol 132, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC (2006). Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci 29, 387–415. [DOI] [PubMed] [Google Scholar]
- Capes DL, Goldschen‐Ohm MP, Arcisio‐Miranda M, Bezanilla F & Chanda B (2013). Domain IV voltage‐sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol 142, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2012). Voltage‐gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590, 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL Jr, Fujimoto E & Bezanilla F (1999). Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron 22, 73–87. [DOI] [PubMed] [Google Scholar]
- Chakrapani S, Cuello LG, Cortes DM & Perozo E (2008). Structural dynamics of an isolated voltage‐sensor domain in a lipid bilayer. Structure 16, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Asamoah OK & Bezanilla F (2004). Coupling interactions between voltage sensors of the sodium channel as revealed by site‐specific measurements. J Gen Physiol 123, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B & Bezanilla F (2002). Tracking voltage‐dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R & Kallen RG (1996). A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol 108, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS & Soderlund DM (2004). Cyclosporin A and deltamethrin block the downregulation of Nav1.8 sodium channels expressed in Xenopus oocytes. Neurosci Lett 367, 389–393. [DOI] [PubMed] [Google Scholar]
- Clairfeuille T, Xu H, Koth CM & Payandeh J (2016). Voltage‐gated sodium channels viewed through a structural biology lens. Curr Opin Struct Biol 45, 74–84. [DOI] [PubMed] [Google Scholar]
- Dutertre S & Lewis RJ (2010). Use of venom peptides to probe ion channel structure and function. J Biol Chem 285, 13315–13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL Jr (2005). Inherited disorders of voltage‐gated sodium channels. J Clin Invest 115, 1990–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist J, Das S, Van Petegem F & Bosmans F (2013). Crystallographic insights into sodium‐channel modulation by the beta 4 subunit. Proc Natl Acad Sci USA 110, E5016–E5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist J, Olivera BM & Bosmans F (2014). Animal toxins influence voltage‐gated sodium channel function. Handb Exp Pharmacol 221, 203–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Zamanian M, Bae C, Milescu M, Krepkiy D, Tilley DC, Sack JT, Yarov‐Yarovoy V, Il Kim J & Swartz KJ (2015). Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. Elife 4, e06774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ion Channels of Excitable Membranes, vol. 1 Sinauer Associates, Inc, MA, USA. [Google Scholar]
- Horn R, Ding S & Gruber HJ (2000). Immobilizing the moving parts of voltage‐gated ion channels. J Gen Physiol 116, 461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, George AL Jr, Horn R & Barchi RL (1996). Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSkM1 sodium channel gating. J Gen Physiol 107, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT & MacKinnon R (2003). X‐ray structure of a voltage‐dependent K+ channel. Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- Jurkat‐Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale‐Santos J, Chauveau D, Fontaine B & Lehmann‐Horn F (2000). Voltage‐sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 97, 11673–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia J, Milescu M, Salvatierra J, Wagner J, Klint JK, King GF, Olivera BM & Bosmans F (2015). From foe to friend: using animal toxins to investigate ion channel function. J Mol Biol 427, 158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA & Hudspeth AJ (2012). Principles of Neural Science. McGraw‐Hill Education/Medical. [Google Scholar]
- Klint JK, Smith JJ, Vetter I, Rupasinghe DB, Er SY, Senff S, Herzig V, Mobli M, Lewis RJ, Bosmans F & King GF (2015). Seven novel modulators of the analgesic target Nav1.7 uncovered using a high‐throughput venom‐based discovery approach. Br J Pharmacol 172, 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ & Goldin AL (1997). Sodium channel inactivation is altered by substitution of voltage sensor positive charges. J Gen Physiol 110, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn FJ & Greeff NG (1999). Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J Gen Physiol 114, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY & MacKinnon R (2004). A membrane‐access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 430, 232–235. [DOI] [PubMed] [Google Scholar]
