

Abstract
Oxygen intermediates in copper enzymes exhibit unique spectroscopic features that reflect novel geometric and electronic structures that are key to reactivity. This perspective will describe: (1) the bonding origin of the unique spectroscopic features of the coupled binuclear copper enzymes and how this overcomes the spin forbiddenness of O2 binding and activates monooxygenase activity, (2) how the difference in exchange coupling in the non-coupled binuclear Cu enzymes controls the reaction mechanism, and (3) how the trinuclear Cu cluster present in the multicopper oxidases leads to a major structure/function difference in enabling the irreversible reductive cleavage of the O–O bond with little overpotential and generating a fully oxidized intermediate, different from the resting enzyme studied by crystallography, that is key in enabling fast PCET in the reductive half of the catalytic cycle.
Graphical Abstract
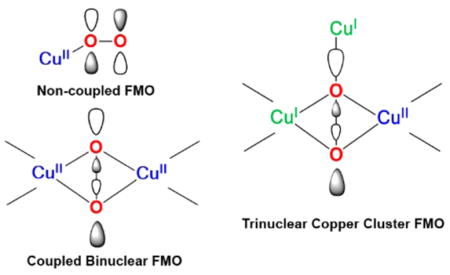
INTRODUCTION
This Article will summarize the three topics covered in my Alfred Bader Lecture (Table 1). The first focus is on O2 binding and activation for electrophilic aromatic attack by the coupled binuclear copper active sites in hemocyanins and tyrosinases. The geometric and electronic structures of these sites will serve as the basis for the other topics in developing structure/function differences. The second topic is O2 activation for Hatom abstraction by the non-coupled binuclear copper enzymes dopamine β-monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM). Here, the focus is on the differences in antiferromagnetic exchange “coupling” that lead to differences in O2 activation. The third topic is the 4e− reduction of dioxygen to water by the trinuclear copper cluster we first showed to be present in biology.1,2 This defined a major structure/function difference between the reversible binding and activation of dioxygen for electrophilic chemistry by the coupled binuclear Cu active sites vs the irreversible reductive cleavage of the O–O bond by the trinuclear Cu cluster in the multicopper oxidases.
Table 1.
Selected Cu/O2 Structure/Function Correlations in Bioinorganic Chemistry

|
The common theme in these three topics is that the O2 intermediates can be trapped in these reactions. These intermediates have unique spectroscopic features that we have shown reflect novel geometric and electronic structures that are key to the different biological functions: O2 binding and activation for electrophilic aromatic substitution (EAS) and H atom abstraction, and its four-electron reduction to water.3
Coupled Binuclear Copper Active Sites
As shown in Table 1, hemocyanin (Hc) and tyrosinase (Ty) have deoxy sites with two Cu(I)’s that reversibly bind dioxygen to generate peroxide bound binuclear cupric Oxy-sites. Since a focus in bioinorganic chemistry has been on understanding the unique spectroscopic features of these Oxy-sites, we first consider normal peroxide–Cu(II) bonding and the associated spectroscopy.
Dioxygen is a triplet because it has two electrons in the doubly degenerate π* set of O 2p orbitals. Reduction by two electrons leads to a (π*)4 HOMO which dominates the bonding of peroxide to the Cu(II) center(s). Figure 1A considers a peroxide end-on bound to a tetragonal Cu(II). One π* orbital, in the CuO2 plane, σ bonds to the half-occupied dx2−y2 on the Cu(II). This gives a half-occupied σ-antibonding Cu dx2−y2 orbital in the ground state, which has the characteristic EPR signal shown in the middle of Figure 1A that serves in this perspective for empirical correlations (vide inf ra). This single Cu(II)–O22− bonding scheme (Figure 1A, top) leads to a peroxide πσ* to Cu(II) charge transfer transition (Figure 1A, bottom) at ~500 nm (~20 000 cm−1) with an ε ~ 5000 M−1 cm−1, where the intensity quantifies the donor interaction of the peroxide to the Cu(II).4 Tuning a laser into this absorption band gives a resonance Raman spectrum with an O–O stretch at 803 cm−1 (inset in Figure 1A, bottom) defining the spectroscopy of peroxide end-on bound to a single Cu(II).5
Figure 1.

Bonding and spectroscopy in “normal” peroxo-Cu(II) complexes. (A) Mononuclear end-on-peroxo-Cu(II). (B) Binuclear end-on bridged peroxo 2 Cu(II). Top: MO diagrams. Middle: EPR spectra. Bottom: Absorption spectra with resonance Raman O–O stretch insets.
In Figure 1B, the peroxide now end-on bridges between two Cu(II)’s. Since the coppers are related by symmetry, one takes symmetric and antisymmetric combinations of their half-occupied dx2−y2 orbitals for bonding to the bridging peroxide. As shown at the top of Figure 1B, the in-plane peroxide π* orbital σ bonds to the symmetric combination of dx2−y2 orbitals on the two Cu(II)’s. This leads to a HOMO/LUMO splitting large enough to overcome electron repulsion resulting in a spin paired ((dx2−y2A − dx2−y2B)2) ground state.
Since this structure has a singlet ground state, there is no EPR signal (Figure 1B middle); this corresponds to an antiferromagnetic “coupled” binuclear Cu(II) site with the πσ* orbital pictured at the top of Figure 1B providing the superexchange pathway for this exchange coupling. The singlet/triplet splitting of the ground state is given by −2J using the Heisenberg, Dirac, Van Vleck Hamiltonian H = −2JS1·S2. Such an end-on peroxide bridged structure has the peroxide to Cu(II) charge transfer spectrum at the bottom of Figure 1B, where the intensity is twice as large as in Figure 1A and reflects the fact that the peroxide has a donor interaction with each of the two Cu(II)’s in the end-on bridged structure. This increased donor interaction removes more electron density from the peroxide π* orbital, leading to a stronger O–O bond. This is reflected in the resonance Raman spectrum (bottom inset Figure 1B) where the O–O stretch has increased to 830 cm−1.6
Oxy hemocyanin has the side-on peroxide bridged structure7 shown in Figure 2B, left, as first anticipated by the model studies of the late Nobu Kitajima.8 Associated with this side-on peroxide bridged structure are the unique spectroscopic features of oxy hemocyanin. From Figure 2A left, oxyHc’s (and oxyTy) have a peroxide to Cu(II) charge transfer transition that is 4 times as intense and at ~8000 cm−1 higher energy than for peroxide end-on bound to a single Cu(II) (i.e., normal Cu in Figure 2A absorbance from Figure 1A). Its resonance Raman spectrum shows a uniquely low energy O–O stretch at ~750 cm−1.9−11 Since there is a peroxide bridge, the two Cu(II)’s are antiferromagnetically coupled; however, the singlet/triplet splitting of oxyHc is very large, −2J > 600 cm−1, the lower limit that can be measured by SQUID magnetic susceptibility on this metalloprotein.12
Figure 2.

