

Abstract
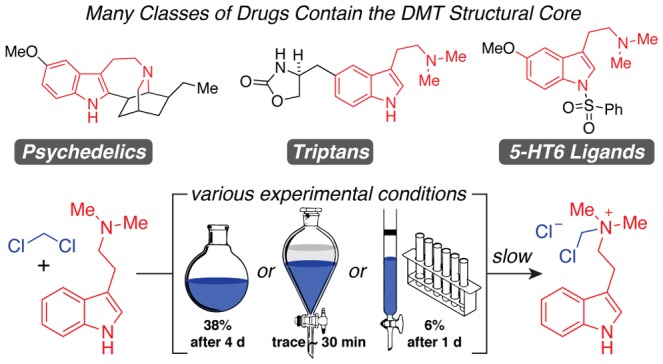
A large number of clinically used drugs and experimental pharmaceuticals possess the N,N-dimethyltryptamine (DMT) structural core. Previous reports have described the reaction of this motif with dichloromethane (DCM), a common laboratory solvent used during extraction and purification, leading to the formation of an undesired quaternary ammonium salt byproduct. However, the kinetics of this reaction under various conditions have not been thoroughly described. Here, we report a series of experiments designed to simulate the exposure of DMT to DCM that would take place during extraction from plant material, biphasic aqueous work-up, or column chromatography purification. We find that the quaternary ammonium salt byproduct forms at an exceedingly slow rate, only accumulates to a significant extent upon prolonged exposure of DMT to DCM, and is readily extracted into water. Our results suggest that DMT can be exposed to DCM under conditions where contact times are limited (<30 min) with minimal risk of degradation and that this byproduct is not observed following aqueous extraction. However, alternative solvents should be considered when the experimental conditions require longer contact times. Our work has important implications for preparing a wide-range of pharmaceuticals bearing the DMT structural motif in high yields and purities.
1. Introduction
A wide-variety of bioactive compounds contain the N,N-dimethyltryptamine (DMT) structural motif (Figure 1). The parent compound DMT (1) is the primary hallucinogenic component of ayahuasca—an Amazonian tisane used in traditional medicine and for religious purposes.1 There is a growing interest in the therapeutic potential of DMT because of recent clinical data demonstrating that ayahuasca produces rapid antidepressant effects in humans that resemble those of the prototypical fast-acting antidepressant ketamine.2−5 Furthermore, DMT is capable of producing behavioral effects in rodents relevant to the treatment of depression and post-traumatic stress disorder.6 Other psychedelics possessing the DMT structural core, such as psilocybin (2) and ibogaine (3), have also demonstrated therapeutic potential for treating various neuropsychiatric disorders including depression, anxiety, and addiction.7,8 Taken together, these data suggest that psychedelics possessing the DMT core structure may have value as medicines and cannot simply be labeled as drugs of abuse.9
Figure 1.

Chemical structures of DMT and related compounds. The DMT structural motif is found in a variety of bioactive natural products, known drugs, and experimental therapeutics.
In addition to the classical serotonergic psychedelics, a variety of other bioactive compounds contain the DMT structural motif. These include the triptan drugs sumatriptan (4), zolmitriptan (5), and rizatriptan (6) (Figure 1)—compounds that are widely prescribed for treating migraines and cluster headaches as well as the 5-HT6 ligands EMDT (7), MS-245 (8), and ST1936 (9).10 The latter compounds are still in the experimental phase but have shown great promise in preclinical models and in clinical trials for improving cognitive deficits associated with Alzheimer’s disease and depression.11 Because of the ubiquity of the DMT structural motif in molecules of medicinal importance, it is imperative to identify methods for their preparation and purification that maximize yield and purity while minimizing cost.
