

Abstract
Purpose
Our preclinical work identified depletion of ATR as a top candidate for topoisomerase 1 (TOP1) inhibitor synthetic lethality and showed that ATR inhibition sensitizes tumors to TOP1 inhibitors. We hypothesized that a combination of selective ATR inhibitor M6620 (previously VX-970) and topotecan, a selective TOP1 inhibitor, would be tolerable and active, particularly in tumors with high replicative stress.
Patients and Methods
This phase I study tested the combination of M6620 and topotecan in 3-week cycles using 3 + 3 dose escalation. The primary end point was the identification of the maximum tolerated dose of the combination. Efficacy and pharmacodynamics were secondary end points.
Results
Between September 2016 and February 2017, 21 patients enrolled. The combination was well tolerated, which allowed for dose escalation to the highest planned dose level (topotecan 1.25 mg/m2, days 1 to 5; M6620 210 mg/m2, days 2 and 5). One of six patients at this dose level experienced grade 4 thrombocytopenia that required transfusion, a dose-limiting toxicity. Most common treatment-related grade 3 or 4 toxicities were anemia, leukopenia, and neutropenia (19% each); lymphopenia (14%); and thrombocytopenia (10%). Two partial responses (≥ 18 months, ≥ 7 months) and seven stable disease responses ≥ 3 months (median, 9 months; range, 3 to 12 months) were seen. Three of five patients with small-cell lung cancer, all of whom had platinum-refractory disease, had a partial response or prolonged stable disease (10, ≥ 6, and ≥ 7 months). Pharmacodynamic studies showed preliminary evidence of ATR inhibition and enhanced DNA double-stranded breaks in response to the combination.
Conclusion
To our knowledge, this report is the first of an ATR inhibitor-chemotherapy combination. The maximum dose of topotecan plus M6620 is tolerable. The combination seems particularly active in platinum-refractory small-cell lung cancer, which tends not to respond to topotecan alone. Phase II studies with biomarker evaluation are ongoing.
INTRODUCTION
Chemotherapy is the standard treatment for a wide range of cancers, but despite initial responses, resistance invariably develops, and most patients die as a result of chemotherapy-resistant cancer. In part, this has been attributed to the presence of a highly effective DNA damage surveillance and repair network in tumors. The DNA damage response (DDR) is a complex signaling mechanism that coordinates activation of cell cycle checkpoints and the appropriate DNA repair pathways.1,2 DDR checkpoint kinases collectively maintain genomic integrity by providing cells time to repair DNA damage before replication or mitosis and initiates an apoptotic response if the damage is beyond repair.
ATR (ataxia-telangiectasia–mutated and rad3-related protein kinase) is a DDR master regulator and plays a key role in stabilizing the genome when DNA replication is compromised.2 ATR is activated by regions of single-stranded DNA, which may arise at DNA double-stranded breaks (DSBs) or as a result of replication stress induced by chemotherapeutic drugs or oncogene activation. In turn, ATR activates the cell cycle kinase Chk1 by phosphorylation, which suppresses the initiation of replication and elongation of replication forks. ATR-mediated S-phase arrest prevents cell division and promotes DNA damage repair, which thereby avoids additional DNA damage and maintains genomic stability.2 Small-molecule ATR inhibitors, therefore, have become attractive as cancer therapeutic agents to target cancer cells under replication stress that result from oncogene addiction, to increase the effectiveness of chemotherapeutic replication inhibitors,3-5 and to exploit defects in DDR mechanisms.6-11
ATR inhibition is particularly toxic in DNA damage–tolerant TP53-deficient cells, an effect also exacerbated by replication stress–inducing states, such as treatment with topoisomerase 1 (TOP1) inhibitors.5,12,13 The primary cytotoxic mechanism of TOP1 inhibitors in dividing cells is the generation of replication fork collisions that convert TOP1 cleavage complexes into irreversible DNA lesions.14 ATR and its downstream target Chk1 are critical for the DDR to TOP1 inhibitors. By using a synthetic lethal short interfering RNA screen, we previously identified depletion of ATR as a top candidate for TOP1 inhibitor synthetic lethality.5 We showed that inhibition of ATR by short interfering RNA or VE-821 and its clinical derivative VX-970 sensitizes tumor cells to TOP1 inhibitors. Mechanistically, VE-821 abrogates the S-phase replication elongation checkpoint and the replication origin-firing checkpoint induced by TOP1 cleavage complexes.5 M6620 (previously VX-970) is an ATP-competitive, highly potent, tightly binding inhibitor of ATR. M6620 blocks ATR with a concentration associated with 50% inhibition of 20 nM.15 In a phase I clinical trial, M6620 monotherapy was well tolerated with no dose-limiting toxicities (DLTs) at doses up to 480 mg/m2 administered weekly.16