Unique geometric and electronic structure of oxyHc/Ty. (A) Absorption, resonance Raman, and EPR spectra; oxyHc/Ty in red, normal Cu(II)–O–O2− in blue. (B) Geometric and electronic structure of oxyHc/Ty. Left: Structure of oxyHc/Ty. Middle: MO diagram. Right: Associated HOMO/LUMO contours.
We developed the electronic structure description associated with these unique spectral features in terms of two special bonding interactions (Figure 2B center and right).13,14 First, the π* orbital in the Cu2O2 plane again undergoes a σ bonding interaction with the symmetric combination of dx2−y2 orbitals on the two coppers. However, in oxy-Hc/Ty, this σ bonding/antibonding interaction is very strong due to the side-on peroxide bridge having two donor interactions with each of the two coppers (Figure 2B right top contour). This leads to the high intensity and high energy of the peroxide πσ* to Cu(II) charge transfer transition of oxy-Hc/Ty in Figure 2A, left. This strong σ donation takes even more electron density out of the π* orbital than in the end-on bridged case in Figure 1B, and thus one might expect the O–O stretch to go up above 830 cm−1. However, it goes down to ~750 cm−1 (Figure 2A center). This reflects a second new bonding interaction for peroxide to Cu(II), where in the side-on case the σ* LUMO on the peroxide undergoes a bonding/antibonding interaction with the HOMO on the coppers. As shown by the contour at the bottom of Figure 2B, right, this shifts electron density from the Cu into the side-on peroxo σ* orbital (i.e., a backbonding interaction), which is strongly antibonding with respect to the O–O bond, hence the low ν(O–O) (Figure 2A, middle). The resultant large HOMO/LUMO splitting in Figure 2B, middle, is responsible for the large antiferromagnetic coupling of the oxyHc site where the πσ* peroxide orbital in the top contour in Figure 2B, right, provides the superexchange pathway for the antiferromagnetic coupling. This exchange coupling overcomes the spin forbiddenness of triplet O2 binding to the singlet deoxyHc active site to form oxyHc, which as described above is a singlet.
As shown in Figure 3A at left and as presented above, both Cu(II)’s overlap the peroxide πσ* orbital, leading to the antiferromagnetic stabilization of the singlet ground state of the oxy site. However, as one proceeds along the reaction coordinate for reversible O2 binding, the structure butterflies (center of Figure 3A).15 In this structure, each Cu(II) overlaps a different π* orbital on the peroxide. This leads to orthogonal magnetic orbitals which brings the triplet down in energy to become the ground state (red curve in Figure 3A). From this structure, one electron of the same spin can be donated from each π* orbital to the coppers leading to the loss of triplet dioxygen. Thus, the exchange coupling which forms along the reaction coordinate for O2 binding to form the oxyHc/Ty site overcomes the spin forbiddenness of O2 binding and leads to the 2e− reduction of O2 to form the peroxide bridge. This side-on peroxide bridge of the oxy-Hc/Ty structure has a σ* frontier molecular orbital activated for the 2e− electrophilic attack on an aromatic ring (Figure 3B).
Figure 3.

Structure/function correlations of coupled binuclear Cu proteins. (A) Reaction coordinate for spin forbidden 3O2 binding to deoxyHc/Ty to produce oxyHc/Ty (singlet). (B) Reaction coordinate for the oxy coupled binuclear Cu site (in Ty) in its monooxygenation of phenol substrate (an electrophilic aromatic substitution (EAS) reaction) with 2e− oxidation of the resultant bound catecholate to produce quinone and the deoxy site. (At right are the two possible reaction intermediates of oxy sites defined by model studies now being evaluated for the oxyTy enzyme/substrate intermediate.)
In early experiments, we showed that oxyTy has the same active site geometric and electronic structure as oxyHc,9 but tyrosinase allows phenol substrate access to the Cu2O2 active site. This led us to the mechanism in Figure 3B where the substrate can directly access the Cu2O2 active site enabling monooxygenation of the ortho carbon of phenolate.16 The resultant bound catecholate is then oxidized to the quinone, and the resulting bicuprous site can again bind O2. An important present issue is whether the side-on peroxide of oxyTy can directly oxygenate the ring (as observed in collaborative model studies with Prof. Ken Karlin17,18) or whether phenolate binding shifts the oxyTy site to a bis(μ-oxo) structure,19 which then oxygenates the ring (as observed in collaborative model studies with Prof. Dan Stack20,21). Studies are now underway to trap and spectroscopically define the ternary complex of oxyTy with substrate bound prior to its mono-oxygenation.22
Non-Coupled Binuclear Copper Active Sites
As shown in the middle of Table 1, non-coupled binuclear copper sites perform H atom abstraction to hydroxylate activated C–H bonds. The enzymes containing these sites include dopamine β-monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM), both important in neurochemistry. These active sites are non-coupled in the sense that the two Cu(II)’s are separated by 11 Å in the enzymes and have no magnetic interaction (Figure 4). The CuM site has two His and one Met ligand and is the metal site involved in O2 activation, and the CuH site, which has three His ligands, provides the second e− required for this chemistry.23
Figure 4.

Structure of the non-coupled binuclear Cu enzyme PHM. In the resting oxidized PHM, CuM has two His, one Met, and an OH− and H2O as ligands, and CuH has three His and a water. CuM activates O2, and CuH supplies the second electron required for the reaction.
Initially, it was thought that dioxygen was reduced by 2e− to form a hydroperoxide at the CuM site for the H atom abstraction. Thus, we studied a hydroperoxide-Cu(II) model (HB(3-tBu-5-iPrpz)3Cu(II)OOH, with Prof. Kyoshi Fujisawa24), performed parallel spectral studies on PHM,25 and combined these to generate a CuM(II)-OOH− model (Figure 5A).26 This had a frontier molecular orbital (FMO) that was a half-occupied orbital on the Cu(II), σ antibonding to a π* orbital on the hydroperoxide (Figure 5A, middle). This wave function had only 2% distal oxygen character (coefficients given in Figure 5A, middle contour) and thus was not activated for H atom abstraction. We then considered O2 activation by 1e− at CuM (Figure 5B). Prof. Fujisawa had a structurally defined complex (HB(3-tBu-5-iPrpz)3Cu(II)(η2-O2 •−) with O2 bound side-on to the Cu.27 Its resonance Raman spectrum showed an O–O stretch at 1043 cm−1; thus it was a superoxide–Cu(II) complex that from SQUID magnetic susceptibility had a singlet ground state.28 Modeling this into the CuM active site (Figure 5B, middle) gave an FMO that was an unoccupied d orbital strongly σ-antibonding to a π* orbital on the side-on superoxide in the CuO2 plane.26 This wave function had 32% character on each oxygen atom and was thus more activated for Hatom abstraction. Thus, a side-on superoxide Cu(II) complex has a singlet ground state with a doubly unoccupied FMO.
Figure 5.