During the course of our studies regarding the synthesis and biological evaluation of DMT derivatives, we became aware of reports detailing the reaction of DMT with dichloromethane (DCM) to form N-chloromethyl-N,N-dimethyltryptamine chloride 10 (Scheme 1).12−14 These findings have caused some chemists to question the appropriateness of using DCM in the synthesis, work-up, or purification of DMT derivatives, despite the fact that DCM possesses many desirable traits like being one of the only commercially available low boiling solvents that is nonflammable.15 One common alternative to DCM is diethyl ether, but the latter can be quite hazardous due to its low flash point (−45 °C), low autoignition temperature (180 °C), and ability to form peroxides.16 Chloroform is another solvent commonly used as a replacement for DCM, but it is far from perfect. In the presence of a base, it can form dichlorocarbene, which is known to react with indoles and amines.17 Additionally, it causes a variety of adverse health effects that are less likely to be caused by DCM.18,19 Finally, both diethyl ether and chloroform are significantly more expensive than DCM (circa 1.5×). Therefore, despite the possibility of amines reacting with DCM through an SN2 reaction, this solvent remains attractive from safety and economic standpoints.19
Scheme 1. Reaction of DMT with DCM.

DMT reacts with DCM via an SN2 reaction to produce N-chloromethyl-N,N-dimethyltryptamine chloride 10.
While there have been several studies reporting the reactivity of DCM with amines, such reactions are highly substrate dependent.20,21 Tertiary amines appear to be the most susceptible to reactions with DCM, though the reactivity of secondary amines has also been reported.20 Secondary amines stored in DCM transiently form an N-chloromethyl product that establishes a rapid equilibrium with the iminium chloride. In the presence of excess amine, the major product formed is the symmetrical aminal.20 As the half-life of triethylamine in DCM is on the order of hours to days,21 we hypothesized that the reaction of DMT with DCM might be negligible under typical conditions used to either extract the natural product from plant sources or produce it synthetically. Here, we confirm that DMT does form 10 when stored in DCM for extended periods of time as previously reported.12−14 However, we find that under typical experimental conditions, the reaction of DMT with DCM does not negatively impact the reaction yield or increase the risk of contamination by 10. We conclude that DCM is a suitable solvent for the work-up and purification of DMT and related analogs and that 10 is unlikely to be useful as a diagnostic impurity characterizing batches of DMT produced by specific clandestine laboratories, as previously suggested.14
2. Results and Discussion
2.1. Synthesis of Analytical Standards
First, we prepared pure DMT and 10 as described previously for use as analytical standards.6,13,14 For reference, the 1H NMR and LC–MS (liquid chromatography–mass spectrometry) spectra are presented in Figure 2. Characterization data for 1 and 10 were consistent with previous reports.6,12
Figure 2.

Characterization data for purified DMT/fumarate (2:1) and 10. 1H NMR spectra taken in DMSO-d6 for DMT (A) and 10 (B). Insets depict LC–MS traces and mass spectra for each compound.
2.2. DMT Degradation in Pure Solvent
Alkaloids are commonly extracted from plant material using DCM.22 To mimic such conditions, we dissolved a DMT free base in DCM and observed the formation of 10 via 1H NMR as a function of time.23 While previous reports have described the formation of 10 after extended reaction times, the rate of its formation was previously unknown.13,14 By comparing the integrations of the peaks corresponding to DMT (1) with those corresponding to 10, we could determine the rate of the reaction. In a pure solvent, the reaction of 1 with DCM is quite clean yielding 10 as the only observable product by 1H NMR (Figure 3A) or LC–MS (data not shown). After 1 h, the percentage of 10 in the mixture was 0.8%, and it reached 38% after 4 days (Figure 3B). Crystals of 10 were observed to precipitate around day 2 when the starting concentration of DMT was 10 mg/mL. The pseudo-first-order rate constant k1 for this reaction was calculated to be 1.21 × 10–5 s–1 and is comparable to that previously reported for the reaction of DCM with trimethylamine.21 Like trimethylamine, DMT reacts approximately 35× faster with DCM than triethylamine, but only 2× as fast as the secondary amine piperidine.21 Compared with the bulkier tertiary amine N-methylpiperidine, DMT reacts nearly 10× as fast highlighting the importance of steric effects in the reaction of amines with DCM.21 Though DMT and trimethylamine react faster with DCM than most amines (with the exception of quinuclidine), this reaction is still quite slow when compared with their reaction with methyl iodide. For example, trimethylamine reacts with methyl iodide approximately 3000× faster than it does with DCM.21,23 Notably, when chloroform was used as the solvent, no appreciable decomposition of DMT (1) was observed (data not shown). When the reaction between DMT and DCM was carried out at −20 °C, the amount of 10 formed was less than 1% as determined by NMR after 4 days. While the rate of formation of 10 is quite slow in DCM, care should be taken when lengthy extraction protocols are required. In such cases, chloroform may be a more appropriate solvent choice.
