

Abstract
The structure of the higher-order chromatin fiber has not been defined in detail. We have used a novel approach based on sucrose gradient centrifugation to compare the conformation of centromeric satellite DNA-containing higher-order chromatin fibers with bulk chromatin fibers obtained from the same mouse fibroblast cells. Our data show that chromatin fibers derived from the centromeric domain of a chromosome exist in a more condensed structure than bulk chromatin whereas pericentromeric chromatin fibers have an intermediate conformation. From the standpoint of current models, our data are interpreted to suggest that satellite chromatin adopts a regular helical conformation compatible with the canonical 30-nm chromatin fiber whereas bulk chromatin fibers appear less regularly folded and are perhaps intermittently interrupted by deformations. This distinctive conformation of the higher-order chromatin fiber in the centromeric domain of the mammalian chromosome could play a role in the formation of heterochromatin and in the determination of centromere identity.
When fragments of chromatin are isolated from cells and maintained under ionic conditions comparable to those in the nucleus, they are invariably found to be folded into higher-order fibers (1–3). Structural studies on such bulk material have formed the basis for a variety of models that are proposed to explain the manner in which chains of nucleosomes are packaged into the higher-order state (3–5). However, the ubiquitous and uniform character for the higher-order chromatin fiber suggested by these models tends to mask the fact that the higher-order chromatin fiber must be an adaptable structure capable of undergoing dynamic structural transitions. Such properties are required to facilitate the unfolding processes presumed to be essential for gene activation and chromosome replication. On the other hand, the chromatin fiber must also have the capacity to adopt an inert character required to maintain genes in a state of sustained repression and to provide local chromosomal domains with distinctive architectures within which specific chromosomal structures, such as the centromere, can exist (6). Despite these expectations, studies on isolated higher-order fibers have failed to reveal a diversity of structure compatible with the diversity of function. For example, chromatin fibers containing globin gene sequences, isolated from erythroid cells in an activated state, have physical properties equivalent to both bulk and transcriptionally inactive chromatin fibers (7, 8). The presence of nucleosome-free hypersensitive sites disrupts the fiber, but between these distinctive regions the chromatin appears to be typically folded. Within the cell, the higher-order chromatin fiber does unfold during transcription, but maintenance of this state is notably transient for the inhibition of polymerase activity leads to a rapid reformation of the folded state (9). Thus, in respect of structural criteria, it appears that chromatin fibers containing active gene sequences cannot be distinguished from bulk or inactive chromatin fibers once they are removed from the nucleus.
Analytical sucrose gradient sedimentation has been widely used to characterize the influence of various components in the establishment of the higher-order chromatin fiber (2, 8, 10, 11). The technique has the unique advantage of permitting the structural analysis of chromatin fibers containing specific DNA sequences which can be directly and simultaneously compared with bulk chromatin fibers (7, 8). In the present study we have used this approach to examine the structure of chromatin fibers released from centromeric heterochromatin. Our results show that fibers containing satellite DNA have a higher sedimentation rate than bulk chromatin fibers containing an equivalent length of DNA. These observations suggests that the higher-order structure of centromeric chromatin is more regularly packaged than the majority of other chromatin fibers, leading us to propose that centromeric chromatin fibers adopt a conformation consistent with the canonical 30-nm chromatin fiber whereas bulk fibers display less regular folding.
Materials and Methods
Cell Culture.
The cell lines used in this study were NIH 3T3, F9 embryonal carcinoma, Ht2, an embryonic stem cell line derived from CGR8 cells (12), and the human cell line HT1080. Routine cell culture was according to Smith (13), and cells were maintained in 5% CO2 at 37°C in a humidified incubator. Embryonic stem cells were propagated in the presence of 100 units/ml leukemia inhibitory factor.
Nuclei and Chromatin Preparations.