Models of possible O2 intermediates in the non-coupled binuclear CuM site in PHM. (A) Two-electron reduced CuM(II)OOH. (B) One-electron reduced side-on superoxide CuM(II). Left: Geometry optimized model. Center: FMO. Right: resonance Raman data on the model complex.
Then, a crystal structure appeared of PHM with a poor substrate that had O2 end-on bound to CuM (Figure 6, left).29 Thus, we performed spectroscopy and electronic structure calculations on the structurally defined end-on superoxide–Cu(II) complex [(TMG3tren)CuO2]+ of Schindler et al.30 (Figure 6, right). This had a triplet ground state,31 which reflects the two half-occupied valence orbitals in Figure 7A.32 One (at left) is a d orbital σ-antibonding to a π* orbital of the superoxide; this parallels the FMO of the side-on superoxide–Cu(II) in Figure 5B center plotted to the right in Figure 7B (LUMO at top). The other half-occupied orbital in the end-on superoxide–Cu(II) is the second π* orbital of the superoxide, perpendicular to the Cu–O2 plane (Figure 7A, right). From Figure 7B, in going from the side-on superoxide–Cu(II) on the right to the end-on superoxide–Cu(II) on the left, the σ bonding/antibonding interaction between the in-plane πσ* of the superoxide and the Cu d valence orbital greatly decreases (i.e., the σ bonding in the end-on case is much weaker), and the splitting between the two highest energy valence orbitals, the d orbital σ-antibonding to the π* of the superoxide (d–π*σ in Figure 7B) and the superoxide π* perpendicular to the CuO2 plane (π*v in Figure 7B), is no longer large enough to overcome e− repulsion and spin pair as in the side-on superoxide–Cu(II) case.33
Figure 6.
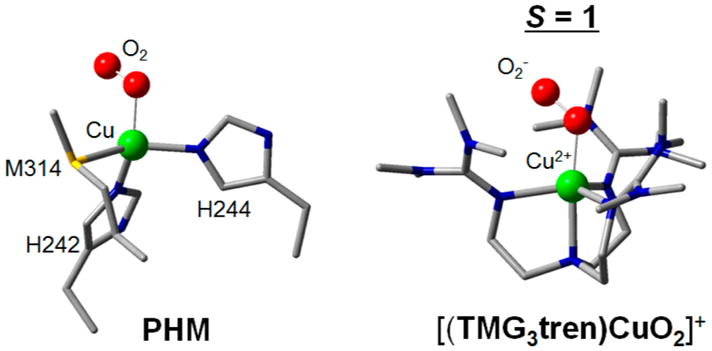
End-on O2− Cu structures. Left: PHM in the presence of the poor substrate N-acetyl-diiodotyrosyl-D-threonine (ref 29). Right: Structurally defined end-on superoxide-Cu(II) model (ref 30).
Figure 7.

Electronic structure of superoxide–Cu(II) sites. (A) FMOs of end-on O2−-Cu(II) triplet. Left: d orbital σ-antibonding to the π* of superoxide in the CuOO plane. Right: superoxide π* orbital perpendicular to CuOO plane. (B) Magneto-structural correlation of side-on singlet (right) to end-on triplet (left).
This limited d–πσ*/πv* splitting in the end-on superoxide–Cu(II) system is key to its reactivity (vide inf ra) and was experimentally probed by resonance Raman excitation profiles.32 Figure 8 gives the absorption spectrum of [(TMG3tren)CuO2]+ (in black); laser excitation into this absorption leads to resonance-enhanced Raman peaks at 1120 cm−1 (the superoxide O–O stretch) and 435 cm−1 (the Cu(II)–O2 •− metal–ligand stretch). Tuning the laser through the absorption spectrum (i.e., resonance Raman excitation profiles (RREP)) gives resonance enhancement in two electronic transitions. At higher energy (~23 000 cm−1), both the Cu–O metal–ligand stretch and the O–O stretch are enhanced. This is characteristic of a superoxide ligand-to-metal charge transfer transition (indicated by the blue arrow in Figure 7B left). At lower energy (~13 500 cm−1) in the [(TMG3tren)-CuO2]+ absorption spectrum, there is an electronic transition which only enhances the O–O stretch. This allows assignment as an intraligand transition (IL, red arrow in Figure 7B left) which involves excitation between the two superoxide π* orbitals, split in energy by bonding to the metal. From Figure 7B, left, the energy difference between these two transitions reflects the splitting of the d–πσ*/πv* valence orbitals and is approximately half of that required to overcome e− repulsion and spin pair. Thus, in an endon superoxide–Cu(II) triplet (Figure 6, [(TMG3tren)CuO2]+ and PHM) there is a low-lying half-occupied πv*FMO(Figure 7B left) which is dominantly on the superoxide with 49% distal oxygen character (Figure 7A right) and thus highly activated for H atom abstraction.
Figure 8.
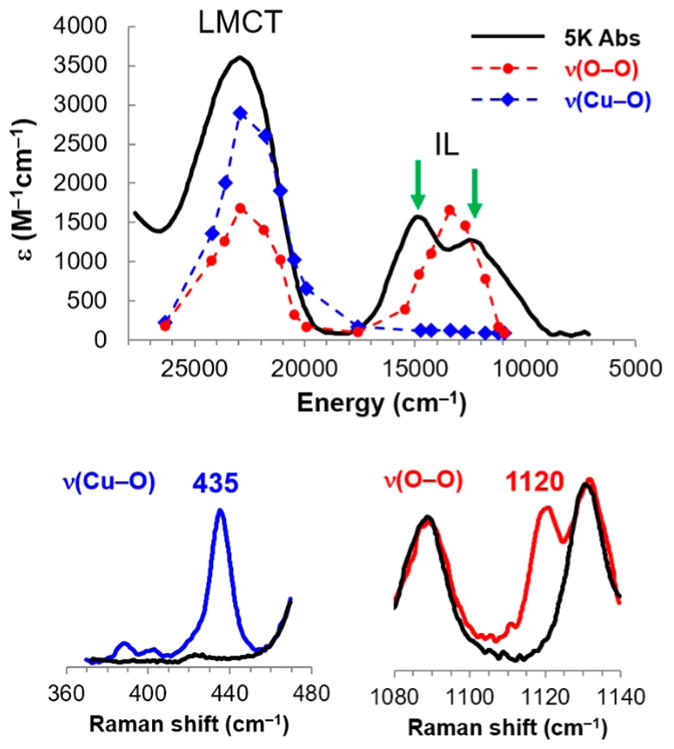
Electronic structure of TMG3tren CuO2−. Top: Absorption spectrum (black) and resonance Raman excitation profiles in the O–O stretch (red, spectrum at bottom right) and Cu–O2− stretch (blue, bottom left). Note that the two peaks indicated by arrows in the absorption spectrum in the IL region are ligand field transitions. (See ref 32 for detailed assignments using MCD spectroscopy.)
These inorganic model studies were then extended to computationally evaluate the reaction coordinate of PHM (Figure 9).34 We first consider O2 reduction by 2e− to form a CuM(II)-OOH intermediate (Figure 9, in blue at left). Reaction with an appropriate model substrate (formylglycine) gave a free energy activation barrier of >40 kcal/mol, which is inaccessible. To the right in Figure 9, we now consider O2 activation by 1e− to form an end-on superoxide–CuM(II) triplet (in red). This has an activation free energy for H atom abstraction from the same substrate of ~20 kcal/mol, consistent with the experimental kinetics.35
Figure 9.

