

Abstract
Hepatitis C virus (HCV) is a major global health concern, and though therapeutic options have improved, no vaccine is available despite decades of research. As HCV can rapidly mutate to evade the immune response, an effective HCV vaccine must rely on identification and characterization of sites critical for broad immune protection and viral neutralization. This knowledge depends on structural and mechanistic insights of the E1 and E2 envelope glycoproteins, which assemble as a heterodimer on the surface of the virion, engage coreceptors during host cell entry, and are the primary targets of antibodies. Due to the challenges in determining experimental structures, structural information on E1 and E2 and their interaction is relatively limited, providing opportunities to model the structures, interactions, and dynamics of these proteins. This review highlights efforts to model the E2 glycoprotein structure, the assembly of the functional E1E2 heterodimer, the structure and binding of human coreceptors, and recognition by key neutralizing antibodies. We also discuss a comparison of recently described models of full E1E2 heterodimer structures, a simulation of the dynamics of key epitope sites, and modeling glycosylation. These modeling efforts provide useful mechanistic hypotheses for further experimental studies of HCV envelope assembly, recognition, and viral fitness, and underscore the benefit of combining experimental and computational modeling approaches to reveal new insights. Additionally, computational design approaches have produced promising candidates for epitope-based vaccine immunogens that specifically target key epitopes, providing a possible avenue to optimize HCV vaccines versus using native glycoproteins. Advancing knowledge of HCV envelope structure and immune recognition is highly applicable toward the development of an effective vaccine for HCV and can provide lessons and insights relevant to modeling and characterizing other viruses.
Keywords: hepatitis C virus, vaccines, modeling, design, E1E2, glycoproteins, antibodies
Introduction
Hepatitis C virus (HCV) is estimated to have infected over 70 million globally, with millions of new cases every year (1). Chronic HCV infection can lead to cirrhosis and hepatocellular carcinoma (HCC) and deaths due to HCV are rising worldwide (1). In the United States, the yearly rate of deaths resulting from HCV infection has surpassed that of human immunodeficiency virus (HIV) and other infectious diseases (2). Direct-acting antivirals (DAA) for treatment of HCV infection have high cure rates, but face major issues: limited patient accessibility due to high costs of treatment (3), little to no awareness of infection in most HCV-positive individuals (4), and neither prevention of reinfection (5) nor elimination of HCC risk (6) in cleared HCV patients following DAA treatments. Thus, there is an ongoing major need for an effective prophylactic vaccine for HCV in order to greatly reduce global disease burden (4, 7).
A major barrier to vaccine and targeted therapeutic efforts is the high sequence variability of HCV, as exemplified by its seven confirmed genotypes, which are subdivided into 86 confirmed subtypes as of June 2017 (8) that can differ by greater than 15% in sequence (9). Furthermore, HCV rapidly mutates to form quasispecies within infected individuals, permitting active escape from neutralizing antibodies; this mechanism was clearly demonstrated in a clinical trial of monoclonal antibody HCV therapy followed by deep sequencing of HCV in patients (10, 11). Effective targeting of this diverse virus would be greatly facilitated by a detailed understanding of the molecular determinants of viral fitness, assembly, and function (12).
The envelope glycoproteins E1 and E2 are targets of anti-HCV antibodies (13), and have been used in numerous B cell vaccine development efforts (14–18) and several clinical trials (19, 20) [reviewed by Fauvelle et al. (21)]. Epitope mapping and other characterization efforts have classified E2 antibody epitopes into five antigenic domains (A–E) (22), a nomenclature that will be used in this review. Alternative definitions such as antigenic regions (antigenic regions 1–3) (23) and epitopes I–III (24) have been used to identify these regions on the E2 surface, in addition to epitopes on E1E2 (antigenic regions 4–5) (25). Despite advances from numerous epitope mapping studies, the overall structure of these glycoproteins and the structural basis of neutralizing antibody engagement of many key epitopes have yet to be determined experimentally. Some structures representing portions of these proteins have been determined to date, spanning a conserved “core” region of E2, portions of E1, and multiple mAb-bound E1 and E2 peptides (Figure 1; Table 1). In contrast, other highly variable viruses, such as HIV and influenza, have likewise been longstanding targets of vaccine design efforts, and the assembly of their envelope glycoproteins, hemagglutinin (HA), and Env have been determined at high resolution (26, 27). Additionally, there are many HA and Env neutralizing antibodies structurally characterized in complex with their epitopes (28–30), providing insights that enabled a number of successful structure-based vaccine design efforts (31–34). Given the relatively limited availability of HCV structural data, as well as the challenges for experimental structure determination presented by innate flexibility (22, 35, 36) and high glycosylation (37) of HCV glycoproteins, there is a major opportunity to bridge gaps in knowledge of current structural and mapping data through computational structural modeling, enabling a comprehensive view of glycoprotein structure, recognition, and dynamics.
Figure 1.

Representative crystallographic and NMR structures of E1E2 regions. Hepatitis C virus (HCV) peptides and proteins in all structures are colored green. Antibody heavy chains are colored gray and light chains are colored light pink. PDB codes of representative structures are 4UOI (192–270), 4N0Y (314–324/IGH526), 2KNU (314–342), 1EMZ (350–370), 4XVJ (412–423/HC33.1), 4DGY (412–423/HCV1), 4MWF (421–645/AR3C), 4JZN (435–446/HC84.1), 5NPJ (532–540/DAO5), and 2KZQ (684–719). Residue ranges of the E1E2 sequence corresponding to specific sites are highlighted by colored bars for reference: H-111 epitope at N-terminus of E1 (aa 192–202, dark blue), E2 hypervariable region 1 (aa 384–410, gray), Domain E (aa 412–423, blue), Domain D/AR3 (aa 434–446, magenta), and Domain B/AR3 (aa 529–535, magenta). Other regions of E1 and E2 with corresponding structures are shown in black bars, and black dashed lines represent gaps in a crystal structure.
Table 1.