- Li‐Smerin Y & Swartz KJ (2000). Localization and molecular determinants of the Hanatoxin receptors on the voltage‐sensing domains of a K+ channel. J Gen Physiol 115, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB & MacKinnon R (2007). Atomic structure of a voltage‐dependent K+ channel in a lipid membrane‐like environment. Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- Marcotte P, Chen LQ, Kallen RG & Chahine M (1997). Effects of Tityus serrulatus scorpion toxin gamma on voltage‐gated Na+ channels. Circ Res 80, 363–369. [DOI] [PubMed] [Google Scholar]
- Martin‐Eauclaire MF, Ferracci G, Bosmans F & Bougis PE (2015). A surface plasmon resonance approach to monitor toxin interactions with an isolated voltage‐gated sodium channel paddle motif. J Gen Physiol 145, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihailescu M, Krepkiy D, Milescu M, Gawrisch K, Swartz KJ & White S (2014). Structural interactions of a voltage sensor toxin with lipid membranes. Proc Natl Acad Sci USA 111, E5463–E5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu M, Bosmans F, Lee S, Alabi AA, Kim JI & Swartz KJ (2009). Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat Struct Mol Biol 16, 1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL & Horn R (2000). Role of domain 4 in sodium channel slow inactivation. J Gen Physiol 115, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X, Castro J, Garcia‐Caraballo S, Grundy L, Rychkov GY, Weyer AD, Dekan Z, Undheim EA, Alewood P, Stucky CL, Brierley SM, Basbaum AI, Bosmans F, King GF & Julius D (2016). Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Sampson K, Iyer V, Julius D & Bosmans F (2017). Pharmacology of the Nav1.1 domain IV voltage sensor reveals coupling between inactivation gating processes. Proc Natl Acad Sci USA 114, 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N & Catterall WA (2011). The crystal structure of a voltage‐gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Elstone FD, Niciforovic AP, Galpin JD, Yang R, Kurata HT & Ahern CA (2014). Asymmetric functional contributions of acidic and aromatic side chains in sodium channel voltage‐sensor domains. J Gen Physiol 143, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T & Catterall WA (1996). Molecular determinants of high affinity binding of alpha‐scorpion toxin and sea anemone toxin in the S3‐S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem 271, 15950–15962. [DOI] [PubMed] [Google Scholar]
- Salari A, Vega BS, Milescu LS & Milescu M (2016). Molecular interactions between tarantula toxins and low‐voltage‐activated calcium channels. Sci Rep 6, 23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Kyle JW, Kallen RG & Hanck DA (1999). The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J 77, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J & Yan N (2017). Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 355 eaal4326. [DOI] [PubMed] [Google Scholar]
- Silva JR & Goldstein SAN (2013). Voltage‐sensor movements describe slow inactivation of voltage‐gated sodium channels I: Wild‐type skeletal muscle Nav1.4. J Gen Phsyiol 141, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KJ (2008). Sensing voltage across lipid membranes. Nature 456, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z, Zhu W, Schubert AR, Pardieck JL, Krumholz A, Hsu EJ, Zaydman MA, Cui J & Silva JR (2015). Direct measurement of cardiac Na+ channel conformations reveals molecular pathologies of inherited mutations. Circ Arrhythm Electrophysiol 8, 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Merkies IS, Gerrits MM, Dib‐Hajj SD, Lauria G, Cox JJ, Wood JN, Woods CG, Drenth JP & Faber CG (2014). Sodium channel genes in pain‐related disorders: phenotype‐genotype associations and recommendations for clinical use. Lancet Neurol 13, 1152–1160. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q & Yan N (2016). Structure of the voltage‐gated calcium channel Cav1.1 at 3.6 Å resolution. Nature 537, 191–196. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M & Yan N (2015). Structure of the voltage‐gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Blumenthal K & Cummins TR (2014). Gating‐pore currents demonstrate selective and specific modulation of individual sodium channel voltage‐sensors by biological toxins. Mol Pharmacol 86, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ramu Y & Lu Z (2010). A shaker K+ channel with a miniature engineered voltage sensor. Cell 142, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J & Yan N (2017). Structure of the Nav1.4‐β1 complex from electric eel. Cell 170, 470–482.e11. [DOI] [PubMed] [Google Scholar]
- Zhang X, Priest BT, Belfer I & Gold MS (2017). Voltage‐gated Na+ currents in human dorsal root ganglion neurons. Elife 6, e23235. [DOI] [PMC free article] [PubMed] [Google Scholar]