Nuclei were prepared by a modification of the method of Cereghini and Yaniv (14). Cell cultures were harvested in PBS containing 0.25 mg/ml trypsin and 1 mM EDTA and were washed in PBS. The cell pellet was resuspended in a small volume of ice-cold NBA [85 mM KCl/10 mM Tris⋅HCl (pH 7.6)/5.5% (wt/vol) sucrose/0.5 mM spermidine/0.2 mM EDTA/0.25 mM PMSF]. To this an equal volume of NBA plus 0.1% (vol/vol) Nonidet P-40 was added, and the cells were incubated on ice for 3 min. Nuclei were collected by centrifugation (360 × g for 3.5 min at 4°C) and washed in NBC (NBA minus EDTA) before resuspending at 20 A260 units/ml in NBC. Soluble chromatin was prepared by digesting nuclei with micrococcal nuclease or a restriction enzyme (MvaI or AluI). To prepare chromatin by using micrococcal nuclease, nuclei in NBC buffer were supplemented with 1.5 mM CaCl2 and digested with 20–60 units micrococcal nuclease per 20 A260 units of nuclei for 10 min at room temperature in the presence of 100 μg/ml RNaseA. The digestion was stopped by adding EDTA to 10 mM. After digestion, the nuclei were pelleted and resuspended in TEP20 (10 mM Tris⋅HCl, pH 7.6/0.1 mM EDTA/0.25 mM PMSF/20 mM NaCl) supplemented with 300 μg/ml lysolecithin and were incubated at 4°C overnight to effect nuclear lysis. Nuclear debris was removed by centrifugation (12,000 × g for 5 min at 4°C), and the soluble chromatin was recovered in the supernatant. To prepare chromatin by using a restriction enzyme, nuclei were suspended in NBC supplemented with 2 mM MgCl2 and 0.1 mM EGTA and digested by using 100 units restriction enzyme per A260 unit chromatin in the presence of 100 μg/ml RNaseA. The digestion was stopped after 8 min by adding EDTA to 10 mM, and the soluble chromatin was recovered as described above.
Analytical Sucrose Gradient Sedimentation.
Aliquots of soluble chromatin were fractionated on 6% to 40% (wt/vol) isokinetic sucrose gradients (15) containing TEP80 (10 mM Tris⋅HCl, pH 7.6/0.1 mM EDTA/0.25 mM PMSF/80 mM NaCl) by centrifugation at 4°C for 2–3 h at 41,000 rpm in a Beckman SW41 rotor. Gradients were fractionated by upward displacement with continuous monitoring of the absorbance profile. The 0.5-ml fractions were collected.
Cesium Chloride Density Gradient Analysis.
Chromatin isolated from a sucrose gradient containing TEP80 was dialysed into TEA80 (10 mM triethanolamine, pH 7.6/0.1 mM EDTA/0.25 mM PMSF/80 mM NaCl) and crosslinked by addition of formaldehyde to 0.5% and incubation overnight at 4°C. Excess crosslinker was removed by dialysis against TEA80. A solution of cesium chloride (density ≈1.7 g/ml; Sigma) was added to the chromatin to give a density of 1.41 g/ml. Centrifugation, at 20°C, was in the Sorvall TV-865 vertical rotor for a minimum of 40 h at 40,000 rpm. Gradients were fractionated and processed as described (7).
Southern Blotting.
DNA from individual sucrose gradient fractions was purified by digestion with proteinase K (100 μg/ml) in the presence of 0.1% SDS for 30 min at 37°C, followed by phenol/chloroform extraction and ethanol precipitation. DNA was resolved by electrophoresis in 0.7 or 1.0% agarose gels in Tris-phosphate buffer containing 0.5 μg/ml ethidium bromide. Suitable DNA size markers (Promega) were included on the gel. The gel was laser scanned at 473 nm by using a Fuji FLA2000. After transfer of DNA to Hybond N (Amersham Pharmacia), the membrane was prehybridized in a buffer containing 0.5 M sodium phosphate, 7% (wt/vol) SDS, 1 mM EDTA, 2% Marvel, and 100 μg/ml salmon sperm DNA for 30 min at 65°C. One to two micrograms of random prime-labeled probe was added per milliliter of prehybridization buffer, and hybridization was continued overnight at 65°C. The membrane was washed for 2 × 20 min with 0.5 M sodium phosphate, 1% SDS and 2 × 20 min 50 mM sodium phosphate, 1% SDS. The membrane was analyzed by phosphorimaging by using a Fuji FLA2000. Quantitative scans of individual lanes from the laser-scanned, ethidium bromide-stained gel and from the phosphorimages of the Southern blots were produced by using aida analysis software (Raytest). From these scans, the DNA size (peak maximum) of individual fractions was determined by reference to the markers. DNA from cesium chloride gradients was analyzed by dot-blotting onto Hybond N and hybridization as described above.