Reaction coordinate for PHM. Two electron O2 activation to form a CuM(II)OOH intermediate has a high barrier for H atom abstraction from formylglycine (FmG; left, blue). One electron O2 activation (center, red) gives a Cu(II)O2− end-on triplet with a low barrier for H atom abstraction from FmG. Radical attack on the nonprotonated O of the resultant Cu(II)OOH (right, green) forms a radical coupled product that drives ET from CuH. Inset: Marcus theory for nonadiabatic ET and the relation of the electronic coupling between the electron donor and acceptor (HDA) to the antiferromagnetic exchange coupling between two Cu(II)’s (−2J).
This leads to the point of emphasis in this section that differences in the antiferromagnetic (AF) exchange coupling between the Cu(II)’s determines the mode of O2 activation.34 As shown in the box at the bottom of Figure 9, the AF coupling between Cu(II)’s, −2J, can be related to the electronic coupling matrix element for ET between the donor and acceptor, HDA, in Marcus theory. As the classification of these copper active sites indicates, in the coupled binuclear Cu sites, the strong electronic coupling leads to 2e− reduction of O2 to form a peroxo intermediate with a peroxo σ* FMO capable of 2e− electrophilic aromatic attack. Alternatively, in the noncoupled binuclear Cu enzymes, the lack of exchange coupling can result in 1e− reduction of O2 to generate an end-on superoxide–CuM(II) triplet with a π* FMO (perpendicular to the CuOO plane) capable of 1e− H atom abstraction from the substrate. Then, at a later stage of the reaction (Figure 9 right, in green), the second e− can transfer from CuH when there is a large driving force to overcome the low HDA. Note that in a recent crystal structure of DβM,36 two site conformations are observed, one similar to PHM with a predicted CuM···CuH distance of ~14 Å and a second “closed” form with a predicted CuM···CuH separation of ~5 Å. While the functional relevance is not yet understood, this suggests that the protein may be able to modulate HDA through conformational flexibility to further control the timing of ET.
The way we envision completion of this reaction is by radical coupling to the nonprotonated oxygen of the bound hydroperoxide. 37 The resultant radical coupled intermediate now has a large driving force enabling intramolecular ET to complete the reaction.
An important point here is that the second e− derives from CuH, which is an unusual ET site in biology (contrast this to the blue Cu site in the next section) in that it has a large innersphere reorganization energy for ET (i.e., it binds a H2O ligand on oxidation, Figure 10). This large reorganization energy is important in controlling ET to occur only at the step in the reaction where there is a large driving force for ET (deriving from the reduction of the coupled radical intermediate at the CuM center), thus avoiding deleterious Fenton chemistry at an earlier stage of the reaction (reduction of the CuM(II)OOH intermediate).37
Figure 10.
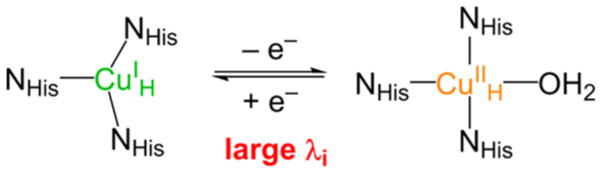
Large geometry change associated with redox at CuH (i.e., water binds to the oxidized site).
Trinuclear Cu Cluster Active Sites
Active sites composed of trinuclear Cu clusters are present in the multicopper oxidases (MCOs; laccases, ceruloplasmins, etc.) that couple four 1e− oxidations of substrates to the 4e− reduction of O2 to water, often with little overpotential (Table 1, bottom). As shown in Figure 11A, MCOs generally contain four copper ions. A type 1 (T1), or blue, Cu center at the surface of the enzyme takes an e− from substrate, which is transferred rapidly over 13 Å through a Cys-His pathway (see ref 38 for details on ET by blue Cu sites) to a trinuclear Cu cluster (TNC) where O2 is reduced to water.39 In the resting enzyme, the TNC is comprised of a type 3 (T3) pair of Cu(II)’s AF coupled through a OH− bridge leading to a singlet ground state with no contribution to the EPR spectrum. This is often incorrectly correlated to the coupled binuclear copper site in Hc and Ty (vide inf ra). Approximately 3.5 Å from the T3 center is a mononuclear type 2 (T2) Cu(II) that exhibits a normal EPR signal. Detailed spectroscopy and calculations on the resting TNC have shown that all three Cu’s (Figure 11B) have open coordination positions oriented toward the center of the cluster, well poised for bridging oxygen intermediates.40
Figure 11.