Experimentally determined structures of E1, E2, and monoclonal antibodies.
| Structure codea | Hepatitis C virus (HCV) glycoproteinb | Residue rangec | Antibody | Reference |
|---|---|---|---|---|
| X-ray crystallography | ||||
| 4UOI | E1 | 192–270 | – | (38) |
| 4N0Y | E1 | 314–324 | IGH526 | (39) |
| 4GAG | E2 | 411–424 | AP33 | (40) |
| 4GAJ | E2 | 412–423 | AP33 | (40) |
| 4GAY | E2 | Unbound mAb | AP33 | (40) |
| 4DGY, 4DGV | E2 | 412–423 | HCV1 | (41) |
| 4G6A | E2 | 412–423 | AP33 | (42) |
| 4HS6 | E2 | 412–423 | MRCT10.362 | (43) |
| 4HS8 | E2 | 412–423 | hu5B3.v3 | (43) |
| 4WHT, 4WHY | E2 | 412–423 | 3/11 | (35) |
| 4XVJ | E2 | 412–423 | HC33.1 | (44) |
| 5FGB | E2 | 417–421 | HC33.4 | (45) |
| 5FGC | E2 | 415–423 | HC33.8 | (45) |
| 5EOC | E2 | 412–422d | C2 | (16) |
| 5KZP | E2 | 412–423d | HCV1 | (17) |
| 5VXR | E2 | 412–423 | MAb24 | (46) |
| 4MWF | E2 | 421–645e | AR3C | (47) |
| 4WEB | E2 | 486–645 | 2A12 | (48) |
| 4Q0X | E2 | 434–442 | mAb#12 | (49) |
| 4HZL | E2 | 430–442 | mAb#8 | (50) |
| 4JZN | E2 | 435–446 | HC84.1 | (51) |
| 4JZO | E2 | 436–446 | HC84.27 | (51) |
| 5ERW | E2 | 438–446 | HC84.26 | –f |
| 5ESA | E2 | Unbound mAb | HC84.26 | –f |
| 4Z0X | E2 | 435–446 | HC84.26.5D | (52) |
| 5NPH, 5NPI, 5NPJ | E2 | 532–540 | DAO5 | (53) |
| 3U6R | E2 | Unbound mAb | 1:7 | (54) |
| 4JVP | E2 | Unbound nanobody | D03 | (55) |
| Nuclear magnetic resonance | ||||
| 1EMZ | E1 | 350–370 | – | (56) |
| 2KNU | E1 | 314–342 | – | (57) |
| 2KZQ | E2 | 684–719 | – | (58) |
| Electron microscopyg | ||||
| 5759 | E2 | 384–717 | AR3A | (47) |
| 5760 | E2 | 384–717 | AR3A, AR2A | (47) |
| 5761 | E2 | 384–717 | AR2A, CD81 | (47) |
| 8338, 8339, 8340 | E2 | 412–645 | AR1B, AR2A, HCV1 | (36) |
aProtein Data Bank (59) or EMDataBank (60) codes shown. Multiple codes are shown in cases with multiple entries reported from same study containing the same residue range and binding partner(s), corresponding to different crystallographic symmetry forms, electron microscopy reconstructions, or HCV isolate sequences.
bIn the case of unbound antibody, glycoprotein target of antibody is given for reference.
cResidue numbering based on H77 isolate. For crystallographic structures, range reflects resolved residues present in coordinates.
dCyclic epitope-based designs are present in these structures.
eThis E2 core construct included engineered deletions of residues.
fThe coordinates for these X-ray structures have been released in the PDB (59) but have no publications associated with them.
gThese negative stain electron microscopy structures have resolutions of 16–30 Å, thus provide approximate envelopes for fitting high-resolution crystallographic or modeled structures.
This review provides an overview of efforts to model HCV envelope structure and recognition, which have collectively yielded many valuable insights into this virus. These efforts include initial work to model the E2 structure, recent modeling of the full-length E1E2 heterodimer, and modeling focused on other aspects of HCV, such as the dynamics of epitopes and recognition of antibodies or coreceptors; a subset of these studies is summarized in Table 2. Models and hypotheses from these studies can be used to inform future experimental and computational modeling efforts, as well as structure-based design of effective vaccines.
Table 2.
Representative modeling studies of hepatitis C virus envelope glycoproteins and receptors.
| Target | Model | Methodsa | Year | Reference |
|---|---|---|---|---|
| E2 | Structure | Homology-based modeling | 2000 | (61) |
| E2 | Structure | Homology-based modeling, disulfide mapping | 2010 | (62) |
| E2-CD81 | Complex structure | Restraints-guided docking | 2013 | (47) |
| E2 | Front layer dynamics | Molecular dynamics simulation | 2016 | (36) |
| E1E2 transmembrane | E1 trimerization, E1E2 heterohexamer | Docking with restraints | 2015 | (63) |
| E1E2 | Structure | Evolutionary constraints-based structure prediction, homology-based modeling, experimental mapping residue constraints | 2017 | (64) |
| E1E2 | Structure, high order assembly | Homology-based modeling, ab initio structure prediction, experimental mapping residue constraints, docking | 2017 | (65) |
| SR-BI | Structure | Homology-based modeling | 2013 | (66) |
| CD81-Claudin | Structure | Homology-based modeling, docking | 2012 | (67) |
aSummary of modeling methods used.
Models of the E2 Structure
Prior to experimentally determined structures of the E2 glycoprotein, computational models were developed to predict its tertiary and quaternary assembly. These efforts used structures of flavivirus and alphavirus class II fusion proteins as modeling templates (61, 62). A crystal structure of the E2 glycoprotein of tick-borne encephalitis virus (PDB code 1SVB) (68) served as the main template for the first of these modeling studies, which was reported over 15 years ago (61). The authors predicted that E2 assembles into an elongated monomer and also described putative E2 homodimerization and a possible site of interaction with E1. Further analysis of this model found that the binding regions predicted for CD81 and multiple E2 mAbs were exposed epitopes on the modeled E2 surface. A more recent E2 modeling study was largely based on the structure of the Semliki Forest virus E1 glycoprotein (PDB code 2ALA) (69), with particular emphasis on shared secondary structure elements, and incorporated nine experimentally determined E2 disulfide bonds as modeling constraints (62). The resulting model included three predicted domains for E2, with domain I (the first in order of amino acid sequence) corresponding to a β-sandwich positioned between the other two domains and forming a tightly packed CD81-binding site that roughly corresponds to antigenic domains B, D, and E. As noted by the authors of the latter modeling study (62), these two E2 models are divergent in several regards, including their predicted disulfide bonds, predicted E2 oligomerization and degree of coverage of the E2 glycoprotein. Subsequent X-ray crystallographic determination of two E2 core crystal structures revealed features distinct from structurally characterized class II fusion proteins (70, 71), including more compactness than the classical three domain organization of class II fusion proteins, despite retaining its immunoglobulin β-sandwich domain (47). Overall differences in architecture presented a likely impediment to template-based modeling, notwithstanding potentially accurate prediction of certain features and secondary structure elements. Regardless, these E2 modeling studies were important first steps in characterizing HCV glycoproteins, providing useful testable hypotheses in the absence of an experimentally determined E2 structure.