We conducted a clinical trial of M6620 combined with topotecan, a highly selective inhibitor of TOP1,14 in patients with advanced solid tumors. The primary objective was to determine the recommended phase II dose by evaluating the feasibility, safety, adverse events, DLT, and maximum tolerated dose (MTD). Secondary objectives were to characterize the pharmacodynamics and to assess preliminary antitumor activity. Peripheral blood mononuclear cells (PBMCs), paired hair follicles, and tumor biopsy specimens obtained after topotecan and M6620 plus topotecan treatment were used to confirm DNA damage by quantifying phosphorylation of the histone protein H2AX (γH2AX). In addition, we studied modulation of topotecan-induced DNA damage and repair by M6620 by measuring markers of cellular response to DNA damage, including γH2AX and pNBS1 (phosphorylated Nijmegan breakage syndrome 1), in tumors.
PATIENTS AND METHODS
Patient Selection
Eligible patients had histologically confirmed metastatic or unresectable malignancy; one or more prior chemotherapy regimens; Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2; and adequate organ and marrow function. Details of patient selection are provided in the Data Supplement.
This trial was conducted under a National Cancer Institute (NCI) Center for Cancer Research–sponsored investigational new drug application with institutional review board approval. Written informed consent was obtained from enrolled patients.
Study Treatment and Design
Patients with advanced malignancies were administered M6620 plus topotecan intravenously (IV) in this open-label, single-arm, phase I study. M6620 was supplied under a Collaborative Research and Development Agreement among NCI, Vertex Pharmaceuticals, and Merck KGaA. Topotecan was obtained from commercial sources.
Treatment cycles were 21 days long (Fig 1). The starting dose of topotecan was 1 mg/m2 IV on days 1 to 5, a dose and schedule that is tolerable in heavily pretreated patients. M6620 was administered IV 15 minutes after completion of topotecan. At the first dose level (DL), a single dose of M6620 was administered at 140 mg/m2 on day 5, whereas at the subsequent DLs, M6620 was administered on days 2 and 5. Details of the study design are provided in the Data Supplement.
Fig 1.

Trial schema and dose escalation schedule. IV, intravenously.
Safety and Efficacy Evaluations
A history and physical examination were conducted at baseline and before each cycle. Complete blood counts and serum chemistries were performed at baseline and weekly thereafter. Radiographic evaluation was performed at baseline and every two cycles for tumor response on the basis of Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Toxicities were graded using NCI Common Terminology Criteria for Adverse Events (version 4.0).
Pharmacodynamics
γH2AX, a marker for the formation of DNA DSB, was assessed in PBMCs, hair follicles, and tumor biopsy specimens. Sampling was designed to assess the effect of topotecan alone (DL1) and topotecan plus M6620 (DL2 to DL4) on γH2AX formation. In addition, biopsy specimens were obtained on day 3, 24 hours after administration of MM6620 and the second dose of topotecan, and prior to administration of a third dose of topotecan. Multiparameter flow cytometric analysis of PBMCs was performed at baseline and before treatment on cycle 1 day 2, cycle 1 day 5, and cycle 2 day 1. Details are provided in the Data Supplement.
Statistical Methods
Differences in correlative parameters were formed by subtraction and tested for significance of the difference by using paired t test when differences were potentially a normal distribution and by using Wilcoxon signed rank test when differences were inconsistent with being from a normal distribution.