Hybridization Probes.
The probes used were mouse minor satellite R198 (16), mouse major satellite (17), and a B2 interspersed repeat (18). The specificity of the satellite probes was confirmed by standard fluorescence in situ hybridization (FISH) analysis (19) of metaphase spreads of NIH 3T3 cells.
Results
Mouse Satellite-Containing Chromatin Fibers Sediment More Rapidly Than Bulk Chromatin Fibers.
By using sucrose gradient sedimentation (2, 7) we have characterized the higher-order chromatin fiber in the centromeric-domain of mammalian chromosomes. Soluble chromatin, released from NIH 3T3 nuclei after digestion with micrococcal nuclease or restriction enzymes, was sedimented through an isokinetic sucrose gradient (see Figs. 7 and 8, which are published as supporting information on the PNAS web site, www.pnas.org) under conditions (80 mM NaCl) that maintained the higher-order structure of the fibers (1, 2). DNA isolated from individual gradient fractions was electrophoresed in an agarose gel. After fluorescence scanning, the DNA in the gel was transferred onto a nylon membrane and hybridized in succession with probes for mouse minor satellite DNA (16), mouse major satellite DNA (17), and a B2 interspersed repeat (18). The size of the DNA contained in the chromatin fibers recovered from each gradient fraction was determined from densitometer traces of lanes from the ethidium bromide-stained gel and from each of the hybridized filters (see Fig. 2e), by reference to DNA size markers. This approach enabled us to relate the sedimentation velocity (gradient fraction number) of particular chromatin fibers to the average length of DNA they contained.
Figure 2.

Mouse centromeric heterochromatin fibers sediment more rapidly than equivalent sized bulk chromatin fibers. Soluble chromatin prepared from NIH 3T3 cells by micrococcal nuclease digestion was fractionated in an isokinetic sucrose gradient, and the DNAs purified from individual fractions were sized on an agarose gel (a). After blotting to a nylon membrane, the samples were probed for mouse minor satellite (b), major satellite (c), and a B2 repeat (d). The size of the DNA contained in each gradient fraction was determined from densitometer traces of lanes from the ethidium bromide-stained gel and from the phosphorimages of hybridized filters, by reference to DNA size markers (e, for example). The relationship between DNA size and sedimentation velocity (fraction number; f) reveals the difference in conformation between satellite and bulk chromatin.
As shown in Fig. 2 the relationship between DNA size and sedimentation velocity for chromatin fibers detected by the B2 probe is the same as that obtained for bulk chromatin fibers (Fig. 2 a, d, and f). Thus, chromatin fibers containing copies of this widely dispersed repeat are not distinct from bulk chromatin in this assay. In contrast, the profiles for chromatin fibers containing either mouse minor or major satellite DNAs, which are derived from the centromeric and pericentromeric chromatin domains, respectively (ref. 6; Fig. 1), diverge from that of bulk chromatin (Fig. 2 a–c and f), revealing that chromatin fibers containing satellite DNA sediment more rapidly than bulk chromatin fibers containing the same length of DNA. This distinction is more pronounced for centromeric (minor satellite) than for non-centromeric (major satellite) heterochromatin fibers (Fig. 2f).
Figure 1.