Geometric and electronic structures of the resting multicopper oxidases. (A) Geometric structure.41 The electron from substrate enters at T1 and is transferred over 13 Å to the TNC where O2 is reduced to water. The TNC has an OH− bridged AF coupled T3 Cu(II) pair with no EPR signal and a localized T2 Cu(II) at 3.5 Å from the T3 Cu’s with a normal EPR signal in the resting enzyme. (B) Results of spectroscopic and electronic structure calculations (ref 40) showing that all three Cu’s of the resting TNC have open coordination positions oriented toward the center of the cluster enabling bridged O2 intermediates.
We have trapped and spectroscopically defined two oxygen intermediates in the reduction of O2 to water by the TNC (Figure 12).42 Reaction of a fully reduced native MCO (reduced T1 + reduced TNC) with O2 occurs with a bimolecular rate constant of ~2 × 106 M−1 s−1 to generate the “native intermediate” (NI; Figure 12, top right). Alternatively, elimination of the T1 Cu (and its e−) by either replacement with Hg2+43 or in a knockout mutant in an expression system44 (in collaboration with Prof. Dan Kosman45) gives a reduced TNC that reacts with O2 at approximately the same rate to generate the “peroxide intermediate” (PI, Figure 12 bottom right),42 where two of the three Cu’s of the TNC are oxidized and have reduced dioxygen by two electrons.46 The peroxide intermediate is a precursor to the native intermediate, but by eliminating the e− from the T1 Cu, the PI to NI conversion is slowed down by >106. Below, we use spectroscopy combined with calculations to define PI, NI, and the reductive cleavage of the O–O bond.
Figure 12.
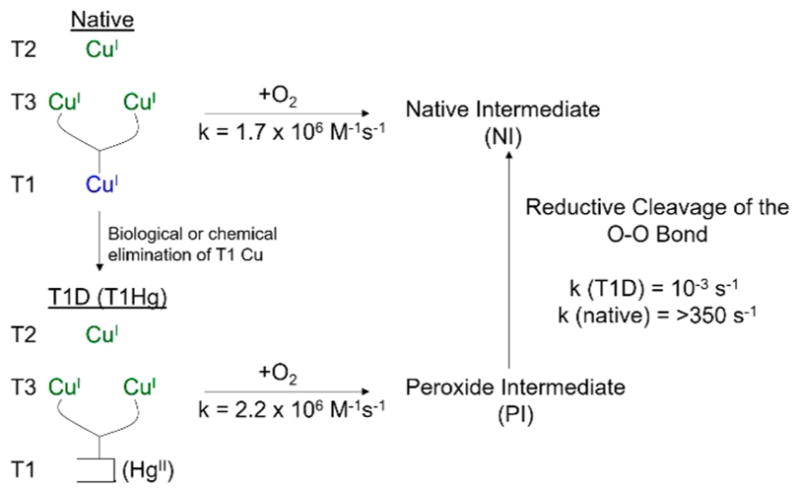
O2 reactions and intermediates of the MCOs. Top: O2 reaction of the fully reduced native enzyme to generate the native intermediate (NI). Bottom: Elimination of the T1 gives a fully reduced TNC which reacts with O2 to generate a peroxide intermediate (PI) with 2Cu(II)/1Cu(I) at the TNC. Right bottom to top: Conversion of PI to NI.
A wide range of studies have defined the nature of PI;46,47 the key result of these studies is nicely illustrated by its peroxide to Cu(II) CT spectrum in Figure 13A. The spectrum of PI (red) is entirely different from that of oxy Hc and Ty (black). The intensity in the 340–350 nm region is greatly reduced, and there are additional lower energy peroxide to Cu(II) CT transitions in PI at ~380 nm and ~490 nm demonstrating that the peroxide intermediate at the TNC has a different electronic and geometric structure from the side-on peroxide bridge in oxyHc/Ty. However, if O2 is computationally added to a reduced TNC and geometry optimized, the structure at the left of Figure 13B is obtained with peroxide binding side-on between two oxidized T3 Cu’s. This O2 binding is uphill. Native MCOs have a carboxylate on the T2/T3B edge, and when this is mutated to a noncharged residue (D94A or D94N in Fet3p), the reduced TNC no longer reacts with O2.48 When the native Asp is replaced by a Glu the reduced TNC again binds O2 to generate PI. Thus, D94 was included in the TNC cluster calculations (Figure 13B right), and a new energetically favorable structure was obtained that is consistent with all of the spectroscopic properties of PI. The PI structure in Figure 13B, right, has peroxide bridging all three Cu’s of the TNC with T2 and T3B oxidized (due to the nearby Asp). The spectral differences in Figure 13A, reflecting the structural differences in Figure 13B, demonstrate a fundamental structure/f unction difference in copper/dioxygen bioinorganic chemistry: reversible binding and activation for electrophilic chemistry by the side-on peroxide bridged coupled binuclear Cu site of oxy Hc/Ty versus the irreversible reductive cleavage of the O–O bond by the all-bridged trinuclear copper cluster.
Figure 13.

Unique geometric and electronic structure of PI and comparison to oxyHc/Ty. (A) Absorption spectrum showing peroxide to Cu(II) CT (PI red, oxyHc/Ty black). (B) Geometry optimization of a reduced TNC with O2, both without (left) and with (right) the Asp present as in the WT enzyme at the T2/T3B edge.
This brings us to the native intermediate (NI) of the MCOs.49 Reaction of the fully reduced native enzyme with both the T1 reduced and the TNC reduced generates the native intermediate with the absorption spectrum shown at the top of Figure 14A. The bands at 365 and 318 nm are CT transitions at the TNC of NI, while the band at ~16 000 cm−1 is the oxidized T1 (blue copper) center. Thus, at least one additional electron has been transferred (from the T1) to O2 relative to PI. We had earlier (with Prof. Keith Hodgson) shown that Cu K edge X-ray absorption spectroscopy could define oxidation states of Cu since reduced Cu has a peak at 8984 eV while oxidized Cu does not.50 NI has no 8984 eV feature reflecting reduced Cu (Figure 14B).49 Thus, all four Cu’s are oxidized in NI, and dioxygen is reduced by 4e− to the water level. NI then contains a fully oxidized TNC, just as present in the resting enzyme (Figure 11). The resting TNC exhibits the EPR signal in black in Figure 14C, which simply reflects the “normal” T2 Cu(II). In striking contrast, NI exhibits the EPR signal in red in Figure 14C, which is very broad, fast relaxing, and most significantly has g values well below 2.00. This is unique for a Cu(II) site. When the ground state is unclear, we can use an excited state to probe the ground state employing temperature- and field-dependent magnetic circular dichroism (MCD) spectroscopy. At the bottom of Figure 14A is the MCD spectrum of a rapid freeze-quench sample of NI. This low temperature, high field MCD spectrum exhibits an intense derivative-shaped signal (a pseudo-A term) associated with the CT transitions at the TNC of NI. Since the TNC of NI is paramagnetic (red spectrum in Figure 14C), its MCD signal intensity should decrease with increasing temperature (at a fixed field) as 1/T (the MCD analog of Curie law paramagnetism in magnetic susceptibility). This is given by the blue curve in Figure 14D. The MCD signal intensity of NI does first decrease with increasing temperature, but then it increases as the temperature is further increased (red dots in Figure 14D). This indicates a Boltzmann population of a low lying excited state with an MCD spectrum different from that of the ground state. From the fit to the MCD data in Figure 14D, this excited state is ~150 cm−1 above the ground state. This is strange for a Cu(II) site and showed us that the TNC of NI was a spin frustrated system. As illustrated in Figure 14E, if Cu(II)A and Cu(II)C have a bridging ligand, their spins should be AF coupled (↑ to ↓ clockwise from top). If Cu(II)C and Cu(II)B are also bridged, their spins should also be AF coupled (↓ to ↑). This leads to up-spins on Cu(II)B and Cu(II)A, but if these also have a bridging ligand (closing the triangle) they are AF coupled (one up, one down). This frustrated spin topology leads to the low lying excited state (Figure 14D) and g values below 2.00 (Figure 14C, red) due to the antisymmetric exchange associated with a frustrated spin system.51 A quantitative analysis of the EPR and pseudo-A term MCD data and QM/MM studies (in collaboration with Profs. Lubomír Rulíšek and Ulf Ryde52) led to the all-bridged NI structure in Figure 14F with a μ3-oxo in the center of the TNC and a μ2-OH at the T3 edge, both oxygens deriving from the reductive cleavage of the O–O bond.47
Figure 14.