RESULTS
Patient Demographics
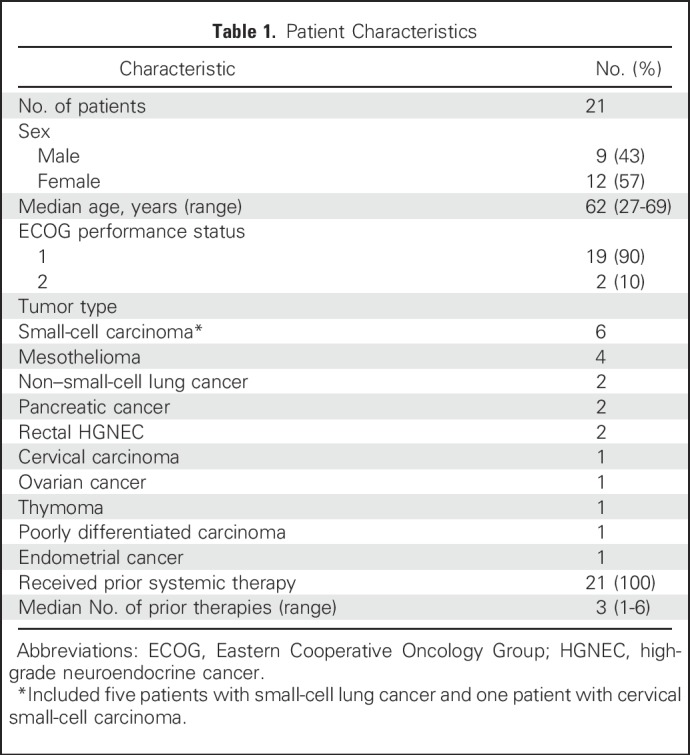
Twenty-one patients were enrolled between September 2016 and February 2017 (Table 1). All patients had received one or more lines of prior systemic therapy and had evidence of disease progression at enrollment. All patients completed at least one cycle (median, two cycles; range, one to 24 cycles) of treatment and were evaluable for MTD. Two patients were not evaluable for response because of rapidly progressive disease (PD).
Table 1.
Patient Characteristics
Dose Escalation and Determination of MTD and Recommended Phase II Dose
During dose escalation, three patients had DLTs (Table 2). One patient at DL1 experienced grade 4 neutropenia that lasted > 7 days, grade 4 thrombocytopenia that required transfusion, and grade 3 elevation of AST and ALT. This cohort was expanded with five additional patients. No other DLTs were observed at DL1. A second DLT, grade 4 thrombocytopenia that required transfusion and grade 3 febrile neutropenia, occurred at DL2, which resulted in expansion of this cohort with five additional patients. No DLTs were observed in the other patients. No DLTs were observed in the three patients treated at DL3. A third DLT, grade 4 thrombocytopenia that required transfusion, occurred at DL4, which resulted in expansion of this cohort with five additional patients. No DLTs were observed in the additional patients, which established DL4, the highest planned dose, as the MTD.
Table 2.
Most Common (≥ 10%) Treatment-Related Adverse Events (maximum grade, all cycles)

Toxicity
The combination of M6620 and topotecan was generally well tolerated (Table 2). The most common toxicities across all DLs (N = 21) were anemia and lymphopenia (100% each), leukopenia (90%), neutropenia (81%), thrombocytopenia (76%), and nausea (62%). The most common grade 3 and 4 toxicities were anemia, leukopenia, and neutropenia (19% each); lymphopenia (14%); and thrombocytopenia (10%).
Eleven patients required dose reductions for neutropenia (n = 6), thrombocytopenia (n = 2), decreased creatinine clearance (n = 5), hypophosphatemia (n = 1), or a combination of these. Eight patients required dose interruption most commonly for recovery of neutropenia (n = 5) and recovery of disease-related complications (bowel obstruction and hyperbilirubinemia). Treatment was interrupted in one patient for grade 2 aortic vasculitis that required high-dose corticosteroids. This event was considered unrelated to the study drugs and possibly related to growth factors. After corticosteroid taper, the patient continued with M6620 monotherapy without recurrence of vasculitis.
Pegfilgrastim was not routinely administered during the first cycle but was used from cycle 2 onward in eight patients and cycle 4 onward in one patient. No instances of neutropenia were observed in patients who received pegfilgrastim. Two patients, both treated at DL1, developed grade 1 infusion reactions to M6620. One patient was rechallenged with no additional reactions; another patient developed an infusion reaction during cycle 2. She elected to continue with topotecan alone, experienced a partial response (PR), and has continued with treatment after 24 cycles.