Chromosomal localization of mouse minor and major satellites as determined by fluorescence in situ hybridization (FISH) analysis of NIH 3T3 metaphase spreads. For the acrocentric mouse chromosomes, the minor satellite (red) and major satellite (light blue) probes used in this study localize to centromeric and pericentromeric regions respectively.
The Distinctive Sedimentation Behavior of Satellite DNA-Containing Chromatin Fibers Depends On Higher-Order Chromatin Folding.
Because the nucleosome repeat lengths of satellite and bulk chromatins are the same (190 bp, Figs. 7 and 8) it follows that, within a given sucrose gradient fraction, the satellite-containing fibers are shorter than the corresponding bulk chromatin fibers (Fig. 2). To corroborate this finding, chromatin fibers were fractionated in a gradient containing 80 mM NaCl, and the individual gradient fractions were then dialysed into 5 mM NaCl to unfold the chromatin fiber into an extended polynucleosome chain (1). This chromatin was then resedimented in this unfolded state, and the gradients were analyzed by blotting. When unfolded, chromatin fibers containing the B2 repeat continued to cosediment with bulk fibers (Fig. 3, a–c). In contrast, chromatin fibers containing mouse minor satellite DNA now sedimented more slowly than bulk fibers (Fig. 3 d–f). Thus, when unfolded, bulk and satellite-containing fibers sediment in a manner consistent with the lengths of DNA, or the number of nucleosomes, they contain. It follows that the distinctive sedimentation behavior of satellite chromatin depends on folding into a higher-order structure.
Figure 3.

The distinctive sedimentation behavior of satellite-containing chromatin depends on higher-order folding. Soluble chromatin prepared from mouse F9 embryonal carcinoma cells by micrococcal nuclease digestion was fractionated as higher-order chromatin fibers in an isokinetic sucrose gradient containing 80 mM NaCl. Chromatin recovered from individual gradient fractions was unfolded by dialysis into 5 mM NaCl and then subjected to a second round of sucrose gradient sedimentation in low-ionic strength (5 mM NaCl). DNAs purified from individual gradient fractions were fractionated on agarose gels (a and d). After blotting to a nylon membrane, the samples were probed for a B2 repeat (b) or for mouse minor satellite (e). The sedimentation profiles (c and f) for bulk (EtBr) and probed chromatins were generated by quantitative densitometry of the gels and phosphorimages.
The Distinctive Structure of Satellite Chromatin Is Observed in Other Mouse and Human Cell Types.
The distinctive higher-order structure of centromeric chromatin was exhibited by NIH 3T3 chromatin isolated by restriction enzyme digestion (Fig. 4 d and e) and by chromatins isolated from a variety of other cell types (Fig. 4 a–c). These latter samples included mouse F9 embryonal carcinoma cells, in which satellite DNA is undermethylated, compared with differentiated cells (20) and mouse embryonic stem cells (ht2) in which the centromeric core histones, which are hypoacetylated in differentiated cells (21), display typical levels of acetylation (22). Finally, analysis of centromeric chromatin fibers in the human cell line HT1080, as detected by an α-satellite DNA probe (Fig. 4c), indicated that the distinctive structure of centromeric heterochromatin is not specific to the mouse.
Figure 4.

Mouse and human centromeric heterochromatin fibers sediment more rapidly than equivalent sized bulk fibers. Soluble chromatins prepared by micrococcal nuclease (a–c) or restriction enzyme (d, MvaI, or e, AluI) digestion of mouse F9 embryonal carcinoma cells (a), mouse ht2 embryonic stem cells (b), human HT1080 cells (c), or mouse NIH 3T3 cells (d and e) were fractionated in isokinetic sucrose gradients, and the DNAs purified from individual gradient fractions were sized on agarose gels. After blotting to a nylon membrane, the samples were probed for mouse minor satellite (a, b, d, and e) or for human α satellite (c). The relationships between DNA size and sedimentation velocity (fraction number) reveal the difference in conformation between satellite and corresponding bulk chromatins. (f) Modeling of the relationship between length and sedimentation coefficient for undisrupted and disrupted helical chromatin fibers. The relative sedimentation coefficients for undisrupted 30-nm diameter chromatin fibers, Su, (■) were derived from Eq. 1 by taking the fibers to comprise 6 nucleosomes per helical turn, a pitch of 11 nm and a repeat length of 190 bp (Figs. 7 and 8). The sedimentation coefficient of disrupted chromatin fibers Sd (●, □), was derived from Sd = Su − (Pdl⋅EdSu), where Pd is the probability of a disruption, l is the fiber length, and Ed is the (negative) effect of the disruption on the sedimentation coefficient. In the examples shown, Pd and Ed were set at 0.166 and −0.01 (●) and at 0.04 and −0.035 (□), respectively.