Models of E1E2 Assembly
Currently, no experimentally determined structure is available for the E1E2 complex, which has led to two recent studies that have presented structural models of this assembly (64, 65). For clarity, they will be referred to as E1E2-C and E1E2-F, after their respective first authors. A third E1E2 model has been proposed, but does not contain a complete heterodimer and, therefore, will not be discussed in detail (72). The E1E2-F and E1E2-C models were generated using distinct methodologies. The E1E2-C model was generated through mapping antibody epitopes with shotgun mutagenesis (73), residue contact prediction with evolutionary coupling analysis (74) supplemented by known contacts of the E2 core crystal structure (47), as well as β-sheet pairing predictions using the bbcontacts algorithm (75). The final E1E2-C model of the heterodimer was generated using the CNS suite (76) and a distance geometry simulated annealing protocol. The E1E2-F model was likewise generated using a detailed computational pipeline, while also ensuring that the model corroborated previous experimental findings. Prediction of the E1 structure combined a partial crystal structure of E1 (38) with structural homolog phosphatidylcholine transfer protein (PDB code: 1LN2) (77) in the Molecular Operating Environment program (78). E2 was modeled in the Robetta server (http://robetta.bakerlab.org/), which added missing loops and termini to the E2 core crystal structure. Following ab initio prediction and molecular dynamics (MD) simulations of E1 and E2 transmembrane regions (TMs), RosettaDock (79) was used to dock the E1 and E2 models to predict their heterodimeric assembly, followed by symmetric docking of the E1E2 model to form heterohexameric E1E2 models (trimers of E1E2).
Comparison of the E1E2-C and E1E2-F models reveals some similarities, but also major distinctions between them (Figure 2). Unsurprisingly, the E2 core region is mostly conserved between the two models, as both E1E2-C and E1E2-F incorporated residue contacts from existing E2 core structures. This conservation includes the overall arrangement of antigenic domains B, D, and E. However, the quaternary structure of the two models display striking differences, with a dramatic change of E1 orientation relative to E2. One notable difference is an inter-chain disulfide bond at C272–C452, which is proposed by E1E2-C on the basis of their antibody epitope mapping data, but is not present in E1E2-F. Additionally, E2 residues 546–547, which are associated with antigenic domain C as well as E1E2 mAb binding based on global epitope mapping studies (80, 81), are located at the predicted interface with E1 in E1E2-F but not E1E2-C. This site has been associated with E1E2 assembly in a recent screening effort, which found that a peptide from JFH-1 (aa 546–560 based on H77 numbering) inhibited HCV entry and bound E1E2 (82). Finally, there are differences in model coverage of E1 and E2 (E1E2-F represents the full glycoprotein sequences), as well as the conformations and orientations of the flexible region at the N-terminus of E2 (HVR1 and antigenic domain E). These models offer intriguing possible modes of E1E2 heterodimerization, providing an avenue to potentially design stabilized vaccines in the absence of an experimentally determined structure, and future studies can confirm (e.g., through structure-guided mutagenesis of predicted interface residues) or refine these models.
Figure 2.

Structural models of E1E2 heterodimeric assembly. (A) E1E2 model from Castelli et al. (E1E2-C) (64) in comparison with (B) E1E2 model from Freedman et al. (E1E2-F) (65), oriented in the same frame of reference based on E2 core regions. E1 and E2 glycoproteins are shown as tan and cyan cartoons, respectively, while key epitopes are colored and labeled, as in Figure 1: H-111 epitope at N-terminus of E1 (“H-111,” aa 192–202, dark blue), E2 hypervariable region 1 (HVR1, aa 384–410, gray), Domain E (aa 412–423, blue), Domain D/AR3 (aa 434–446, magenta), Domain B/AR3 (aa 529–535, magenta). Additionally, selected features of modeled E1E2 are highlighted: the predicted E1–E2 disulfide bond of E1E2-C (C272–C452), shown as yellow sticks, and E2 residues L546–G547, predicted to interact with E1 in E1E2-F model, are shown in spacefill on both models. C-terminal residues of E1 and E2 are also labeled for both models (H312, S711 for E1E2-C, A383, A746 for E1E2-F).
Recent Modeling Studies of E1 and E2
Other studies have used existing crystal structures to explore conformational flexibility and assembly, capturing the dynamic properties of E2. Flexibility of the CD81-binding site (CD81bs) has been examined in a recent study using MD simulations, hydrogen–deuterium exchange (HDX), and calorimetry (36). The MD simulations suggested that the helical region near residue 434 displays a pronounced tendency to “drift” away from the E2 core, which is supported by crystallographic studies of multiple antibodies bound to the corresponding epitope of the peptide (49). Mobility of these regions has also been examined using an E2 core crystal structure plus modeled domain E, finding a broad range of conformations that occasionally resembled those observed in X-ray structures of the antibody-domain E complex (83).
Studies focused on modeling E1E2 TM domains have provided insights into determinants of E1E2 heterodimerization and assembly. Following descriptions of SDS-resistant E1E2 TM heterodimers and E1 trimers, a trimeric model of E1 TM domains was generated (63). This model was partially based on an experimentally determined structure of the monomeric E1 TM (PDB code: 1EMZ) (56) and also included constraints to enforce putative inter-helical interactions between G354xxxG358 residues, a motif essential for E1 TM assembly and conserved in other helix–helix interactions (84). Critical charged and polar residues were exposed in the trimeric model, allowing E1 trimers to form key interactions with E2 such as the putative K370–D728 salt bridge, which was also observed in a separate study that performed MD simulations of the E1E2 TM heterodimer (85, 86). These studies and others (87, 88) have used modeling on this small yet critical region to gain a clearer picture of E1E2 association.
In combination with experimental mutagenesis data, modeling has been used to explore how residue substitutions affect glycoprotein stability and structural integrity. Using the program Rosetta, in silico alanine mutagenesis of all E2 residues available in one of the E2 core crystal structures predicted changes in protein stability for each mutant (80). Alanine mutants with greatest predicted destabilizing effects on E2 corresponded to those with experimentally measured loss of binding for 14 conformationally sensitive HCV mAbs during global alanine scanning mutagenesis of E2. In the same study, alanine scanning data from each mAb was analyzed by hierarchical clustering to form groups of residues that delineated energetically linked regions on the E2 surface and core. These studies highlight how the incorporation of experimental mutagenesis data and other techniques (e.g., HDX) with modeling methods can reveal key aspects of glycoprotein flexibility and structural determinants.