Efficacy
Of 19 evaluable patients, two had confirmed PR, eight had stable disease (SD), and nine had PD (Fig 2A). One patient with stage IVB endometrioid-endometrial carcinoma treated at DL1 had a PR that is continuing at 18 months (Fig 2B). M6620 was discontinued after the second cycle, during which she had an infusion reaction. Her CA-125 declined steeply after the first cycle, continued to decrease after the second cycle, and has remained low over the duration of treatment now with topotecan alone (Fig 2C). One patient with refractory small-cell lung cancer (SCLC) treated at DL4 had a PR that is ongoing at > 7 months.
Fig 2.

Efficacy in the overall population. (A) The waterfall plot shows responses in evaluable patients (n = 19). (B) Computed tomography scans from before and 6 weeks after treatment that show a partial response; area of tumor indicated by circle and (C) CA-125 trend in a patient with previously treated endometrial cancer. (D) The swimmer plot shows responses and durations of response in evaluable patients. (*) Patient came off treatment because of brain metastases. DL, dose level; HGNEC, high-grade neuroendocrine cancer; NSCLC, non–small-cell lung cancer; SCLC, small-cell lung cancer.
Seven of the eight patients with SD had prolonged SD (≥ 3 months; median, 9 months; range, 3 to 12 months; Fig 2D). These patients included one with rectal high-grade neuroendocrine cancer refractory to platinum plus etoposide and temozolomide plus capecitabine combinations who had SD that lasted > 9 months, one with squamous non–small-cell lung cancer with SD that lasted 6 months, and one with peritoneal mesothelioma with SD that is ongoing at 10 months.
Of five patients with SCLC, all of whom had platinum-refractory disease, three derived durable clinical benefit (one PR and two prolonged SD; Fig 3). All three were diagnosed with extensive-stage SCLC, had received four cycles of platinum and etoposide, and were found to have SD or near-complete responses to first-line therapy but had PD soon after first-line chemotherapy. With the current treatment, one patient with refractory SCLC developed a striking decrease in size and number of splenic metastases (Fig 3A) accompanied by a marked decrease in metabolic activity of the lesions, which is ongoing at 7 months. A second patient had SD as the best response (−25%), with a marked decrease of a left-sided adrenal mass (Fig 3B), which is maintained with treatment at 6 months. A third patient had −26% shrinkage in tumor and achieved marked improvement in cancer-related symptoms (Fig 3C). He came off treatment after 10 months because of brain metastases. The median progression-free survival of patients with SCLC (n = 5) was 10.2 months (95% CI, 1.4 to 10.2 months), and 6-month progression-free survival probability was 60.0% (95% CI, 12.6% to 88.2%).
Fig 3.

Tumor responses in patients with refractory small-cell lung cancer (SCLC). (A) Computed tomography (CT) scans from before treatment and at 5 months after treatment show partial response in a patient with refractory SCLC, with a striking decrease in size and number of splenic metastases accompanied by a marked decrease in metabolic activity of the lesions, which is ongoing at 7 months. (B) CT scans from before treatment and 6 months after treatment that show regression of a left-sided adrenal metastasis in a patient with refractory SCLC. Best tumor response in this patient was stable disease (−25% decrease in sum of target lesions), and the patient continues with treatment at 6 months. (C) CT scans from before and 9 months after treatment that show a stable subcarinal lymph node in a patient with refractory SCLC. Best tumor response in this patient was stable disease (−26% decrease in sum of target lesions). This patient continued with treatment for 10 months and discontinued because of brain metastases. Tumor areas are indicated by arrows.
Pharmacodynamics
PBMC and hair samples were obtained from all 21 patients (Fig 4A). Hair samples from 17 patients had follicles that could be analyzed. The number and intensity of γH2AX foci increased in follicles obtained 5 to 10 minutes after topotecan administration on day 2 from all patients, which indicated DNA DSBs after topotecan alone. Patients 7 to 21 who received M6620 plus topotecan on day 2 (Fig 4B, right) had significantly higher DNA damage levels on day 3 than on day 2, which was not the case for patients 1 to 6 who received topotecan alone on day 2 (Fig 4B, left).