Figure 3.

Formation of 10 in pure solvent. (A) Reaction of DMT (1) with DCM to produce 10 monitored by 1H NMR over time. The peaks highlighted in gray were used to quantify the molar ratio of 1 to 10. (B) Quantification of the peaks in A demonstrates that 10 reaches 38 mol % after 4 days.
2.3. DMT Degradation under Biphasic Basic Work-Up Conditions
While the rate of formation of 10 in DCM is relatively slow, appreciable amounts were observed after several days. However, it is quite uncommon for reactive tertiary amines to be left in solvent for multiple days at a time. Therefore, we were interested in assessing the formation of 10 under more typical experimental conditions, namely, those corresponding to a biphasic aqueous work-up. Because of the partitioning between the organic and aqueous phases, one might expect to form 10 at an accelerated rate because of the dynamic removal of the ammonium salt product from the organic layer where the SN2 reaction occurs. In fact, Brandt and co-workers have reported that a basic aqueous work-up using DCM following the synthesis of DMT (1) resulted in the formation of 10, which constituted 4% of the total peak area in the LC-UV trace.13 As Brandt and co-workers did not precisely quantify the formation of 10, we subjected DMT to DCM/NaOHaq biphasic work-up conditions and monitored the formation of 10 in both layers as a function of time using LC–MS with selective ion monitoring (SIM). In our hands, we observed that the rate of DMT degradation under biphasic conditions is comparable to that in pure DCM (Figure 4A,B). By 24 h, 5% of 10 was observed in the aqueous layer, comparable to the 4% observed in pure DCM conditions. Importantly, we discovered that 10, likely because of its ionic nature, partitions almost exclusively into the aqueous phase, resulting in undetectable amounts of 10 contaminating the DMT dissolved in the organic phase (Figure 4B).
Figure 4.

Formation of 10 under basic biphasic work-up conditions. (A) Formation of 10 in the aqueous phase as a function of time. Quantification was achieved by determining relative peak ratios of 10 and an internal standard (caffeine) and comparing those values to a calibration curve. (B) Combined LC–MS traces using SIM (masses = 189, DMT; 237, 10; 195, caffeine) of the aqueous and organic phases after 24 h of biphasic work-up conditions. The presence of 10 was only detected in the aqueous layer. (C) Unpurified 1H NMRs taken in CDCl3 following the synthesis and 30 min biphasic basic aqueous work-up of DMT (1) using three different solvents. Comparable purity of DMT was obtained, regardless of the solvent used during the work-up. Isolated yields following crystallization as the fumarate salt (2:1 DMT/fumarate) are indicated.
Because of fear of forming impurity 10, others have preferred the use of chloroform to DCM for extracting DMT free base. However, chloroform is known to form dichlorocarbene under basic conditions, such as those employed in aqueous work-ups involving amines.17 Therefore, we wished to directly compare the yields and purities of synthetic DMT subjected to basic biphasic work-up conditions utilizing either DCM, chloroform, or diethyl ether. The work-ups were carefully timed such that the solvent was completely removed 30 min following the initial exposure of DMT to solvent. Inspection of the unpurified 1H NMRs revealed that 10 was unobservable following aqueous work-up regardless of the organic solvent employed (Figure 4C). This fact makes it unlikely that 10 can be used as a diagnostic impurity when trying to identify DMT produced by clandestine laboratories using DCM in their syntheses. After crystallization as the fumarate salt, the synthetic DMT produced using these three distinct work-up conditions was judged to be analytically pure (via 1H NMR, and LC–MS). Furthermore, the yields of the final product were comparable (79–86%), regardless of the solvent used during the work-up. Thus, we conclude that DCM/NaOHaq is a perfectly suitable extraction system for the work-up of synthetically produced DMT, as long as the contact times are minimized (<30 min).