Modeling Antibody Recognition
Modeling conserved epitopes of HCV glycoproteins has been valuable for elucidating the structural basis of broadly neutralizing antibody (bnAb) recognition. Crystal structures for the domain E peptide (E2 residues 412–423) bound to HCV1 (41), HC33.1 (44), 3/11 (35), and AP33 (40, 42) established different conformations of the same conserved epitope. Understanding the structural basis of these variable conformations was critical for determining why rare domain E mutations evaded neutralization by some of these antibodies, but not all (43, 89). Computational alanine scanning of antigenic domain E bound to HC33.1 predicted a decrease in antibody affinity when key binding residues were mutated, but no change in affinity when a “glycan shift” viral escape mutation was modeled (44). The program GlyProt (90) was used to model E2 glycosylation in the HCV1 and HC33.1 complexes, showing that glycosylated N415 in domain E would be sterically unfavorable for binding by HCV1, which like AP33 engages the β hairpin form of the epitope, but it would be permitted at the exposed N415 residue in the extended conformation bound by HC33.1 (44). Additional modeling of domain E structures in the same study used the PEP-FOLD server (91) to generate ab initio peptide models that largely matched a β-hairpin conformation, suggesting that this folding pattern is preferred for domain E in the absence of antibody engagement and that this conformation can be disrupted by several domain E mAbs (35, 44). Computational mutagenesis and modeling not only helped to delineate domain E antibody recognition, but also domain D recognition by an affinity-matured antibody (52). These techniques can be used to build on structural knowledge of other antibody epitopes to E1, E2, or the E1E2 heterodimer, especially if similar crystal structures of antibody–antigen complexes provide informative comparisons.
Modeling Receptor Structure and Recognition
Although many E1E2 modeling efforts have focused on antibody–antigen interactions or heterodimerization, some studies have examined the structures of host entry receptors and their interactions. The tetraspanin CD81 (92), scavenger receptor class B type I (SR-BI) (93), and tight junction proteins claudin-1 (CLDN1) (94) and occludin (OCLN) (95) represent the minimal set of HCV coreceptors and together are sufficient for HCV entry (96). Determinants of E1E2, glycoprotein–receptor, and receptor–receptor interactions are shown in Figure 3, summarizing current knowledge through high resolution or homologous protein structures that may inform prospective modeling studies. CD81 and SR-BI bind directly to E2 (92, 93) and CLDN1 associates with CD81 to permit HCV entry (97), but the basis of OCLN viral engagement is unknown. CD81 has been characterized the most among these receptors, due to its critical role in HCV entry, infection, and cell-to-cell transmission (98). Kong et al. modeled the CD81–E2 interface using restraints-guided docking using restraints-guided docking (47) with the HADDOCK modeling program (99), which incorporated mutagenesis data into structure prediction. The model was corroborated by a negative stain electron microscopy structure containing E2 and CD81 large extracellular loop (LEL) reported in the same study; the interface contained the CD81-LEL C and D helices, which are implicated in E2 binding (100). To validate this model experimentally, the authors generated E2 mutants based on their docking model that disrupted CD81 binding. A subsequent study (101) concentrated on the interface between CD81-LEL and antigenic domain D, using PEP-FOLD (91) to model the peptide and the AutoDock Vina program (102) for docking to a CD81-LEL crystal structure. CD81 MD have also explored CD81-LEL flexibility, and several crystal structures found pH-dependent conformational changes in these loops (103). The CD81-E2 interface could soon be resolved in greater detail through additional modeling or experimental studies, given that new CD81 crystal structures are available (103, 104) and that CD81 binding determinants on E1E2 have recently been fully delineated through global alanine scanning (81).
Figure 3.
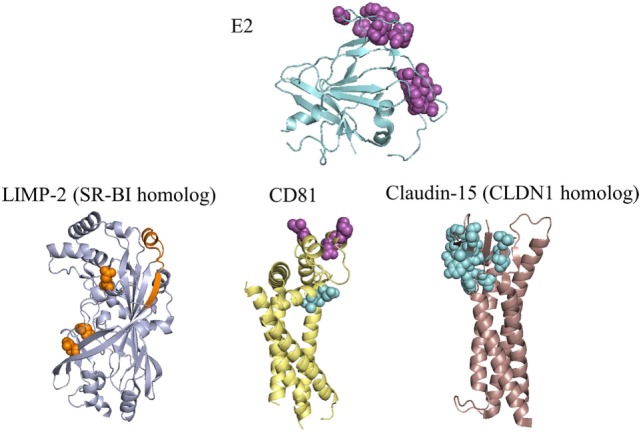
Residues of E2 and coreceptors that influence hepatitis C virus (HCV) entry and infection. E2 and three receptors are depicted with the most complete crystal structure available, or with a crystal structure of a homologous receptor. Purple spacefill residues on E2 showed <20% binding to CD81 when substituted to alanine (81, 105); residues with the same color and representation on CD81 showed reduced or eliminated binding to soluble E2 during random mutagenesis (100). Orange spacefill residues on LIMP-2 showed reduced binding to soluble E2 when mutated to a non-synonymous coding variant or the corresponding residue for mouse SR-BI (106, 107). Binding determinants of E2 to SR-BI are present in HVR1 (108), and are not present on the E2 crystal structure. Cyan spacefill residues on CD81 showed reduced association with CLDN1 when mutated to alanine (67), while cyan spacefill residues on CLDN1 showed either reduced binding to CD81 or decreased entry of HCVpp in alanine substitutions (67, 97, 109). PDB codes used are: 4MWF (E2), 5TCX (CD81), 4F7B (LIMP-2, representing SR-BI), and 4P79 (mouse claudin-15, representing claudin-1). SR-BI and CLDN1 have only moderate sequence identities to their structurally characterized homologs (LIMP-2 has 34% identity with SR-BI, mouse claudin-15 has 35% identity with human CLDN1), thus structures of these receptors may differ from the homologs shown.
Although fewer modeling studies have focused on other HCV receptors, these provide important insights into the structure and recognition of these molecules. SR-BI does not have a reported X-ray structure, making its interactions with E2 relatively challenging to model with protein docking methods. However, the crystal structure of the closely related LIMP-2 (PDB code: 4F7B) led to a homology model of SR-BI, which was then used to elucidate the structural basis of its role in cholesterol uptake (66). Related scavenger receptor CD36 also has a crystal structure available (PDB code: 5LGD) (110), and was recently proposed as an additional coreceptor that binds E1 (111). Several studies have examined the structural determinants of the CLDN1–CD81 interface (97, 112, 113). In silico mutagenesis of this interface revealed key binding residues (67), and MD simulations of CLDN1 point mutations showed disruptions of receptor structure thought to diminish HCV entry (114). There is no reported X-ray crystal structure of CLDN1, but several claudin family members have solved structures (115, 116).
Discussion
Given the numerous unknown aspects of the structural basis of HCV envelope glycoprotein assembly, as well as uncertainties regarding antibody and receptor recognition, there is a unique opportunity to leverage modern computational modeling and design algorithms to provide insights and testable mechanistic hypotheses for this system. Based on the challenges inherent in modeling this unique and dynamic viral envelope, future studies can utilize iterative experimental, and modeling approaches, where data-driven modeling is validated through experiments suggested by a model or sets of models. This paradigm has been utilized in previous studies to select and confirm models of antibody–antigen complexes (117, 118), as well as a modeled coiled coil assembly (119).