Unique spectroscopic features, electronic structure, and geometric structure of NI. (A) Absorption (top) and low temperature MCD (bottom). (B) X-ray absorption K-edge of NI (red) compared to those of reduced (green) and oxidized Cu (blue). (C) EPR spectrum of NI (red) compared to that of the resting TNC(black, T2 normal EPR, T3 AF coupled S = 0). (D) Variable temperature (at high field) MCD of NI (blue gives 1/T behavior of normal paramagnetic Cu site). The arrow extending from the MCD spectrum in A to the intensity vs temperature plot in D indicates that the intensity plotted is for the 27 560 cm−1 band in A. (E) Spin topology of three all-bridged Cu(II)’s leading to spin frustration. (F) Structure of NI from spectroscopies and calculations.
These studies have led to the structures of the peroxide intermediate (PI) at the left of Figure 15 and native intermediate (NI) at the right of Figure 15. The addition of an electron to PI (from the T1 Cu) gives the structure in the center of Figure 15 with T2 and T3A reduced and T3B oxidized, which is the starting point for reductive cleavage of the O–O bond.47 The description of this cleavage requires a two-dimensional potential energy surface (Figure 16A). One dimension is O–O bond elongation (coming out on the left), and the second (derived from mutagenesis studies48) involves proton transfer from a carboxylate at the T3 edge (coming out on the right in Figure 16A). Two low energy paths are found on this surface. Path 2 involves proton transfer before the transition state (TS2) and predicts an inverse KIE that is observed experimentally in the decay of PI at low pH.53 Path 1 involves O–O bond cleavage before proton transfer. Importantly, both have very low barriers of 5–6 kcal/mol, reflecting the FMOs of the TNC. As shown in Figure 16B, both reduced Cu’s (T2 and T3A) have good overlap with the peroxide σ* electron pair acceptor orbital, and the third Cu (T3B) which is oxidized acts like a proton in lowering the energy of the σ* orbital and activating O–O cleavage.
Figure 15.

Reaction coordinate for the reductive cleavage of the O–O bond by the TNC. Left: PI. Center: reduction of PI by one electron (from T1 Cu). Right: NI.
Figure 16.

Reductive cleavage of the O–O bond by the TNC. (A) 2D potential energy surface (path 1 for high pH and path 2 for low pH). (B) FMOs associated with the TNC leading to the low barrier for O–O cleavage. T2 and T3A are reduced, and T3B is oxidized.
Our studies on the multicopper oxidases have led to the molecular mechanism in Figure 17.54 Starting from the top left and proceeding clockwise, dioxygen is reduced in two 2e− steps, the second having a low barrier due to the FMOs of the TNC (Figure 16B). This is effectively a 4e− process producing NI. NI is a fully oxidized form of the MCOs, just as is the case for the resting enzyme that is studied by X-ray crystallography. 41 However, NI and not the resting enzyme is the catalytically relevant fully oxidized form of the MCOs.55,56 Its decay to resting is too slow (0.06 s−1) to be in the catalytic cycle (TON ~ 560 s−157). Also the rate of reduction of the resting TNC by the T1 is too slow to be catalytically relevant (1 s−158). In contrast, the rate of reduction of NI is >700 s−1,55,56 as it involves a proton coupled electron transfer process (PCET) that is driven by the high basicity of the μ3-oxo at the center of the trinuclear Cu cluster that results from the reductive cleavage of the O–O bond.
Figure 17.
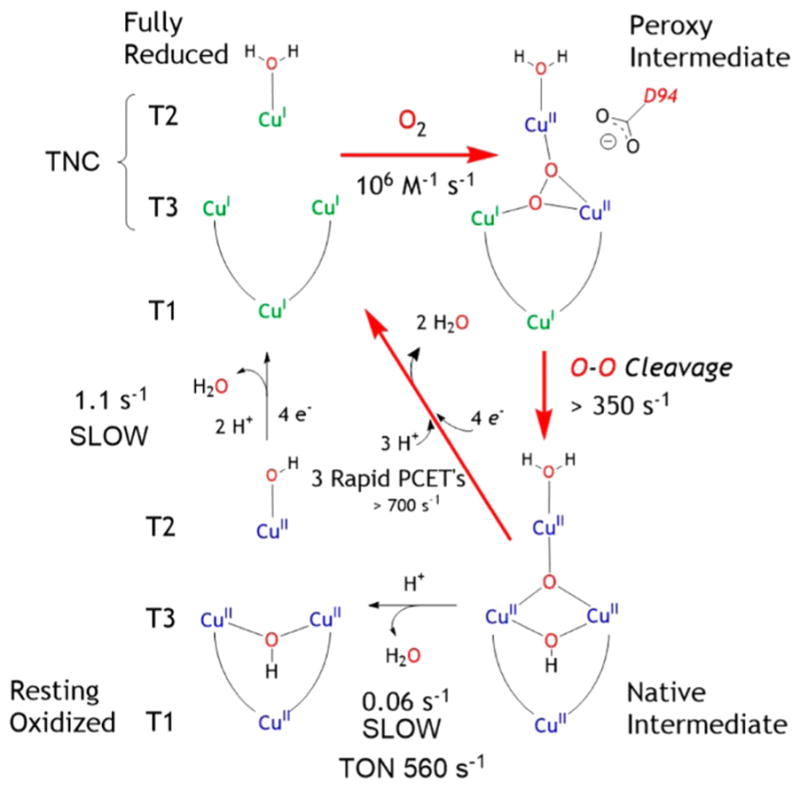
Molecular mechanism for the reduction of O2 to water and the reduction of the oxidized MCO in turnover. Red arrows give the reaction cycle. Black arrows give off turnover pathways leading to the resting enzyme (bottom) and its slow reduction on the left.
Acknowledgments
First, I thank Stephen Jones, Ryan Cowley, and Greg Schneider for assistance in preparing this Award Article. I would like to thank all my past graduate students and postdocs (listed above) who have made the past 40 years an amazing scientific adventure, and my close friends and collaborators, many having participated in the Bioinorganic Symposium associated with the Alfred Bader Award. I thank the NIH (DK31450) for 34 years of continued support of this research.
Biography