2.4. DMT Degradation Following Purification via Chromatography
Basic amines such as DMT (1) are often purified via flash chromatography with the most common solvent system being 9:1 DCM/MeOH with 1% NH4OHaq. In fact, similar conditions produced 10 from an extract of Acacia confusa, and the authors concluded that its formation was an artifact of the isolation conditions.12 Following chromatography using this solvent system, amines are often left to sit in the collected fractions for a period of time, and thus, we were interested to measure the conversion of DMT to 10 in 9:1 DCM/MeOH with 1% NH4OHaq (Figure 5A,B). After 1 h, the percentage of 10 in the mixture was 2% and had reached 6% by 24 h, comparable to previously seen quantities at 24 h. The reaction proceeded under pseudo-first-order conditions at a comparable rate to that of DMT in pure DCM, and thus, it would appear that the addition of MeOH and NH4OHaq has very little effect on the rate of the reaction of DMT with DCM. However, we recommend that fractions containing DMT be concentrated within 1 h or stored at −20 °C following column chromatography using 9:1 DCM/MeOH with 1% NH4OHaq as the solvent system to minimize any potential for degradation.
Figure 5.

Formation of 10 under purification conditions. (A) Reaction of DMT (1) with 9:1 DCM/MeOH with 1% NH4OHaq to produce 10 monitored by 1H NMR over time. The peaks highlighted in gray were used to quantify the molar ratio of 1 to 10. (B) Quantification of the peaks in A demonstrates that 10 reaches 6 mol % after 24 h.
3. Conclusions
Here, we have demonstrated that 1 will form 10 in the presence of DCM as described previously.12−14 However, the rate of this reaction is exceedingly slow under most common experimental conditions, and significant amounts of this impurity do not accumulate when contact times are limited (<30 min) or when cryogenic temperatures are used. Therefore, concerns regarding the use of DCM during the preparation of DMT (and likely other related dimethyltryptamine analogs) are not warranted. It has been previously suggested that forensic teams could use 10 as a diagnostic impurity to identify clandestine laboratories using DCM in their syntheses of DMT. However, we think that this possibility is unlikely due to the readily extractable nature of 10 in water as well as the slow rate of reaction between 1 and DCM. Although there are still environmental concerns with using chlorinated solvents such as DCM, this study has shown that its use has no effect on the purity or yield of the final DMT product as compared to other common laboratory solvents. Dimethyltryptamine and related analogs have high medicinal value, and our work answers some fundamental questions regarding their economical preparation in excellent yields and purities.
4. Methods
4.1. Materials
The DCM (ACS reagent grade, stabilized), chloroform (HPLC grade), and all other reagents used in these studies were purchased from Fisher Scientific and used without purification. The concentration of the ammonium hydroxide used in the chromatography experiments was 28–30%.
4.2. Synthesis of DMT (1)
To an ice-cold solution of tryptamine (0.60 g, 3.1 mmol) and glacial acetic acid (0.88 mL, 15 mmol, 5.0 equiv) in MeOH (48 mL) was added sodium cyanoborohydride (0.40 g, 6.4 mmol, 2.1 equiv) followed by 37% formaldehydeaq (0.64 mL, 7.9 mmol, 2.6 equiv). The reaction was stirred at room temperature for 5 h, at which time the mixture was transferred equally to three separate vials. Concentration under reduced pressure afforded an unpurified solid, which was dissolved in either DCM, diethyl ether, or CHCl3 (25 mL) and diluted with 1.0 M NaOHaq (75 mL). The phases were separated and the aqueous phase was extracted twice with solvent (25 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield a colorless oil. The unpurified oil was dissolved in acetone (5 mL) and added dropwise to a boiling solution of fumaric acid (0.059 g, 0.50 mmol, 0.50 equiv) in acetone (15 mL). A precipitate formed immediately. The solution was allowed to cool to room temperature and filtered. The white solid was dried under reduced pressure to yield the pure compound as the fumarate salt (2:1 DMT/fumaric acid) (DCM: 0.207 g, 0.084 mmol, 83%; CHCl3: 0.199 g, 0.081 mmol, 79%; diethyl ether: 0.215 g, 0.87 mmol, 86%). TLC Rf (DMT free base) = 0.35 (9:1 DCM/MeOH 1% NH4OHaq); mp 140–142 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.8 (br s, 1H), 7.5 (d, 1H, J = 7.9 Hz), 7.3 (d, 1H, J = 7.9 Hz), 7.1 (s, 1H), 7.0 (t, 1H, J = 7.9 Hz), 6.9 (t, 1H, J = 7.9 Hz), 6.5 (s, 1H), 2.9 (t, 2H, J = 8.0 Hz), 2.8 (t, 2H, J = 8.0 Hz), 2.4 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD): δ 173.8, 138.2, 136.9, 128.1, 124.2, 122.7, 120.1, 119.0, 112.5, 110.0, 59.2, 43.4, 21.9 ppm; IR (diamond, ATR) ν̅max: 3483, 3146, 3107, 3045, 2927, 2881, 1561, 1226, 749 cm–1. LC–MS (ES+) calcd for C12H16N2+, 188.13; found, 189.32 (MH+).