One additional area of recent interest has been the use of computational structure-based methods to design optimized protein and epitope-based immunogens for vaccines to better engage and elicit neutralizing antibodies, also known as “reverse vaccinology” (120). As seen for modeling, recent work has shown that iterative computational and experimental approaches are quite effective for vaccine design (121). Some have noted that HCV is a promising potential target for structure-based vaccine design (122), and early efforts have shown promise (15–17). Such work includes the design of scaffolded constructs based on the β hairpin form of antigenic domain E and an epitope from E1 (aa 314–324), and the display of these designs on protein nanoparticles, which showed maintained binding to the epitope-specific antibodies HCV1 and IGH526. In another study, a cyclic peptide design based on antigenic domain E, stabilized with a disulfide bond, was found to be immunogenic in mice; the X-ray structure of an induced murine antibody in complex with this design was determined (16), but no neutralizing antibodies were detected. A more recent vaccine design study reported two other cyclic antigenic domain E peptide designs, as well as a design of E2 with two copies of the antigenic domain E epitope based on structural similarity of a site on the E2 back layer to the β hairpin domain E structure (17). These designs elicited neutralizing antibodies in mice, but varied in H77 neutralization potency and showed limited response to the two non-H77 isolates tested (17). Follow-up studies as well as additional novel designs are needed to demonstrate the potential of rational vaccine design approaches for this virus. Furthermore, though cellular immunology is outside the scope of this review, fine mapping and molecular characterization of T cell epitopes may provide useful information to optimize vaccine constructs that will enhance or focus cellular immune responses, possibly in the context of a B-cell-based vaccine. The recently described structure of a T cell receptor engaging an immunodominant epitope from the HCV NS3 protein (123) is a compelling example for such a strategy.
The increasing application of powerful computational structural modeling techniques has led to a number of insights into HCV and its envelope glycoproteins. With the rapidly growing amount of data, including epitope mapping, structural characterization, and immune repertoire sequencing (124), there will be many opportunities to utilize these methods, to contribute further to the understanding of HCV immunogens, and to design an HCV vaccine. Centralized and up-to-date databases, resources, and standards for those focused on HCV research should facilitate these efforts. Effective resources may be analogous to a database developed for HIV bnAbs (125) or an existing database on HCV sequences and immunology (126). These resources will in turn permit the development of improved algorithms, more accurate models, and additional collaborative efforts focused on elucidating the native assembly and key features of the HCV envelope and eradicating HCV through an effective vaccine.
Author Contributions
Both authors wrote and edited this work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the submitted work was carried out in the absence of any personal, professional, or financial relationships that could potentially be construed as a conflict of interest. The handling Editor declared a past co-authorship with one of the authors BP.
Acknowledgments
The authors are grateful to Matteo Castelli and Nicasio Mancini (Vita-Salute San Raffaele University) for kindly sharing the coordinates of their E1E2 model, as well as Holly Freedman and Michael Houghton (University of Alberta) for providing a figure representing their E1E2 model. The authors also thank Eric Toth (University of Maryland) for comments on Table 1. This work was supported by NIH grants R21AI126582 and R01AI132213 to BP, as well as startup funding from the University of Maryland to BP. JG was supported by the University of Maryland Virology Program graduate training grant (NIH T32AI125186).
References
- 1.World Health Organization. Global Hepatitis Report 2017. Geneva, Switzerland: Global Hepatitis Programme, Department of HIV/AIDS, WHO; (2017). [Google Scholar]
- 2.Ly KN, Hughes EM, Jiles RB, Holmberg SD. Rising mortality associated with hepatitis C virus in the United States, 2003–2013. Clin Infect Dis (2016) 62:1287–8. 10.1093/cid/ciw111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callaway E. Hepatitis C drugs not reaching poor. Nature (2014) 508:295–6. 10.1038/508295a [DOI] [PubMed] [Google Scholar]
- 4.Cox AL. MEDICINE. Global control of hepatitis C virus. Science (2015) 349:790–1. 10.1126/science.aad1302 [DOI] [PubMed] [Google Scholar]
- 5.Bartenschlager R, Baumert TF, Bukh J, Houghton M, Lemon SM, Lindenbach BD, et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: considerations for scientists and funding agencies. Virus Res (2018) 248:53–62. 10.1016/j.virusres.2018.02.016 [DOI] [PubMed] [Google Scholar]
- 6.Roche B, Coilly A, Duclos-Vallee JC, Samuel D. The impact of treatment of hepatitis C with DAAs on the occurrence of HCC. Liver Int (2018) 38(Suppl 1):139–45. 10.1111/liv.13659 [DOI] [PubMed] [Google Scholar]
- 7.Walker CM, Grakoui A. Hepatitis C virus: why do we need a vaccine to prevent a curable persistent infection? Curr Opin Immunol (2015) 35:137–43. 10.1016/j.coi.2015.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. A web resource to manage the classification and genotype and subtype assignments of hepatitis C virus. (2017). Available from: https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/56/hcv-classification (Accessed: April, 2018).