Fig 4.

Pharmacodynamic assessment of γH2AX formation in plucked hair bulbs and peripheral blood mononuclear cells (PBMCs) after topotecan and M6620. (A) Diagram depicting the collection of PBMCs and hair follicles. All patients received topotecan on days 1, 2, and 3, with patients 7 to 21 receiving M6620 on day 2. Samples were collected before infusions of topotecan and M6620 (D1-pre), 5 to 30 minutes after day 2 topotecan (D2-post 1), 30 minutes to 1 hour after day 2 M6620 (D2-post 2), before day 3 topotecan (D3-post 1), and 5 minutes after day 3 topotecan (D3-post 2). (B) γH2AX in plucked hairs with all data plotted as γH2AX signal intensities. γH2AX induction in patients who received topotecan only (patients 1 to 6) shows DNA double-stranded breaks after topotecan alone (left; n = 6). Patients who received M6620 plus topotecan on day 2 (patients 7 to 21) had increased DNA damage levels on day 3 compared with day 2, which was not the case for those who received topotecan alone on day 2 (right; n = 12). To understand the kinetics of γH2AX formation further, we obtained hair follicles at additional time points: 30 minutes to 1 hour after day 2 M6620 infusion (D2-post 2) and before day 3 topotecan (D3-post 1). (C) γH2AX in plucked hairs with all data plotted as γH2AX signal intensities in six patients who had additional sampling. Although follicles obtained 30 to 60 minutes after M6620 infusion on day 2 showed a decrease in γH2AX signal, an increase in H2AX phosphorylation was observed in follicles obtained 24 hours after M6620 infusion (before any additional topotecan). (D) Representative images of γH2AX staining in plucked hair bulbs collected from patient 14 (green, γH2AX; red, DNA). Original magnification x63. (E) γH2AX in PBMCs, with all data plotted as γH2AX foci per cells in patients who received topotecan only (left; n = 6) and in patients who received both topotecan and M6620 (right; n = 15). (F) Representative images of γH2AX staining in PBMCs (green, γH2AX; red, DNA). Original magnification x63. P values for the paired differences appear on the plots. The γH2AX signal intensities are expressed in arbitrary unit.
To understand the kinetics of γH2AX formation further, we obtained follicles at additional time points. Although an increase in γH2AX signal was detected after topotecan on day 2, follicles collected 30 to 60 minutes after M6620 infusion on day 2 harbored reduced γH2AX signals (Fig 4C). However, an increase in H2AX phosphorylation in follicles obtained 24 hours after M6620 (before additional topotecan) was observed. These results suggest that M6620 infusion on day 2 inhibited the ATR-dependent phosphorylation of H2AX17 within minutes. Inhibition of H2AX phosphorylation (and the likely inhibition of phosphorylation of other DNA repair proteins), which is critical for recognition and repair of DNA DSB, subsequently led to an altered DDR that translated into accrued DNA damage and increased γH2AX 24 hours after M6620 treatment.
The DNA damage pattern also changed overtime. Although the γH2AX signal was limited to the bottom end of hair bulbs at day 2, this signal was broader on day 3, with more γH2AX-positive cells appearing toward the top of the hair bulbs (Fig 4D). Increases in γH2AX signals were much less pronounced in PBMCs obtained at the corresponding time points than in plucked hair bulbs (Figs 4E and 4F), which is consistent with observations that cytotoxicity by TOP1 poisoning is primarily related to replication18 and that minimal ATR expression exists in quiescent and noncycling cells.19
Evaluable pre- and post-treatment tumor samples were available from two patients treated at DL3 and DL4, respectively (Data Supplement). Although TOP1 expression decreased in both post-treatment tumors (obtained on cycle 1 day 3 24 hours after day 2 of M6620 and before day 3 topotecan), no change was observed in pNBS1 or γH2AX likely because of delayed sampling of post-treatment tumor.
