

Abstract
Gnathodiaphyseal dysplasia (GDD; OMIM #166260) is an ultra-rare autosomal dominant disorder caused by heterozygous mutation in the anoctamin 5 (ANO5) gene and features fibro-osseous lesions of the jawbones, bone fragility with recurrent fractures, and bowing/sclerosis of tubular bones. The physiologic role of ANO5 is unknown. We report a 5-year-old boy with a seemingly atypical and especially severe presentation of GDD and unique ANO5 mutation. Severe osteopenia was associated with prenatal femoral fractures, recurrent postnatal fractures, and progressive bilateral enlargement of his maxilla and mandible beginning at ~ 2 months-of-age that interfered with feeding and speech and required four debulking operations. Histopathological analysis revealed benign fibro-osseous lesions resembling cemento-ossifying fibromas of the jaw without psammomatoid bodies. A novel, de novo, heterozygous, missense mutation was identified in exon 15 of ANO5 (c.1553G>A; p.Gly518Glu). Our findings broaden the phenotypic and molecular spectra of GDD. Fractures early in life with progressive facial swelling are key features. We assessed his response to a total of 7 pamidronate infusions commencing at age 15 months. Additional reports must further elucidate the phenotype, explore any genotype-phenotype correlation, and evaluate treatments.
Keywords: autosomal dominant, bisphosphonates, cemento-ossifying fibroma, cherubism, diaphyseal sclerosis, fracture, psammomatoid bodies
II) Introduction
Gnathodiaphyseal dysplasia (GDD) (OMIM #166260) is the autosomal dominant (AD) disorder characterized by cemento-osseous lesions of the mandible and maxilla, facial deformity, bowing/sclerosis of tubular bones, and generalized skeletal fragility with recurrent fractures (1–3). GDD was first described in 1969 in 21 individuals from a four-generation Japanese kindred suffering frequent bone fractures at young ages and osteomyelitis of their jaws during adult life (4). In 2001, ‘gnathodiaphyseal dysplasia’ was coined to capture the characteristic abnormalities of the jaw containing fibro-osseous lesions with a prominent psammomatoid body component and the skeletal disease (1). To date, ~80 affected individuals in ~14 families have been reported (1,2,4–10). Table 1 summarizes the principal disease features recognized for GDD.
Table 1.
Clinical, radiological and genetic characteristics of GDD patients with molecular diagnosis or full phenotypic characterization
| Family 1 (21 pts) |
Family 2 (17 pts) |
Patient 3 | Patient 4 | Patient 5 | Family 6 (16 pts) |
Family 7 (2 pts) |
Family 8 (8 pts) |
Family 9 (2 pts) |
Family 10 (1 pt) |
Family 11 (7 pts) |
Family 12 (2 pts) |
This case (1 pt) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Tsutsumi et al., 2004 | Tsutsumi et al., 2004 | Riminucci et al., 2001 | Ahluwalia et al. 2007 | Roginsky et al., 2010 | Marconi et al. 2013 | Vengoech ea 2015 | Doung et al, 2016 | Andreeva et al. 2016 | Rolvien et al. 2016 | Jin et al. 2017 | Jin et al. 2017 | Otaify et al. |
| Ethnicity | Japanese | African American | Caucasian | N/A | Russian | Italian | N/A | German/It alian | Russian | N/A | Caucasian | Chinese | Caucasian |
| ANO5 Mutation | c.1067T> A (p.Cys356 Arg) exon 11 | c.1067T>G (p.Cys356G ly) exon 11 | c.643 A > G (p.Arg215 Gly) exon 7 | Not done | Not done | c.1538C>T, (p.Thr513Il e) exon 15 | c.1067 G>A (p.Cys356 Tyr) exon 11 | c. 1067 G>A (p. Cys356 Tyr) exon 11 | c.1067G > A (p.Cys356 Tyr) exon 11 | c.1499C> T (p.Ser500 Phe) Exon 15 | c.1067G> A (p.Cys356 Tyr) exon 11 | c.1079G > A (p.Cys360 Tyr) exon 11 | c.1553G>A, (p.Gly518Gl u) exon 15 |