Edward I. Solomon grew up in North Miami Beach, Florida, received his Ph.D. at Princeton (with D.S. McClure), and was a postdoctoral fellow at The Ørsted Institute in Denmark (with C.J. Ballhausen) and then at Caltech (with H.B. Gray). He was a Professor at the Massachusetts Institute of Technology until 1982, when he joined the faculty at Stanford University, where he is now the Monroe E. Spaght Professor of Humanities and Sciences and Professor of Photon Science at SLAC National Accelerator Lab. He has been an Invited Professor in Argentina, Australia, Brazil, China, France, India, and Japan. Professor Solomon’s research is in the fields of Physical–Inorganic and Bioinorganic Chemistry with emphasis on the application of a wide range of spectroscopic methods combined with QM calculations to elucidate the electronic structure of transition metal sites and its contribution to physical properties and reactivity. He has received a range of medals and awards including the ACS National Awards in Inorganic Chemistry, Distinguished Service in the Advancement of Inorganic Chemistry, and the Alfred Bader Award in Bioinorganic or Bioorganic Chemistry. He is a member of the National Academy of Sciences, the American Academy of Arts and Sciences, and a Fellow of the American Association for the Advancement of Science and of the American Chemical Society.
Group Alumni. Mark Allendorf, T. J. Augustine, Michael Baldwin, Lipika Basumallick, Caleb Bell, Christina Brown, Carl Brown, Thomas Brunold, Kristine Butcher, Cecelia Campochiaro, Peng Chen, Marina Chow, Jordi Cirera, Pat Clark, Michael Clay, Susan Cohen, Jim Cole, Kevin D’Amico, Mindy Davis, Joseph Deaton, Serena DeBeer, Andrea Decker, Sylvia Desjardins, Abhishek Dey, Steve Didziulis, Adrienne Diebold, Diego Diaz-Jara, Nancy Eickman, Mathieu Frenette, Daniel Gamelin, Robert Gay, Matthew Gebhard, Andrew Gewirth, Somdatta Ghosh, Jake Ginsbach, Thorsten Glaser, Serge Gorelsky, Jeffrey Guckert, Ryan Hadt, Mark Hensen, Dave Heppner, Richard Himmelwright, Rosalie Hocking, O. Horner, Yeong Tsyr Hwang, Esther Johnston, Paul Jones, Lung Kau, Jyllian Kemsley, Pierre Kennepohl, Matthew Kieber-Emmons, Marty Kirk, Christian Kjaergaard, Thomas Kroll, Yeonju Kwak, Lou LaCroix, Sang-Kyu Lee, Nicolai Lehnert, Barry Liboiron, Kenneth Light, Jianyi Lin, Lei Liu, John Long, Mike Lowery, Cynthia LuBien, Marcus Lundberg, Pam Mabrouk, Timothy Machonkin, Jim McCormick, Markus Metz, Natasa Mitic, Frank Neese, Mike Neidig, Takhiro Ohta, Bryan Op’t Holt, Amy Palmer, Kiyoung Park, Jim E. Pate, Monita Pau, Elizabeth Pavel, Mark Pavlosky, Kevin Penfield, James Penner-Hahn, Elna Pidcock, Arturo G. Porras, Sabine Pulver Coates, Munzarin Qayyum, Liliana Quntanar, David Randall, Richard Reem, Brad Reitz, David Richardson, David Root, Kendra Rose, Paul Ross, P.O. Sandusky, Ritimukta Sarangi, Gary Schenk, Jennifer Schwartz, Tami Seddon (Westre), Susan Shadle, Woonsup Shin, Andrew Skulan, Pieter Smeets, Rae Ana Snyder, Darlene Solomon (Spira), Martin Srnec, Grit Straganz, Uma Sundarum, Robert Szilagyi, Federico Tasca, Adam Tenderholt, Tom Thamann, Li Tian, Ming-Li Tsai, Felix Tuczek, Michael Vance, Erik Wasinger, Pinpin Wei, T. David Westmoreland, James Whittaker, Dean Wilcox, Randy Wilson, Sam Wilson, Marge Winkler, Julia Woertink, Shaun Wong, Winson Xie, Yi-Shan Yang, Jungjoo Yoon, Jeffrey Zaleski, Kelly Zaleski (Loeb), Yan Zhang, and Jing Zhou
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Allendorf MD, Spira DJ, Solomon EI. Proc Natl Acad Sci U S A. 1985;82(10):3063–3067. doi: 10.1073/pnas.82.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spira-Solomon DJ, Allendorf MD, Solomon EI. J Am Chem Soc. 1986;108:5318–5328. [Google Scholar]
- 3.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114(7):3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldwin MJ, Root DE, Pate JE, Fujisawa K, Kitajima N, Solomon EI. J Am Chem Soc. 1992;114(26):10421–10431. [Google Scholar]
- 5.Pate JE, Cruse RW, Karlin KD, Solomon EI. J Am Chem Soc. 1987;109(9):2624–2630. [Google Scholar]
- 6.Baldwin MJ, Ross PK, Pate JE, Tyeklar Z, Karlin KD, Solomon EI. J Am Chem Soc. 1991;113(23):8671–8679. [Google Scholar]
- 7.Magnus KA, Hazes B, Ton-That H, Bonaventura C, Bonaventura J, Hol WGJ. Proteins: Struct, Funct, Genet. 1994;19(4):302–309. doi: 10.1002/prot.340190405. [DOI] [PubMed] [Google Scholar]
- 8.Kitajima N, Fujisawa K, Morooka Y, Toriumi K. J Am Chem Soc. 1989;111(24):8975–8976. [Google Scholar]
- 9.Eickman NC, Solomon EI, Larrabee JA, Spiro TG, Lerch K. J Am Chem Soc. 1978;100(20):6529–6531. [Google Scholar]
- 10.Loehr JS, Freedman TB, Loehr TM. Biochem Biophys Res Commun. 1974;56(2):510–515. doi: 10.1016/0006-291x(74)90872-9. [DOI] [PubMed] [Google Scholar]
- 11.Larrabee JA, Spiro TG, Ferris NS, Woodruff WH, Maltese WA, Kerr MS. J Am Chem Soc. 1977;99(6):1979–1980. doi: 10.1021/ja00448a053. [DOI] [PubMed] [Google Scholar]
- 12.Solomon EI, Dooley DM, Wang RH, Gray HB, Cerdonio M, Mogno F, Romani GL. J Am Chem Soc. 1976;98(4):1029–1031. doi: 10.1021/ja00420a035. [DOI] [PubMed] [Google Scholar]
- 13.Ross PK, Solomon EI. J Am Chem Soc. 1990;112(15):5871–5872. [Google Scholar]
- 14.Ross PK, Solomon EI. J Am Chem Soc. 1991;113(9):3246–3259. [Google Scholar]
- 15.Metz M, Solomon EI. J Am Chem Soc. 2001;123(21):4938–4950. doi: 10.1021/ja004166b. [DOI] [PubMed] [Google Scholar]
- 16.Wilcox DE, Porras AG, Hwang YT, Lerch K, Winkler ME, Solomon EI. J Am Chem Soc. 1985;107(13):4015–4027. [Google Scholar]
- 17.Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. J Am Chem Soc. 1998;120(31):7841–7847. [Google Scholar]
