

To the Editor: Glycogen storage disease (GSD) is a group of inherited disorders with abnormal glycogen deposition.[1] Pulmonary hypertension (PH) is a rare complication of GSD with unclear etiology. Since the pioneer description in 1980,[2] only a few cases of GSD-associated PH have been reported. Here, we reported a Chinese case with informed consent by the patient and the guardian.
A 16-year-old girl was admitted to China-Japan Friendship Hospital with cyanosis and dyspnea on exertion for 3 months. The patient was found with growth retardation from 5 years of age. She was 153 cm in height and 34 kg in body weight on admission. Physical examination revealed that her skeletal muscles were generally weak and atrophic. On auscultation, an accentuated pulmonic second heart sound was noted. Serum creatine kinase concentration was 238 U/L. Electrocardiogram showed right-axis deviation. Echocardiogram showed right ventricular enlargement and tricuspid regurgitation with systolic pulmonary artery pressure of 65 mmHg (<36 mmHg in normal, 1 mmHg = 0.133 kPa), and no intracardiac shunt was found. The arterial blood gas analysis in room air revealed pH 7.325, PCO283.2 mmHg, PO242.3 mmHg, and BE 11.7 mmol/L. Pulmonary function test showed forced expiratory volume in 1 s (FEV1) 0.5 L (20.6% of predicted value), forced vital capacity (FVC) 0.5 L, and FEV1/FVC 100%. Based on the clinical information, neuromuscular diseases were suspected. Activity of acid-α-glucosidase was then detected with 5.4 nmol·h−1· mg protein−1 (normal, 62.3–301.7 nmol·h − 1·mg protein − 1). Muscle biopsy of left biceps brachii was performed which showed several fibers with vacuolar appearance and accumulation of PAS-positive granules [Figure 1]. Genetic analysis revealed compound heterozygous mutations in GAAat.1309C>T (p.R437C) and c.1562A>T (p.E521V). These findings confirmed that the patient had GSD Type II. After treatment with noninvasive positive pressure ventilation, diuretics, antibiotics, and supportive care, she got improved and discharged.
Figure 1.
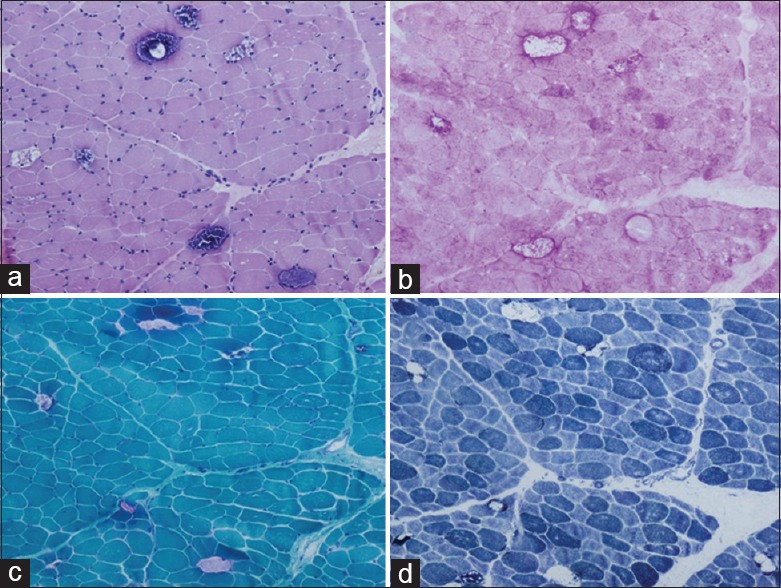
Histopathological findings from muscle biopsy. (a) Variation in fiber size and sarcoplasmic vacuolar appearance with basophilic granule deposits (H and E, original magnification, ×100). (b) Accumulated glycogen was identified in vacuoles (PAS, original magnification, ×100). (c) The disrupted fibers contain excess glycogen with trichrome reaction (MGT, original magnification, ×100). (d) Irregular loss of enzyme staining was observed in vacuolated fibers (NADH-TR, original magnification, ×100). PAS: Periodic acid-Schiff; MGT: Modified Gomori trichrome; NADH-TR: Nicotinamide adenine dinucleotide-tetrazolium reductase.
GSD-associated PH is poorly understood. So far, GSD Types I, II, and III have all been reported to be associated with PH. Most patients with PH complicating the course of GSD were in their second or third decade of life, suggesting that long-term metabolic abnormalities may contribute to the long latency of PH.[3] Endothelial dysfunction secondary to the metabolic disturbances of glucose has been speculated as a mechanism of PH. In our case, muscle weakness is one of the main features, including the skeletal and respiratory muscles, causing severely impaired pulmonary function and respiratory failure, which may lead to PH. Some suggested that smooth muscle cells in blood vessels might also be involved as another mechanism for PH.[4,5]
In conclusion, GSD-associated PH is rare with unclear etiology and/or multifactorial factors. Respiratory muscle weakness and reduced pulmonary function may be the main factors for PH in GSD Type II.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has given her consent for her images and other clinical information to be reported in the journal. The patient understand that her name and initials will not be published and due efforts will be made to conceal her identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This work was supported by grants from the National Natural Science Foundation of China (No. 81570049), Beijing Natural Science Foundation (No. 7152062), and the National Key Research and Development Program of China (No. 2016YFC0905600).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.MENA Pompe Working Group, Al Jasmi F, Al Jumah M, Alqarni F, Al-Sanna'a N, Al-Sharif F, et al. Diagnosis and treatment of late-onset Pompe disease in the Middle East and North Africa region: Consensus recommendations from an expert group. BMC Neurol. 2015;15:205. doi: 10.1186/s12883-015-0412-3. doi: 10.1186/s12883-015-0412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pizzo CJ. Type I glycogen storage disease with focal nodular hyperplasia of the liver and vasoconstrictive pulmonary hypertension. Pediatrics. 1980;65:341–3. [PubMed] [Google Scholar]
- 3.Humbert M, Labrune P, Simonneau G. Severe pulmonary arterial hypertension in type 1 glycogen storage disease. Eur J Pediatr. 2002;161(Suppl 1):S93–6. doi: 10.1007/s00431-002-1012-y. doi: 10.1007/s00431-002-1012-y. [DOI] [PubMed] [Google Scholar]
- 4.Katona I, Weis J, Hanisch F. Glycogenosome accumulation in the arrector pili muscle in Pompe disease. Orphanet J Rare Dis. 2014;9:17. doi: 10.1186/1750-1172-9-17. doi: 10.1186/1750-1172-9- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Beek NA, van Capelle CI, van der Velden-van Etten KI, Hop WC, van den Berg B, Reuser AJ, et al. Rate of progression and predictive factors for pulmonary outcome in children and adults with Pompe disease. Mol Genet Metab. 2011;104:129–36. doi: 10.1016/j.ymgme.2011.06.012. doi: 10.1016/j.ymgme. 2011.06.012. [DOI] [PubMed] [Google Scholar]