4.3. Synthesis of N-Chloromethyl-N,N-dimethyltryptamine Chloride (10)
The DMT free base [0.050 g, 0.26 mmol; see below for a procedure for preparing the free base from the DMT/fumarate (2:1) salt] was dissolved in DCM (5 mL) to give a concentration of 10 mg/mL (0.05 M) The vial was blanketed with N2 and stored in the dark. After 5 days, the precipitate that had formed was filtered, rinsed with DCM and dried under reduced pressure to yield 10 as an off-white solid (0.027 g, 0.076 mmol, 19%). 1H NMR (400 MHz, DMSO-d6): δ 11.1 (br s, 1H), 7.6 (d, 1H, J = 8.0 Hz), 7.4 (d, 1H, J = 8.0 Hz), 7.3 (s, 1H), 7.1 (t, 1H, J = 8.0 Hz), 7.0 (t, 1H, J = 8.0 Hz), 5.5 (s, 2H), 3.7 (t, 2H, J = 8.1 Hz), 3.3 (s, 6H) 3.2 (t, 2H, J = 8.1 Hz) ppm; 13C NMR (100 MHz, DMSO-d6): δ 136.2, 126.5, 123.7, 121.3, 118.6, 118.2, 111.7, 107.7, 68.1, 62.1, 48.9, 18.3 ppm; IR (diamond, ATR) ν̅max: 3142, 3047, 3017, 2999, 2923, 2863, 866, 755, 727 cm–1. LC–MS (ES+) calcd for C13H18Cl2N2+, 237.12; found, 237.22 (M+).
4.4. Conversion of DMT/Fumarate (2:1) to the Free Base
DMT/fumarate (2:1) (0.20 g, 0.82 mmol) was basified using 1.0 M NaOHaq (30 mL) and extracted twice with diethyl ether (20 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield DMT free base (0.11 g, 0.58 mmol, 71%).
4.5. DMT Degradation in Pure Solvent
The free base of DMT (as prepared above) was dissolved in either DCM or CHCl3 to give a concentration of 10 mg/mL (0.05 M). The vial was blanketed with N2 and stored in the dark. Aliquots were taken at various time points (1 h, 1 day, 2 days, 3 days, and 4 days) and concentrated under reduced pressure. Care was taken to ensure that the mixture was homogenous before aliquots were taken. The unpurified samples were dissolved in 0.6 mL of DMSO-d6 and 1H NMR spectra were obtained. The aliquot taken after 4 days was also subjected to analysis by LC–MS to confirm the formation of 10.
4.6. DMT Degradation under Biphasic Basic Work-Up Conditions
The free base of DMT (as prepared above) was dissolved in DCM to give a concentration of 10 mg/mL (0.05 M). To this solution was added an equal volume of 1.0 M NaOHaq. The vial was blanketed with N2 and stored in the dark. Aliquots were taken at 5 min, 15 min, 30 min, 1 h, and 1 day from the aqueous layer, neutralized with 2.0 M HClaq, and subjected to analysis by SIM LC–MS using caffeine as an internal standard. Aliquots from the organic layer were taken at the same time points, concentrated under reduced pressure, diluted with methanol, and subjected to analysis by SIM LC–MS using caffeine as an the internal standard. Standard curves were prepared by comparing SIM peak integrations for increasing concentrations of purified DMT/fumarate (2:1) or 10 relative to a constant concentration of caffeine as an internal standard.