Immunophenotyping of PBMCs showed a significant decrease in the frequency of myeloid-derived suppressor cells after the combination relative to baseline (Data Supplement). Total CD14+ monocytes increased significantly after topotecan but markedly decreased after the combination. A marked redistribution was observed among the monocyte subsets. The more immunosuppressive-classic monocytes were the major population at baseline, whereas intermediate and nonclassical monocytes, which tend to promote antitumor activity, were the minor populations. After the combination, classic monocytes significantly decreased, whereas intermediate and nonclassical monocytes significantly increased. Both monocytes and myeloid-derived suppressor cells returned to near-baseline levels in most patients 3 weeks after treatment. No significant changes in T-cell subsets, including regulatory T cells, conventional CD4+ T cells, and CD8+ T cells, were observed.
Response by Genetic Alterations
In a post hoc exploratory analysis among patients with such data available, we correlated somatic mutations in DDR pathways and chromatin modifiers with clinical responses. The patient with SCLC and a PR had an ARID1A mutation that was likely deleterious (Q528*). Two other patients with prolonged SD, one with peritoneal mesothelioma and one with ovarian cancer, had pathogenic mutations in CHEK2 and BRCA1, respectively.
DISCUSSION
We explored the tolerability, safety, and antitumor activity of the ATR inhibitor M6620 in combination with the TOP1 inhibitor topotecan. This study was conducted on the basis of our previous work that identified ATR as a top candidate for TOP1 inhibitor synthetic lethality and the antitumor activity of the combination observed both in vitro and in vivo.5 In general, the combination of M6620 and topotecan was well tolerated, which allowed dose escalation to DL4, the highest planned dose.
The toxicity profile of the combination largely mirrored that of topotecan, which as monotherapy is associated with a high frequency of myelosuppression. The most common reason for dose reduction was neutropenia, which did not recur with pegfilgrastim prophylaxis. Given the fundamental role of DDR, one would have expected a higher frequency of toxicities with ATR inhibition plus chemotherapy. On the contrary, we did not observe additive toxicities. The differentiating features of rapidly dividing cancer cells, including higher replicative stress and increased DNA damage,20,21 might render them more reliant on the DDR pathways and, consequently, more sensitive to DDR inhibitors than normal cells.
Although the patient population was heavily pretreated, PRs and instances of prolonged SD were observed. Of eight patients with SD, seven had prolonged SD (≥ 12 weeks) associated with improvement or stability of symptoms. ATR inhibition is believed to be particularly active in the setting of high replication stress, and therapeutic approaches that exacerbate this stress could selectively kill tumors by replicative damage. Replication stress is a common feature of SCLC, driven by its many hallmarks.22 Although the number of patients with SCLC treated in this study was small, an important clinical activity signal was noted; all five patients with SCLC had chemotherapy-refractory disease defined as disease progression during or within 60 to 90 days of completing first-line chemotherapy. Three of these patients had a PR or prolonged SD (≥ 6, ≥ 7, and 10 months; Fig 3). Chemotherapy-refractory SCLC is considered a highly aggressive type of SCLC, and previous studies of topotecan alone have reported response rates < 5% and 1-year survival rates < 10%.23
We provide proof of concept of pharmacodynamic modulation of ATR by M6620 with enhanced DNA DSBs seen in response to the combination of topotecan and M6620 relative to topotecan alone. The kinetics of γH2AX induction and its release from chromatin correlate with the rate of DSB rejoining, which allows the use of γH2AX as a sensitive marker of DSB repair.24,25 Plucked hair bulbs, which contain replicating cells, can be obtained noninvasively and may be efficiently used to monitor in vivo DSB formation in real-time.26
Preclinical studies have identified vulnerabilities that may drive a reliance on the DDR, and ATR specifically, for cell survival. These include alterations in DDR/cell cycle checkpoint genes ERCC1,7 XRCC1,8 CDC25A,9 and ATM10,27; processes beyond DDR (eg, loss of tumor suppressor proteins of the switch/sucrose nonfermentable chromatin remodeling complex11); and high replicative stress driven by expression of MYC and RAS, high APOBEC3A and/or APOBEC3B activities, and a hypoxic microenvironment.4,28-31 In this study, response and prolonged SD were observed in patients with pathogenic mutations in DNA repair genes and chromatin modifiers. Larger patient sample sizes and a more-detailed characterization of DDR and chromatin remodeling complex components in resistant and sensitive tumors will be necessary to optimize the identification of patients most likely to derive benefit from M6620 and chemotherapy combinations.