- 9.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology (2014) 59:318–27. 10.1002/hep.26744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant (2013) 13:1047–54. 10.1111/ajt.12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babcock GJ, Iyer S, Smith HL, Wang Y, Rowley K, Ambrosino DM, et al. High-throughput sequencing analysis of post-liver transplantation HCV E2 glycoprotein evolution in the presence and absence of neutralizing monoclonal antibody. PLoS One (2014) 9:e100325. 10.1371/journal.pone.0100325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuerst TR, Pierce BG, Keck ZY, Foung SKH. Designing a B cell-based vaccine against a highly variable hepatitis C virus. Front Microbiol (2017) 8:2692. 10.3389/fmicb.2017.02692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ball JK, Tarr AW, McKeating JA. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res (2014) 105:100–11. 10.1016/j.antiviral.2014.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong JA, Bhat R, Hockman D, Logan M, Chen C, Levin A, et al. Recombinant hepatitis C virus envelope glycoprotein vaccine elicits antibodies targeting multiple epitopes on the envelope glycoproteins associated with broad cross-neutralization. J Virol (2014) 88:14278–88. 10.1128/JVI.01911-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He L, Cheng Y, Kong L, Azadnia P, Giang E, Kim J, et al. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci Rep (2015) 5:12501. 10.1038/srep12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandomenico A, Leonardi A, Berisio R, Sanguigno L, Foca G, Foca A, et al. Generation and characterization of monoclonal antibodies against a cyclic variant of hepatitis C virus E2 epitope 412-422. J Virol (2016) 90:3745–59. 10.1128/JVI.02397-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierce BG, Boucher EN, Piepenbrink KH, Ejemel M, Rapp CA, Thomas WD, Jr, et al. Structure-based design of hepatitis C virus vaccines that elicit neutralizing antibody responses to a conserved epitope. J Virol (2017) 91:e01032–17. 10.1128/JVI.01032-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruwona TB, Giang E, Nieusma T, Law M. Fine mapping of murine antibody responses to immunization with a novel soluble form of hepatitis C virus envelope glycoprotein complex. J Virol (2014) 88:10459–71. 10.1128/JVI.01584-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frey SE, Houghton M, Coates S, Abrignani S, Chien D, Rosa D, et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine (2010) 28:6367–73. 10.1016/j.vaccine.2010.06.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colombatto P, Brunetto MR, Maina AM, Romagnoli V, Almasio P, Rumi MG, et al. HCV E1E2-MF59 vaccine in chronic hepatitis C patients treated with PEG-IFNalpha2a and ribavirin: a randomized controlled trial. J Viral Hepat (2014) 21:458–65. 10.1111/jvh.12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fauvelle C, Colpitts CC, Keck ZY, Pierce BG, Foung SK, Baumert TF. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev Vaccines (2016) 15(12):1535–44. 10.1080/14760584.2016.1194759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keck ZY, Li TK, Xia J, Bartosch B, Cosset FL, Dubuisson J, et al. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. J Virol (2005) 79:13199–208. 10.1128/JVI.79.21.13199-13208.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med (2008) 14:25–7. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- 24.Drummer HE. Challenges to the development of vaccines to hepatitis C virus that elicit neutralizing antibodies. Front Microbiol (2014) 5:329. 10.3389/fmicb.2014.00329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A (2012) 109:6205–10. 10.1073/pnas.1114927109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature (1981) 289:366–73. 10.1038/289366a0 [DOI] [PubMed] [Google Scholar]
- 27.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science (2013) 342:1477–83. 10.1126/science.1245625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu NC, Wilson IA. Structural insights into the design of novel anti-influenza therapies. Nat Struct Mol Biol (2018) 25:115–21. 10.1038/s41594-018-0025-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol (2013) 13:693–701. 10.1038/nri3516 [DOI] [PubMed] [Google Scholar]
- 30.Zhou T, Lynch RM, Chen L, Acharya P, Wu X, Doria-Rose NA, et al. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell (2015) 161:1280–92. 10.1016/j.cell.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med (2015) 21:1065–70. 10.1038/nm.3927 [DOI] [PubMed] [Google Scholar]
- 32.Impagliazzo A, Milder F, Kuipers H, Wagner MV, Zhu X, Hoffman RM, et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science (2015) 349:1301–6. 10.1126/science.aac7263 [DOI] [PubMed] [Google Scholar]
- 33.Steichen JM, Kulp DW, Tokatlian T, Escolano A, Dosenovic P, Stanfield RL, et al. HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity (2016) 45:483–96. 10.1016/j.immuni.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulp DW, Steichen JM, Pauthner M, Hu X, Schiffner T, Liguori A, et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat Commun (2017) 8:1655. 10.1038/s41467-017-01549-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meola A, Tarr AW, England P, Meredith LW, McClure CP, Foung SK, et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J Virol (2015) 89:2170–81. 10.1128/JVI.02190-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong L, Lee DE, Kadam RU, Liu T, Giang E, Nieusma T, et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc Natl Acad Sci U S A (2016) 113:12768–73. 10.1073/pnas.1609780113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goffard A, Callens N, Bartosch B, Wychowski C, Cosset FL, Montpellier C, et al. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J Virol (2005) 79:8400–9. 10.1128/JVI.79.13.8400-8409.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, et al. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun (2014) 5:4874. 10.1038/ncomms5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong L, Kadam RU, Giang E, Ruwona TB, Nieusma T, Culhane JC, et al. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314-324 in complex with antibody IGH526. J Mol Biol (2015) 427:2617–28. 10.1016/j.jmb.2015.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potter JA, Owsianka AM, Jeffery N, Matthews DJ, Keck ZY, Lau P, et al. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J Virol (2012) 86:12923–32. 10.1128/JVI.02052-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A (2012) 109:9499–504. 10.1073/pnas.1202924109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong L, Giang E, Nieusma T, Robbins JB, Deller MC, Stanfield RL, et al. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol (2012) 86:13085–8. 10.1128/JVI.01939-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pantua H, Diao J, Ultsch M, Hazen M, Mathieu M, McCutcheon K, et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol (2013) 425:1899–914. 10.1016/j.jmb.2013.02.025 [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Pierce BG, Wang Q, Keck ZY, Fuerst TR, Foung SK, et al. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J Biol Chem (2015) 290:10117–25. 10.1074/jbc.M115.643528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keck ZY, Girard-Blanc C, Wang W, Lau P, Zuiani A, Rey FA, et al. Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. J Virol (2016) 90:3112–22. 10.1128/JVI.02458-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu J, Hardy J, Boo I, Vietheer P, McCaffrey K, Alhammad Y, et al. Escape of hepatitis C virus from epitope I neutralization increases sensitivity of other neutralization epitopes. J Virol (2018) 92:e02066–17. 10.1128/JVI.02066-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science (2013) 342:1090–4. 10.1126/science.1243876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature (2014) 509:381–4. 