4.7. DMT Degradation under Column Chromatography Conditions
The free base of DMT (as prepared above) was dissolved in 9:1 DCM/MeOH with 1% NH4OHaq to give a concentration of 10 mg/mL (0.05 M). The vial was blanketed with N2 and stored in the dark. Aliquots were taken at 5 min, 15 min, 30 min, 1 h, and 1 day, and then concentrated under reduced pressure. The unpurified samples were dissolved in 0.6 mL of DMSO-d6, and 1H NMR spectra were obtained. An aliquot taken after 1 day was also subjected to analysis by LC–MS to confirm the formation of 10.
Acknowledgments
We thank members of the Olson Laboratory for helpful discussions regarding the manuscript.
This work was supported by funds from the UC Davis Department of Chemistry, Department of Biochemistry & Molecular Medicine, and a UC Davis Science Translation and Innovative Research (STAIR) Grant.
The authors declare no competing financial interest.
References
- McKenna D. J. Clinical investigations of the therapeutic potential of ayahuasca: rationale and regulatory challenges. Pharmacol. Ther. 2004, 102, 111–129. 10.1016/j.pharmthera.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Dos Santos R. G.; Osório F. L.; Crippa J. A. S.; Hallak J. E. C. Antidepressive and anxiolytic effects of ayahuasca: a systematic literature review of animal and human studies. Rev. Bras. Psiquiatr. 2016, 38, 65–72. 10.1590/1516-4446-2015-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Clavé E.; Soler J.; Elices M.; Pascual J. C.; Álvarez E.; de la Fuente Revenga M.; Friedlander P.; Feilding A.; Riba J. Ayahuasca: Pharmacology, neuroscience and therapeutic potential. Brain Res. Bull. 2016, 126, 89–101. 10.1016/j.brainresbull.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Osório F. d. L.; Sanches R. F.; Macedo L. R.; dos Santos R. G.; Maia-de-Oliveira J. P.; Wichert-Ana L.; de Araujo D. B.; Riba J.; Crippa J. A.; Hallak J. E. Antidepressant effects of a single dose of ayahuasca in patients with recurrent depression: a preliminary report. Rev. Bras. Psiquiatr. 2015, 37, 13–20. 10.1590/1516-4446-2014-1496. [DOI] [PubMed] [Google Scholar]
- Berman R. M.; Cappiello A.; Anand A.; Oren D. A.; Heninger G. R.; Charney D. S.; Krystal J. H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Cameron L. P.; Benson C. J.; Dunlap L. E.; Olson D. E. Effects of N,N-Dimethyltryptamine (DMT) on Rat Behaviors Relevant to Anxiety and Depression. ACS Chem. Neurosci. 2018, 10.1021/acschemneuro.8b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]; accepted
- dos Santos R. G.; Osório F. L.; Crippa J. A. S.; Riba J.; Zuardi A. W.; Hallak J. E. C. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther. Adv. Psychopharmacol. 2016, 6, 193–213. 10.1177/2045125316638008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper K. R.Ibogaine: a review. The Alkaloids: Chemistry and Biology; Academic Press, 2001; Vol. 56, pp 1–38. [DOI] [PubMed] [Google Scholar]
- Kyzar E. J.; Nichols C. D.; Gainetdinov R. R.; Nichols D. E.; Kalueff A. V. Psychedelic Drugs in Biomedicine. Trends Pharmacol. Sci. 2017, 38, 992–1005. 10.1016/j.tips.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Cameron C.; Kelly S.; Hsieh S.-C.; Murphy M.; Chen L.; Kotb A.; Peterson J.; Coyle D.; Skidmore B.; Gomes T.; Clifford T.; Wells G. Triptans in the Acute Treatment of Migraine: A Systematic Review and Network Meta-Analysis. Headache 2015, 55, 221–235. 10.1111/head.12601. [DOI] [PubMed] [Google Scholar]