To our knowledge, this study is the first published of a combination of an ATR inhibitor with chemotherapy. Combinations with platinum, gemcitabine, irinotecan, poly (ADP-ribose) polymerase inhibitors, and radiation are being explored in clinic. Of note, PBMC immune phenotyping data from this study have shown evidence of a favorable immunomodulatory effect. Indeed, emerging preclinical data suggest that both cell cycle checkpoint inhibitors32 and topotecan33 can enhance antitumor immunity by reducing proliferation of immunosuppressive T cells and by generating an innate immune response to tumor-derived DNA. Additional studies are needed to confirm the effect we observed, determine the mechanisms, and elucidate implications for combinations with immune checkpoint inhibitors.
In summary, we established tolerable doses of M6620 in combination with topotecan that show preliminary pharmacodynamic evidence of ATR inhibition and enhanced DNA DSBs. The results are consistent with preclinical data that support a synergistic interaction of TOP1 inhibition with ATR inhibition. The study also demonstrated interesting clinical activity signals in refractory SCLC and instances of prolonged SD. A phase II efficacy arm of this clinical trial that combines M6620 and topotecan in SCLC currently is enrolling patients.
ACKNOWLEDGMENT
We gratefully acknowledge the efforts of the following individuals in the design and conduct of this clinical trial: Riquita Pollard, NCI Technology Transfer Office; Hilda Grabczewski and Eugene Bond; NCI Protocol Support Office; Alejandra Chipana Cordero, data manager; Daniel Zlott and Stacey McAdams, NIH Clinical Center pharmacists; and Susan Sansone, patient care coordinator Thoracic and GI Oncology Branch. We are grateful to Drs. Susan Garfield, Poonam Mannan, Langston Lim (Confocal Microscopy Core Facility, CCR, NCI, NIH) for providing a productive environment for confocal microscopy analyses.
Footnotes
Supported by the Center for Cancer Research, Intramural Program of the National Cancer Institute (ZIA BC 011793, Z01 BC 0016150, and ZIA BC 011746). M6620 was supplied by Vertex Pharmaceuticals (Boston, MA) and Merck KGaA (Darmstadt, Germany) under a Cooperative Research and Development Agreement with the National Cancer Institute.
Clinical trial information: NCT02487095.
See accompanying article on page 1628
AUTHOR CONTRIBUTIONS
Conception and design: Anish Thomas, Christophe E. Redon, Susan E. Bates, Seth M. Steinberg, Jane B. Trepel, Yves Pommier
Collection and assembly of data: Anish Thomas, Christophe E. Redon, Linda Sciuto, Emerson Padiernos, Jiuping Ji, Min-Jung Lee, Akira Yuno, Sunmin Lee, Yiping Zhang, Lan Tran, William Yutzy, Arun Rajan, Udayan Guha, Haobin Chen, Raffit Hassan, Christine C. Alewine, Eva Szabo, Mirit I. Aladjem, Jane B. Trepel, Yves Pommier
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Anish Thomas
No relationship to disclose
Christophe E. Redon
Patents, Royalties, Other Intellectual Property: Methods and kits (enzyme-linked immunosorbent assay) for measuring and quantifying DSBs by using γH2AX and H2AX; the methods or kit was not used in the submitted manuscript (Inst)
Linda Sciuto
No relationship to disclose
Emerson Padiernos
No relationship to disclose
Jiuping Ji
No relationship to disclose
Min-Jung Lee
No relationship to disclose
Akira Yuno
No relationship to disclose
Sunmin Lee
No relationship to disclose
Yiping Zhang
No relationship to disclose
Lan Tran
No relationship to disclose
William Yutzy
No relationship to disclose
Arun Rajan
No relationship to disclose
Udayan Guha
Research Funding: AstraZeneca
Haobin Chen
No relationship to disclose
Raffit Hassan
Research Funding: Aduro Biotech (Inst), Morphotek (Inst), Bayer AG (Inst)
Christine C. Alewine
No relationship to disclose
Eva Szabo
No relationship to disclose
Susan E. Bates
No relationship to disclose
Robert J. Kinders
No relationship to disclose
Seth M. Steinberg
No relationship to disclose
James H. Doroshow
No relationship to disclose
Mirit I. Aladjem
No relationship to disclose
Jane B. Trepel
Research Funding: Syndax Pharmaceuticals (Inst), AstraZeneca (Inst)
Yves Pommier
No relationship to disclose
REFERENCES