- 18.Qayyum MF, Sarangi R, Fujisawa K, Stack TDP, Karlin KD, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2013;135(46):17417–17431. doi: 10.1021/ja4078717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbuhler AD, Tolman WB. Science. 1996;271(5254):1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 20.Mirica LM, Vance M, Jackson Rudd D, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308(5730):1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 21.Op’t Holt BT, Vance MA, Mirica LM, Heppner DE, Stack TDP, Solomon EI. J Am Chem Soc. 2009;131(18):6421–6438. doi: 10.1021/ja807898h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ginsbach JW, Kieber-Emmons MT, Schneider GL, Andrikopoulos P, Rulíšek L, Solomon EI. Unpublished results. [Google Scholar]
- 23.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278(5341):1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 24.Chen P, Fujisawa K, Solomon EI. J Am Chem Soc. 2000;122(41):10177–10193. [Google Scholar]
- 25.Chen P, Bell J, Eipper BA, Solomon EI. Biochemistry. 2004;43(19):5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 26.Chen P, Solomon EI. J Am Chem Soc. 2004;126(15):4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 27.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. J Am Chem Soc. 1994;116(26):12079–12080. [Google Scholar]
- 28.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J Am Chem Soc. 2003;125(2):466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 29.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304(5672):864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 30.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem, Int Ed. 2006;45(23):3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 31.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. J Am Chem Soc. 2007;129(47):14697–14709. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 32.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg Chem. 2010;49(20):9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Inorg Chem. 2013;52(22):12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen P, Solomon EI. Proc Natl Acad Sci U S A. 2004;101(36):13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francisco WA, Merkler DJ, Blackburn NJ, Klinman JP. Biochemistry. 1998;37(22):8244–8252. doi: 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]
- 36.Vendelboe TV, Harris P, Zhao Y, Walter TS, Harlos K, El Omari K, Christensen HEM. Sci Adv. 2016;2(4):e1500980–e1500980. doi: 10.1126/sciadv.1500980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowley RE, Tian L, Solomon EI. To be published. [Google Scholar]
- 38.Hadt RG, Gorelsky SI, Solomon EI. J Am Chem Soc. 2014;136(42):15034–15045. doi: 10.1021/ja508361h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole JL, Tan GO, Yang EK, Hodgson KO, Solomon EI. J Am Chem Soc. 1990;112(6):2243–2249. [Google Scholar]
- 40.Quintanar L, Yoon J, Aznar CP, Palmer AE, Andersson KK, Britt RD, Solomon EI. J Am Chem Soc. 2005;127(40):13832–13845. doi: 10.1021/ja0421405. [DOI] [PubMed] [Google Scholar]
- 41.Messerschmidt A, Rossi A, Ladenstein R, Huber R, Bolognesi M, Gatti G, Marchesini A, Petruzzelli R, Finazzi-Agró A. J Mol Biol. 1989;206(3):513–529. doi: 10.1016/0022-2836(89)90498-1. [DOI] [PubMed] [Google Scholar]
- 42.Cole JL, Ballou DP, Solomon EI. J Am Chem Soc. 1991;113(22):8544–8546. [Google Scholar]
- 43.Morie-Bebel MM, Morris MC, Menzie JL, McMillin DR. J Am Chem Soc. 1984;106(12):3677–3678. [Google Scholar]
- 44.Blackburn NJ, Ralle M, Hassett R, Kosman DJ. Biochemistry. 2000;39(9):2316–2324. doi: 10.1021/bi992334a. [DOI] [PubMed] [Google Scholar]
- 45.Palmer AE, Quintanar L, Severance S, Wang TP, Kosman DJ, Solomon EI. Biochemistry. 2002;41(20):6438–6448. doi: 10.1021/bi011979j. [DOI] [PubMed] [Google Scholar]
- 46.Shin W, Sundaram UM, Cole JL, Zhang HH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1996;118:3202–3215. [Google Scholar]
- 47.Yoon J, Solomon EI. J Am Chem Soc. 2007;129(43):13127–13136. doi: 10.1021/ja073947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Augustine AJ, Quintanar L, Stoj CS, Kosman DJ, Solomon EI. J Am Chem Soc. 2007;129(43):13118–13126. doi: 10.1021/ja073905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SK, George SD, Antholine WE, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2002;124(21):6180–6193. doi: 10.1021/ja0114052. [DOI] [PubMed] [Google Scholar]
- 50.Kau L, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. J Am Chem Soc. 1987;109(9):6433–6442. [Google Scholar]
- 51.Yoon J, Mirica LM, Stack TDP, Solomon EI. J Am Chem Soc. 2005;127(39):13680–13693. doi: 10.1021/ja0525152. [DOI] [PubMed] [Google Scholar]
- 52.Rulíšek L, Solomon EI, Ryde U. Inorg Chem. 2005;44(16):5612–5628. doi: 10.1021/ic050092z. [DOI] [PubMed] [Google Scholar]
- 53.Palmer AE, Lee SK, Solomon EI. J Am Chem Soc. 2001;123(27):6591–6599. doi: 10.1021/ja010365z. [DOI] [PubMed] [Google Scholar]
- 54.Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew Chem, Int Ed. 2001;40(24):4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 55.Heppner DE, Kjaergaard CH, Solomon EI. J Am Chem Soc. 2013;135(33):12212–12215. doi: 10.1021/ja4064525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heppner DE, Kjaergaard CH, Solomon EI. J Am Chem Soc. 2014;136(51):17788–17801. doi: 10.1021/ja509150j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petersen LCHR, Degn H. Biochimica et Biophysica Acta (BBA) - Enzymology. 1978;526:85–92. doi: 10.1016/0005-2744(78)90292-9. [DOI] [PubMed] [Google Scholar]
- 58.Farver O, Wherland S, Koroleva O, Loginov DS, Pecht I. FEBS J. 2011;278(18):3463–3471. doi: 10.1111/j.1742-4658.2011.08268.x. [DOI] [PubMed] [Google Scholar]