- Karila D.; Freret T.; Bouet V.; Boulouard M.; Dallemagne P.; Rochais C. Therapeutic Potential of 5-HT6 Receptor Agonists. J. Med. Chem. 2015, 58, 7901–7912. 10.1021/acs.jmedchem.5b00179. [DOI] [PubMed] [Google Scholar]
- Buchanan M. S.; Carroll A. R.; Pass D.; Quinn R. J. NMR spectral assignments of a new chlorotryptamine alkaloid and its analogues from Acacia confuse. Magn. Reson. Chem. 2007, 45, 359–361. 10.1002/mrc.1959. [DOI] [PubMed] [Google Scholar]
- Brandt S. D.; Martins C. P. B.; Freeman S.; Dempster N.; Wainwright M.; Riby P. G.; Alder J. F. N,N-Dimethyltryptamine and dichloromethane: rearrangement of quaternary ammonium salt product during GC-EI and CI-MS-MS analysis. J. Pharm. Biomed. Anal. 2008, 47, 207–212. 10.1016/j.jpba.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Brandt S. D.; Martins C. P. B.; Freeman S.; Dempster N.; Riby P. G.; Gartz J.; Alder J. F. Halogenated solvent interactions with N,N-dimethyltryptamine: Formation of quaternary ammonium salts and their artificially induced rearrangements during analysis. Forensic Sci. Int. 2008, 178, 162–170. 10.1016/j.forsciint.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Prat D.; Pardigon O.; Flemming H.-W.; Letestu S.; Ducandas V.; Isnard P.; Guntrum E.; Senac T.; Ruisseau S.; Cruciani P.; Hosek P. Sanofi’s Solvent Selection Guide: A Step Toward More Sustainable Processes. Org. Process Res. Dev. 2013, 17, 1517–1525. 10.1021/op4002565. [DOI] [Google Scholar]
- Henderson R. K.; Jiménez-González C.; Constable D. J. C.; Alston S. R.; Inglis G. G. A.; Fisher G.; Sherwood J.; Binks S. P.; Curzons A. D. Expanding GSK’s solvent selection guide – embedding sustainability into solvent selection starting at medicinal chemistry. Green Chem. 2011, 13, 854–862. 10.1039/c0gc00918k. [DOI] [Google Scholar]
- Rees C. W.; Smithen C. E.. The Reactions of Heterocyclic Compounds with Carbenes. Advances in Heterocyclic Chemistry; Academic Press, 1964; Vol. 3, pp 57–78. [Google Scholar]
- Alfonsi K.; Colberg J.; Dunn P. J.; Fevig T.; Jennings S.; Johnson T. A.; Kleine H. P.; Knight C.; Nagy M. A.; Perry D. A.; Stefaniak M. Green chemistry tools to influence a medicinal chemistry and research chemistry based organization. Green Chem. 2008, 10, 31–36. 10.1039/b711717e. [DOI] [Google Scholar]
- Rossberg M.; Lendle W.; Pfleiderer G.; Tögel A.; Dreher E. L.; Langer E.; Rassaerts H.; Kleinschmidt P.; Strack H.; Cook R.; Beck U.; Lipper K. A.; Torkelson T. R.; Löser E.; Beutel K. K.; Mann T.. Chlorinated Hydrocarbons. In Ullmann’s Encyclopedia of Industrial Chemistry; Ullmann F., Gerhartz W., Eds.; Wiley-VCH: Weinheim, 2006, pp 1–186. [Google Scholar]
- Mills J. E.; Maryanoff C. A.; McComsey D. F.; Stanzione R. C.; Scott L. Reaction of amines with methylene chloride. Evidence for rapid aminal formation from N-methylenepyrrolidinium chloride and pyrrolidine. J. Org. Chem. 1987, 52, 1857–1859. 10.1021/jo00385a038. [DOI] [Google Scholar]
- Nevstad G. O.; Songstad J. Solvent Properties of Dichloromethane. II. The Reactivity of Dichloromethane Toward Amines. Acta Chem. Scand., Ser. B 1984, 38, 469–477. 10.3891/acta.chem.scand.38b-0469. [DOI] [Google Scholar]
- El Jaber-Vazdekis N.; Gutierrez-Nicolás F.; Ravelo A. G.; Zárate R. Studies on tropane alkaloid extraction by volatile organic solvents: dichloromethane vs. chloroform. Phytochem. Anal. 2006, 17, 107–113. 10.1002/pca.893. [DOI] [PubMed] [Google Scholar]
- Fahim R. B.; Moelwyn-Hughes E. A. Kinetics of the Reaction between Methyl Iodide and Trimethylamine in Carbon Tetrachloride Solution. J. Chem. Soc. 1956, 1035–1041. 10.1039/jr9560001035. [DOI] [Google Scholar]