- 1.Mazouzi A, Velimezi G, Loizou JI: DNA replication stress: Causes, resolution and disease. Exp Cell Res 329:85-93, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK, Cimprich KA: Causes and consequences of replication stress. Nat Cell Biol 16:2-9, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reaper PM, Griffiths MR, Long JM, et al. : Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7:428-430, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Pires IM, Olcina MM, Anbalagan S, et al. : Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br J Cancer 107:291-299, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jossé R, Martin SE, Guha R, et al. : ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase I inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res 74:6968-6979, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flynn RL, Cox KE, Jeitany M, et al. : Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347:273-277, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohni KN, Kavanaugh GM, Cortez D: ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res 74:2835-2845, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sultana R, Abdel-Fatah T, Perry C, et al. : Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One 8:e57098, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruiz S, Mayor-Ruiz C, Lafarga V, et al. : A genome-wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Mol Cell 62:307-313, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwok M, Davies N, Agathanggelou A, et al. : ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 127:582-595, 2016 [DOI] [PubMed] [Google Scholar]
- 11.Williamson CT, Miller R, Pemberton HN, et al. : ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun 7:13837, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toledo LI, Murga M, Zur R, et al. : A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 18:721-727, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall AB, Newsome D, Wang Y, et al. : Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 5:5674-5685, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pommier Y: Topoisomerase I inhibitors: Camptothecins and beyond. Nat Rev Cancer 6:789-802, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Fokas E, Prevo R, Pollard JR, et al. : Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis 3:e441, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Carrigan B, de Miguel Luken MJ, Papadatos-Pastos D, et al: Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J Clin Oncol 34, 2016 (suppl; abstr 2504)
- 17.Ward IM, Chen J: Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276:47759-47762, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Lowe M, Herliczek T, et al. : Protection of normal proliferating cells against chemotherapy by staurosporine-mediated, selective, and reversible G(1) arrest. J Natl Cancer Inst 92:1999-2008, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Jones GG, Reaper PM, Pettitt AR, et al. : The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene 23:1911-1921, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Técher H, Koundrioukoff S, Nicolas A, et al. : The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 18:535-550, 2017 [DOI] [PubMed] [Google Scholar]
- 21.Brown JS, O’Carrigan B, Jackson SP, et al. : Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov 7:20-37, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas A, Pommier Y: Small cell lung cancer: Time to revisit DNA-damaging chemotherapy. Sci Transl Med 8:346fs12, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horita N, Yamamoto M, Sato T, et al. : Topotecan for relapsed small-cell lung cancer: Systematic review and meta-analysis of 1347 patients. Sci Rep 5:15437, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothkamm K, Löbrich M: Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A 100:5057-5062, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redon CE, Nakamura AJ, Martin OA, et al. : Recent developments in the use of γ-H2AX as a quantitative DNA double-strand break biomarker. Aging (Albany NY) 3:168-174, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fong PC, Boss DS, Yap TA, et al. : Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361:123-134, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Kwok M, Davies N, Agathanggelou A, et al. : Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet 385:S58, 2015. (suppl 1) [DOI] [PubMed] [Google Scholar]
- 28.Gilad O, Nabet BY, Ragland RL, et al. : Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res 70:9693-9702, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoppy DW, Ragland RL, Gilad O, et al. : Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest 122:241-252, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murga M, Campaner S, Lopez-Contreras AJ, et al. : Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol 18:1331-1335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buisson R, Lawrence MS, Benes CH, et al. : APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res 77:4567-4578, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goel S, DeCristo MJ, Watt AC, et al. : CDK4/6 inhibition triggers anti-tumour immunity. Nature 548:471-475, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitai Y, Kawasaki T, Sueyoshi T, et al. : DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol 198:1649-1659, 2017 [DOI] [PubMed] [Google Scholar]