Discussion
Nature of the Distinctive Centromeric Chromatin Structure.
For the idealized, canonical higher-order 30-nm chromatin fiber (1, 5, 23), the sedimentation coefficients of fibers with the same nucleosomal repeat length will be a function of their molecular weights (M), fiber lengths (l), and frictional ratios (f/fo) as given by the following equation (24):
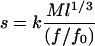 |
1 |
Our analyses (Figs. 2 and 3) provide values for the lengths of DNA associated with satellite and bulk higher-order chromatin fibers, which have the same sedimentation velocity (coefficient), allowing us to derive a relationship (2) that can be used to investigate how the equivalence in sedimentation coefficient could be explained in terms of the molecular weights or lengths of the chromatin fibers.
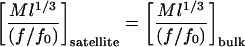 |
2 |
If the higher sedimentation velocity of satellite chromatin were attributable to additional bound protein, with no change in fiber shape, this would require a 40–50% increase in the protein to DNA ratio, compared with bulk chromatin. Although this result is not supported by current evidence (21, 25), we investigated this possibility by analyzing our chromatin, after crosslinking, by cesium chloride density gradient centrifugation (Fig. 5). Bulk mouse chromatin bands mainly as a single peak, with a buoyant density (1.443 g/ml) appropriate for a chromatin of this repeat length (7). The majority of chromatin containing the B2 repeat bands at the same density as bulk chromatin. In contrast, chromatin containing either minor and major satellite DNA appears to band as two peaks, both shifted to lighter densities relative to bulk. One expects a small shift in this direction because of the lower buoyant density of satellite DNA (1.69 as opposed to 1.70 for bulk). Taking this fact into account, the satellite chromatin peak close to bulk has a density (1.437 g/ml) that suggests a protein to DNA ratio 1.05 greater than bulk, which could be within experimental error. For the other peak, which is shifted to an even lower density (1.417 g/ml), the protein to DNA ratio is about 1.24 greater than bulk. Minor and major satellite chromatins display the same behavior although the relative amounts in the two peaks are a little different.
Figure 5.

Chromatin containing satellite DNAs have a higher protein to DNA ratio than bulk chromatin. Chromatin fragments isolated from an 80-mM NaCl sucrose gradient were crosslinked with formaldehyde and then fractionated in a cesium chloride density gradient. The distribution profile for bulk chromatin (■) was determined by measuring the A260 of individual fractions whereas the profiles for minor satellite (●), major satellite (○), and the B2 repeat (□) chromatins were determined by dot-blot analysis of DNA recovered from the gradient fractions. The density profile for the cesium chloride gradient is also indicated (+).
Overall, the average protein to DNA ratios for minor and major chromatins are 15 and 13% greater than bulk, respectively. This amount is substantially less than that required to explain the sedimentation behavior of satellite chromatin simply on the basis of an increase in molecular weight. Therefore, the distinctive sedimentation property of satellite chromatin must at least in part reflect a difference in shape or conformation.