10.1038/nature13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng L, Ma L, Virata-Theimer ML, Zhong L, Yan H, Zhao Z, et al. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc Natl Acad Sci U S A (2014) 111:10690–5. 10.1073/pnas.1411317111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng L, Zhong L, Struble E, Duan H, Ma L, Harman C, et al. Structural evidence for a bifurcated mode of action in the antibody-mediated neutralization of hepatitis C virus. Proc Natl Acad Sci U S A (2013) 110:7418–22. 10.1073/pnas.1305306110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog (2013) 9:e1003364. 10.1371/journal.ppat.1003364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keck ZY, Wang Y, Lau P, Lund G, Rangarajan S, Fauvelle C, et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute HCV infection. Hepatology (2016) 64:1922–33. 10.1002/hep.28850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasiliauskaite I, Owsianka A, England P, Khan AG, Cole S, Bankwitz D, et al. Conformational flexibility in the immunoglobulin-like domain of the hepatitis C virus glycoprotein E2. MBio (2017) 8:e00382–17. 10.1128/mBio.00382-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilmartin AA, Lamp B, Rumenapf T, Persson MA, Rey FA, Krey T. High-level secretion of recombinant monomeric murine and human single-chain Fv antibodies from Drosophila S2 cells. Protein Eng Des Sel (2012) 25:59–66. 10.1093/protein/gzr058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tarr AW, Lafaye P, Meredith L, Damier-Piolle L, Urbanowicz RA, Meola A, et al. An alpaca nanobody inhibits hepatitis C virus entry and cell-to-cell transmission. Hepatology (2013) 58:932–9. 10.1002/hep.26430 [DOI] [PubMed] [Google Scholar]
- 56.Op De Beeck A, Montserret R, Duvet S, Cocquerel L, Cacan R, Barberot B, et al. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J Biol Chem (2000) 275:31428–37. 10.1074/jbc.M003003200 [DOI] [PubMed] [Google Scholar]
- 57.Spadaccini R, D’Errico G, D’Alessio V, Notomista E, Bianchi A, Merola M, et al. Structural characterization of the transmembrane proximal region of the hepatitis C virus E1 glycoprotein. Biochim Biophys Acta (2010) 1798:344–53. 10.1016/j.bbamem.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 58.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, et al. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J Virol (2011) 85:1777–92. 10.1128/JVI.02170-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res (2000) 28:235–42. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawson CL, Patwardhan A, Baker ML, Hryc C, Garcia ES, Hudson BP, et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res (2016) 44:D396–403. 10.1093/nar/gkv1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yagnik AT, Lahm A, Meola A, Roccasecca RM, Ercole BB, Nicosia A, et al. A model for the hepatitis C virus envelope glycoprotein E2. Proteins (2000) 40:355–66. [DOI] [PubMed] [Google Scholar]
- 62.Krey T, d’Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog (2010) 6:e1000762. 10.1371/journal.ppat.1000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Falson P, Bartosch B, Alsaleh K, Tews BA, Loquet A, Ciczora Y, et al. Penin, hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J Virol (2015) 89:10333–46. 10.1128/JVI.00991-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castelli M, Clementi N, Pfaff J, Sautto GA, Diotti RA, Burioni R, et al. A biologically-validated HCV E1E2 heterodimer structural model. Sci Rep (2017) 7:214. 10.1038/s41598-017-00320-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freedman H, Logan MR, Hockman D, Koehler Leman J, Law JL, Houghton M. Computational prediction of the heterodimeric and higher-order structure of gpE1/gpE2 envelope glycoproteins encoded by hepatitis C virus. J Virol (2017) 91:e02309–16. 10.1128/JVI.02309-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature (2013) 504:172–6. 10.1038/nature12684 [DOI] [PubMed] [Google Scholar]
- 67.Davis C, Harris HJ, Hu K, Drummer HE, McKeating JA, Mullins JG, et al. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell Microbiol (2012) 14:1892–903. 10.1111/cmi.12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature (1995) 375:291–8. 10.1038/375291a0 [DOI] [PubMed] [Google Scholar]
- 69.Roussel A, Lescar J, Vaney MC, Wengler G, Wengler G, Rey FA. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure (2006) 14:75–86. 10.1016/j.str.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 70.Khan AG, Miller MT, Marcotrigiano J. HCV glycoprotein structures: what to expect from the unexpected. Curr Opin Virol (2015) 12:53–8. 10.1016/j.coviro.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sabahi A, Uprichard SL, Wimley WC, Dash S, Garry RF. Unexpected structural features of the hepatitis C virus envelope protein 2 ectodomain. J Virol (2014) 88:10280–8. 10.1128/JVI.00874-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nayak A, Pattabiraman N, Fadra N, Goldman R, Kosakovsky Pond SL, Mazumder R. Structure-function analysis of hepatitis C virus envelope glycoproteins E1 and E2. J Biomol Struct Dyn (2015) 33:1682–94. 10.1080/07391102.2014.967300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davidson E, Doranz BJ. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology (2014) 143:13–20. 10.1111/imm.12323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol (2017) 13:e1005324. 10.1371/journal.pcbi.1005324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andreani J, Soding J. bbcontacts: prediction of beta-strand pairing from direct coupling patterns. Bioinformatics (2015) 31:1729–37. 10.1093/bioinformatics/btv041 [DOI] [PubMed] [Google Scholar]
- 76.Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc (2007) 2:2728–33. 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 77.Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, et al. Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat Struct Biol (2002) 9:507–11. [DOI] [PubMed] [Google Scholar]
- 78.Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Current topics in medicinal chemistry (2008) 8(18):1555–72. [DOI] [PubMed] [Google Scholar]
- 79.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, et al. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol (2003) 331:281–99. 10.1016/S0022-2836(03)00670-3 [DOI] [PubMed] [Google Scholar]
- 80.Pierce BG, Keck ZY, Lau P, Fauvelle C, Gowthaman R, Baumert TF, et al. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc Natl Acad Sci U S A (2016) 113:E6946–54. 10.1073/pnas.1614942113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gopal R, Jackson K, Tzarum N, Kong L, Ettenger A, Guest J, et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog (2017) 13:e1006735. 10.1371/journal.ppat.1006735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin P, Zhang L, Ye F, Deng Y, Lu S, Li YP, et al. A screen for inhibitory peptides of hepatitis C virus identifies a novel entry inhibitor targeting E1 and E2. Sci Rep (2017) 7:3976. 10.1038/s41598-017-04274-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barone D, Balasco N, Autiero I, Vitagliano L. The dynamic properties of the hepatitis C virus E2 envelope protein unraveled by molecular dynamics. J Biomol Struct Dyn (2017) 35:805–16. 10.1080/07391102.2016.1162198 [DOI] [PubMed] [Google Scholar]
- 84.Russ WP, Engelman DM. The GxxxG Motif: a framework for transmembrane helix-helix association. J Mol Biol (2000) 296:911–9. 10.1006/jmbi.1999.3489 [DOI] [PubMed] [Google Scholar]
- 85.Jusoh SA, Welsch C, Siu SW, Bockmann RA, Helms V. Contribution of charged and polar residues for the formation of the E1-E2 heterodimer from hepatitis C virus. J Mol Model (2010) 16:1625–37. 10.1007/s00894-010-0672-1 [DOI] [PubMed] [Google Scholar]