The divergent relationship between the curves relating DNA size (fiber length) and sedimentation velocity for centromeric heterochromatin and for bulk chromatin (Figs. 2 and 4) suggests that the satellite-containing chromatin may adopt a more regular helical structure than bulk chromatin. By using Eq. 1 (24), we calculated the relative sedimentation coefficients (S/k) for a range of lengths of a regular 30-nm diameter chromatin fiber comprising six nucleosomes per helical turn, a pitch of 11 nm, and a repeat length of 190 bp. The effect of higher-order fiber irregularity was then incorporated into this relationship by assuming that a point of disruption increases the frictional coefficient, and therefore reduces the sedimentation coefficient, of a chromatin fiber (7) and that the probability of such a disruption is a function of fiber length. The results presented in Fig. 4f were generated by using this approach, and the relationship between the curves obtained for regular and disrupted fibers bears a close resemblance to the experimental data collected (Fig. 4, compare f with a–e). Thus, our sedimentation analyses are consistent with the interpretation that satellite DNA-containing chromatin fibers adopt an orderly helical structure whereas bulk higher-order fibers are not regular structures but are occasionally interrupted by a disruption. This proposal is schematically represented in Fig. 6.
Figure 6.

Schematic representation of proposed higher-order structures of satellite and bulk chromatin fibers.
Determinants of Centromeric Chromatin Structure.
The compact state of the higher-order fiber of centromeric heterochromatin may reflect the influence of proteins that are targeted to the centromere and/or the nature of its constituent DNA sequence. Because the extent of higher-order folding of chromatin is a function of the amount of linker histone associated with the fiber (1, 2), the distinctive sedimentation properties of satellite chromatin could be a consequence of a relatively high linker histone to nucleosome ratio. However, previous studies tend to suggest that these proteins are actually less abundant in satellite chromatin (25–27). Numerous other proteins specifically associate with the centromere throughout the cell cycle. These proteins include centromere-associated protein (CENP)-A, a variant of the core histone H3, which is incorporated into nucleosomes but does not appear to confer any distinctive features at this level of structure (28, 29). CENP-B, pJα, and high mobility group (HMG)-I, which recognize sequence motifs contained in mouse minor and/or human α-satellite DNAs, have been implicated in the assembly of satellite DNA into chromatin (6). Centromeric satellite DNAs are also usually extensively methylated (30) and bind MeCP2 (31), mbd2, and mbd4 (32). Heterochromatin protein 1 (HP1) localizes at heterochromatin (33–35) and consequently is concentrated at centromeres (36, 37). At present, the role of all of these chromatin-associated proteins as determinants of higher-order chromatin structure remains to be characterized.
The tandemly repeated nature of satellite DNA is also likely to influence higher-order chromatin structure. In vivo studies (38) have shown that nucleosomes adopt precise positions with respect to the underlying mouse satellite DNA sequence. This is in part due to the fact that multiple overlapping positions, invariably related to each other by an ≈10-bp periodicity, are available for occupation by nucleosomes within the satellite DNA repeat (39). Furthermore, those mouse genomic DNA fragments that form the most stable, naturally occurring, positioned nucleosomes come from the centromeric regions of mouse chromosomes (40). Thus, mouse minor satellite DNA could establish a regular array of strongly positioned nucleosomes (41) with the potential to fold into a higher-order fiber of almost paracrystalline order (42). This proposal is supported by the observation that the related, but more diverged, mouse major satellite sequence adopts a higher-order chromatin structure intermediate between that of bulk and centromeric minor satellite (Fig. 2).
Concluding Remarks.
Our observations demonstrate that higher-order chromatin fibers derived from functionally distinct regions of the chromosome can be distinguished by structural criteria. The regular, and therefore more condensed, folding of centromeric chromatin fibers is intuitively compatible with the distinctly heterochromatic nature of centromeres. The formation of the centromere and the assembly of a competent kinetochore may be facilitated by a particular type of local chromosomal architecture (6), and the capacity of satellite DNA to adopt a uniform higher-order fiber could contribute to this process. A regularly folded higher-order fiber could constitute a structure predisposed to the establishment of stable, higher levels of chromosomal packaging, partly, perhaps, as a consequence of presenting itself as a preferred substrate for the binding of heterochromatin-specific proteins.
Supplementary Material
Acknowledgments
We are grateful to Drs. Wendy Bickmore, Colin Davey, Marieta Gencheva, and Gary Felsenfeld for their comments and advice on the manuscript. We also thank Professor Peter Stockley for providing access to analytical ultracentrifuge facilities. This work was supported by a Wellcome Trust project grant to J.A. and a Medical Research Council studentship to N.G.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Thoma F, Koller T H, Klug A. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allan J, Cowling G J, Harborne N, Cattini P, Craigie R, Gould H. J Cell Biol. 1981;90:279–288. doi: 10.1083/jcb.90.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Holde K E. Chromatin. New York: Springer; 1988. [Google Scholar]
- 4.Felsenfeld G, McGhee J D. Cell. 1986;44:375–377. doi: 10.1016/0092-8674(86)90456-3. [DOI] [PubMed] [Google Scholar]
- 5.Ramakrishnan V. Crit Rev Eukaryot Gene Express. 1997;7:215–230. doi: 10.1615/critreveukargeneexpr.v7.i3.20. [DOI] [PubMed] [Google Scholar]
- 6.Choo K H A. The Centromere. Oxford: Oxford Univ. Press; 1997. [Google Scholar]
- 7.Caplan A, Kimura T, Gould H, Allan J. J Mol Biol. 1987;193:57–69. doi: 10.1016/0022-2836(87)90626-7. [DOI] [PubMed] [Google Scholar]
- 8.Fisher E A, Felsenfeld G. Biochemistry. 1986;25:8010–8016. doi: 10.1021/bi00372a033. [DOI] [PubMed] [Google Scholar]
- 9.Andersson K, Mahr R, Bjorkroth B, Daneholt B. Chromosoma. 1982;87:33–48. doi: 10.1007/BF00333508. [DOI] [PubMed] [Google Scholar]
- 10.McGhee J D, Nickol J M, Felsenfeld G, Rau D C. Nucleic Acids Res. 1983;11:4065–4075. doi: 10.1093/nar/11.12.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allan J, Harborne N, Rau D C, Gould H. J Cell Biol. 1982;93:285–297. doi: 10.1083/jcb.93.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mountford P, Zevnik B, Duwel A, Nichols J, Li M, Dani C, Robertson M, Chambers I, Smith A. Proc Natl Acad Sci USA. 1994;91:4303–4307. doi: 10.1073/pnas.91.10.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith A G. J Tissue Cult Methods. 1991;13:89–94. [Google Scholar]
- 14.Cereghini S, Yaniv M. EMBO J. 1984;3:1243–1253. doi: 10.1002/j.1460-2075.1984.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noll H. In: Techniques in Protein Biosynthesis. Campbell P N, Sargent J R, editors. II. London: Academic; 1969. pp. 101–178. [Google Scholar]
- 16.Kipling D, Wilson H E, Mitchell A R, Taylor B A, Cooke H J. Chromosoma. 1994;103:46–55. doi: 10.1007/BF00364725. [DOI] [PubMed] [Google Scholar]
- 17.Lewis J D, Meehan R R, Henzel W J, Maurerfogy I, Jeppesen P, Klein F, Bird A. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 18.Chambers I, Cozens A, Broadbent J, Robertson M, Lee M, Li M, Smith A. Biochem J. 1997;328:879–888. doi: 10.1042/bj3280879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fantes J A, Oghene K, Boyle S, Danes S, Fletcher J M, Bruford E A, Williamson K, Seawright A, Schedl A, Hanson I, et al. Genomics. 1995;25:447–461. doi: 10.1016/0888-7543(95)80045-n. [DOI] [PubMed] [Google Scholar]