- 86.Ciczora Y, Callens N, Penin F, Pecheur EI, Dubuisson J. Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J Virol (2007) 81:2372–81. 10.1128/JVI.02198-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shalom-Elazari H, Zazrin-Greenspon H, Shaked H, Chill JH. Global fold and backbone dynamics of the hepatitis C virus E2 glycoprotein transmembrane domain determined by NMR. Biochim Biophys Acta (2014) 1838:2919–28. 10.1016/j.bbamem.2014.07.023 [DOI] [PubMed] [Google Scholar]
- 88.Zazrin H, Shaked H, Chill JH. Architecture of the hepatitis C virus E1 glycoprotein transmembrane domain studied by NMR. Biochim Biophys Acta (2014) 1838:784–92. 10.1016/j.bbamem.2013.10.021 [DOI] [PubMed] [Google Scholar]
- 89.Keck ZY, Angus AG, Wang W, Lau P, Wang Y, Gatherer D, et al. Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids 412-423. PLoS Pathog (2014) 10:e1004297. 10.1371/journal.ppat.1004297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bohne-Lang A, von der Lieth CW. GlyProt: in silico glycosylation of proteins. Nucleic Acids Res (2005) 33:W214–9. 10.1093/nar/gki385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res (2012) 40:W288–93. 10.1093/nar/gks419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, et al. Binding of hepatitis C virus to CD81. Science (1998) 282:938–41. 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 93.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J (2002) 21:5017–25. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature (2007) 446:801–5. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 95.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature (2009) 457:882–6. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miao Z, Xie Z, Miao J, Ran J, Feng Y, Xia X. Regulated entry of hepatitis C virus into hepatocytes. Viruses (2017) 9:100. 10.3390/v9050100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, et al. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem (2010) 285:21092–102. 10.1074/jbc.M110.104836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Feneant L, Levy S, Cocquerel L. CD81 and hepatitis C virus (HCV) infection. Viruses (2014) 6:535–72. 10.3390/v6020535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc (2003) 125:1731–7. 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- 100.Drummer HE, Wilson KA, Poumbourios P. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J Virol (2002) 76(21):11143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harman C, Zhong L, Ma L, Liu P, Deng L, Zhao Z, et al. A view of the E2-CD81 interface at the binding site of a neutralizing antibody against hepatitis C virus. J Virol (2015) 89:492–501. 10.1128/JVI.01661-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem (2010) 31:455–61. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cunha ES, Sfriso P, Rojas AL, Roversi P, Hospital A, Orozco M, et al. Mechanism of structural tuning of the hepatitis C virus human cellular receptor CD81 large extracellular loop. Structure (2017) 25:53–65. 10.1016/j.str.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 104.Zimmerman B, Kelly B, McMillan BJ, Seegar TCM, Dror RO, Kruse AC, et al. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell (2016) 167:1041–51.e11. 10.1016/j.cell.2016.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T, et al. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol (2011) 85:10451–63. 10.1128/JVI.05259-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol (2010) 84:34–43. 10.1128/JVI.02199-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Westhaus S, Deest M, Nguyen ATX, Stanke F, Heckl D, Costa R, et al. Scavenger receptor class B member 1 (SCARB1) variants modulate hepatitis C virus replication cycle and viral load. J Hepatol (2017) 67:237–45. 10.1016/j.jhep.2017.03.020 [DOI] [PubMed] [Google Scholar]
- 108.Bankwitz D, Vieyres G, Hueging K, Bitzegeio J, Doepke M, Chhatwal P, et al. Role of hypervariable region 1 for the interplay of hepatitis C virus with entry factors and lipoproteins. J Virol (2014) 88:12644–55. 10.1128/JVI.01145-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cukierman L, Meertens L, Bertaux C, Kajumo F, Dragic T. Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell–cell contacts. J Virol (2009) 83:5477–84. 10.1128/JVI.02262-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hsieh FL, Turner L, Bolla JR, Robinson CV, Lavstsen T, Higgins MK. The structural basis for CD36 binding by the malaria parasite. Nat Commun (2016) 7:12837. 10.1038/ncomms12837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cheng JJ, Li JR, Huang MH, Ma LL, Wu ZY, Jiang CC, et al. CD36 is a co-receptor for hepatitis C virus E1 protein attachment. Sci Rep (2016) 6:21808. 10.1038/srep21808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bonander N, Jamshad M, Hu K, Farquhar MJ, Stamataki Z, Balfe P, et al. Structural characterization of CD81-Claudin-1 hepatitis C virus receptor complexes. Biochem Soc Trans (2011) 39:537–40. 10.1042/BST0390537 [DOI] [PubMed] [Google Scholar]
- 113.Bonander N, Jamshad M, Oberthur D, Clare M, Barwell J, Hu K, et al. Production, purification and characterization of recombinant, full-length human claudin-1. PLoS One (2013) 8:e64517. 10.1371/journal.pone.0064517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vipperla B, Dass JF, Jayanthi S. Insilico modeling and molecular dynamic simulation of claudin-1 point mutations in HCV infection. J Biomol Struct Dyn (2014) 32:1443–55. 10.1080/07391102.2013.823624 [DOI] [PubMed] [Google Scholar]
- 115.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science (2014) 344:304–7. 10.1126/science.1248571 [DOI] [PubMed] [Google Scholar]
- 116.Suzuki H, Tani K, Fujiyoshi Y. Crystal structures of claudins: insights into their intermolecular interactions. Ann N Y Acad Sci (2017) 1397:25–34. 10.1111/nyas.13371 [DOI] [PubMed] [Google Scholar]
- 117.Sivasubramanian A, Chao G, Pressler HM, Wittrup KD, Gray JJ. Structural model of the mAb 806-EGFR complex using computational docking followed by computational and experimental mutagenesis. Structure (2006) 14:401–14. 10.1016/j.str.2005.11.022 [DOI] [PubMed] [Google Scholar]
- 118.Tharakaraman K, Robinson LN, Hatas A, Chen YL, Siyue L, Raguram S, et al. Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc Natl Acad Sci U S A (2013) 110:E1555–64. 10.1073/pnas.1303645110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhu J, Yu Y, Ulbrich MH, Li MH, Isacoff EY, Honig B, et al. Structural model of the TRPP2/PKD1 C-terminal coiled-coil complex produced by a combined computational and experimental approach. Proc Natl Acad Sci U S A (2011) 108:10133–8. 10.1073/pnas.1017669108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rappuoli R, Bottomley MJ, D’Oro U, Finco O, De Gregorio E. Reverse vaccinology 2.0: human immunology instructs vaccine antigen design. J Exp Med (2016) 213:469–81. 10.1084/jem.20151960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Joyce MG, Zhang B, Ou L, Chen M, Chuang GY, Druz A, et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat Struct Mol Biol (2016) 23:811–20. 10.1038/nsmb.3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kong L, Jackson KN, Wilson IA, Law M. Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr Opin Virol (2015) 11:148–57. 10.1016/j.coviro.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Y, Singh NK, Spear TT, Hellman LM, Piepenbrink KH, McMahan RH, et al. How an alloreactive T-cell receptor achieves peptide and MHC specificity. Proc Natl Acad Sci U S A (2017) 114:E4792–801. 10.1073/pnas.1700459114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boyd SD, Crowe JE, Jr. Deep sequencing and human antibody repertoire analysis. Curr Opin Immunol (2016) 40:103–9. 10.1016/j.coi.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Eroshkin AM, LeBlanc A, Weekes D, Post K, Li Z, Rajput A, et al. bNAber: database of broadly neutralizing HIV antibodies. Nucleic Acids Res (2014) 42:D1133–9. 10.1093/nar/gkt1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics (2005) 21:379–84. 10.1093/bioinformatics/bth485 [DOI] [PubMed] [Google Scholar]