- 20.Selig S, Ariel M, Goitein R, Marcus M, Cedar H. EMBO J. 1988;7:419–426. doi: 10.1002/j.1460-2075.1988.tb02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pashev I G, Dimitrov S I, Ivanov I G, Markov G G. Eur J Biochem. 1983;133:379–382. doi: 10.1111/j.1432-1033.1983.tb07473.x. [DOI] [PubMed] [Google Scholar]
- 22.Keohane A M, O'Neill L P, Belyaev N D, Lavender J S, Turner B M. Dev Biol. 1996;180:618–630. doi: 10.1006/dbio.1996.0333. [DOI] [PubMed] [Google Scholar]
- 23.McGhee J D, Nickol J M, Felsenfeld G, Rau D C. Cell. 1983;33:831–841. doi: 10.1016/0092-8674(83)90025-9. [DOI] [PubMed] [Google Scholar]
- 24.Harrison B D, Klug A. Virology. 1966;30:738–740. doi: 10.1016/0042-6822(66)90179-6. [DOI] [PubMed] [Google Scholar]
- 25.Jasinskas A, Hamkalo B A. Chromosome Res. 1999;7:341–354. doi: 10.1023/a:1009211929408. [DOI] [PubMed] [Google Scholar]
- 26.Mazrimas J A, Balhorn R, Hatch F T. Nucleic Acids Res. 1979;7:935–946. doi: 10.1093/nar/7.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathew C G P, Goodwin G H, Igokemenes T, Johns E W. FEBS Lett. 1981;125:25–29. doi: 10.1016/0014-5793(81)80988-x. [DOI] [PubMed] [Google Scholar]
- 28.Shelby R D, Vafa O, Sullivan K F. J Cell Biol. 1997;136:501–513. doi: 10.1083/jcb.136.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoda K, Ando S, Morishita S, Houmura K, Hashimoto K, Takeyasu K, Okazaki T. Proc Natl Acad Sci USA. 2000;97:7266–7271. doi: 10.1073/pnas.130189697. . (First Published June 6, 2000; 10.1073/pnas.130189697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchell A R, Jeppesen P, Nicol L, Morrison H, Kipling D. J Cell Sci. 1996;109:2199–2206. doi: 10.1242/jcs.109.9.2199. [DOI] [PubMed] [Google Scholar]
- 31.Nan X S, Tate P, Li E, Bird A. Mol Cell Biol. 1996;16:414–421. doi: 10.1128/mcb.16.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendrich B, Bird A. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eissenberg J C, Elgin S C. Curr Opin Genet Dev. 2000;10:204–210. doi: 10.1016/s0959-437x(00)00058-7. [DOI] [PubMed] [Google Scholar]
- 34.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Nature (London) 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 35.Bannister A J, Zegerman P, Partridge J F, Miska E A, Thomas J O, Allshire R C, Kouzarides T. Nature (London) 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 36.Aagaard L, Schmid M, Warburton P, Jenuwein T. J Cell Sci. 2000;113:817–829. doi: 10.1242/jcs.113.5.817. [DOI] [PubMed] [Google Scholar]
- 37.Taddei A, Maison C, Roche D, Almouzni G. Nat Cell Biol. 2001;3:114–120. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X Y, Horz W. J Mol Biol. 1984;176:105–129. doi: 10.1016/0022-2836(84)90384-x. [DOI] [PubMed] [Google Scholar]
- 39.Linxweiler W, Horz W. Cell. 1985;42:281–290. doi: 10.1016/s0092-8674(85)80123-9. [DOI] [PubMed] [Google Scholar]
- 40.Widlund H R, Cao H, Simonsson S, Magnusson E, Simonsson T, Nielsen P E, Kahn J D, Crothers D M, Kubista M. J Mol Biol. 1997;267:807–817. doi: 10.1006/jmbi.1997.0916. [DOI] [PubMed] [Google Scholar]
- 41.Sun F L, Cuaycong M H, Elgin S C. Mol Cell Biol. 2001;21:2867–2879. doi: 10.1128/MCB.21.8.2867-2879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson R T. Prog Nucleic Acid Res Mol Biol. 1991;40:143–184. doi: 10.1016/s0079-6603(08)60841-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.