| Jaw bone findings (age of onset) | Growth &purulent osteomyel itis (adulthood ) | N/A | Maxillary growth &sinus infection; debulking (13 mo) | Multicentr ic sclerotic expansion & calcificati on involving palate & maxillary sinuses (14 yrs) | Mandibula r & maxillary growth 3 debulking operations & subtotal mandibule ctomy | Growth & purulent osteomyelit is (25 yrs; 1 pt) | Growth dental abscesses & recurrent jaw osteomyel itis (6 & 54 yrs) | Mandibula r osteomyel itis & impacted teeth (21& 41 yrs; 2 pts) | Mandibula r & maxillary growth; debulking & subtotal mandibule ctomy(3, 10 yo) | Mandibula r growth; impacted teet; subtotal mandibule ctomy | Mandibula r mass (lytic & sclerotic); partial mandible & maxilla resection (7-21yrs) | Enlargeme nt and infections in mandible and maxilla; subtotal Mandibule ctomy (43 yrs) | Growth & debulking (2 months) |
| Age of 1st fracture | childhood &adolesce nce | N/A | 14 months | 12 years | 13 years | 8-50 yrs | 6 yrs-late teens | Childhood | 14 yrs | N/A | Multiple | No fractures | Intrauterine |
| Diaphyseal thickening of long bones | N/A | N/A | N/A | N/A | 2/5 | 1/2 | N/A | N/A | Yes | N/A | 2/2 | No | |
| Generalized reduced bone density | Yes | N/A | N/A | N/A | N/A | Yes | N/A | No | N/A | Yes | N/A | No | Yes |
| Histology of jaw bone lesion | Cemento-ossifying fibroma | N/A | Cemento-ossifying fibroma, psammom atoid variant | Cemento-ossifying fibromas | Cemento-ossifying fibroma, psammom atoid variant | Cemento-ossifying fibroma | N/A | N/A | Cemento-ossifying fibroma, psammom atoid variant | N/A | Juvenile florid osseous dysplasia, psammom atoid type | Purulent osteomyel itis-like lesions, cementom a | Cemento- ossifying fibroma |
The genetic basis of GDD commenced discovery in 2003 when Tsutsumi et al.(11) used linkage analysis of the Japanese family to map the GDD locus to chromosome 11p14.3p15.1. Then in 2004, in this kindred and in an African-American family with GDD, additional studies identified a novel gene initially called GDD1 (5) that harbored a different heterozygous missense mutation within the same codon in the two families.
GDD1, re-named in 2009 anoctamin 5 (ANO5) (12), encodes a 913 amino-acid member of the eight transmembrane-spanning domain TMEM16/anoctamin protein family. ANO5 is an integral membrane glycoprotein found predominantly in intracellular vesicles, most abundantly in skeletal and cardiac muscle but also in growth-plate chondrocytes and in osteoblasts (13). This suggests importance for ANO5 in muscle and bone development (5,13). Nevertheless, the biochemical properties and physiological function of ANO5 are incompletely understood (3,14,15).
Herein, we report comprehensive clinical, biochemical, radiological, histopathological, and molecular characterization of a 5-year-old boy with severe and atypical GDD (including findings from serum multiplex biomarker profiling, experience with bisphosphonate treatment, and multiple debulking procedures of his jaw lesions) who harbors a novel, heterozygous, missense mutation in ANO5.
III) Materials and Methods
The Human Research Protection Office, Washington University School of Medicine, St. Louis, MO, USA approved all research studies. Informed written consent to publish the clinical data and the photographs of the patient was obtained from his parents.
A) Case Report
This 5-year-old boy was referred at 3 weeks-of-age for prenatally diagnosed bilateral femur fractures first identified on a 20-week prenatal ultrasound study and presumed to be from osteogenesis imperfecta (OI). Amniocentesis had demonstrated a normal male karyotype and negative mutation studies of the COL1A1 and COL1A2 genes (Matrix DNA Diagnostic, New Orleans, LA) commonly the cause of OI (16).
He was born to non-consanguineous healthy Caucasian parents via C-section at 39 weeks-of-gestation. There were no maternal illnesses or exposures during the pregnancy. Birth weight was 3.34 kg (26-50th centile), length 47 cm (4-10th centile), and head circumference 34.3 cm (26-50th centile). There were no other perinatal or neonatal complications. The family’s medical history was unremarkable until a younger sister manifested an unrelated multi-system syndrome.
Physical examination at 3 weeks-of-age revealed shortened femurs and skin dimpling over the lateral thighs, but his mandible and maxilla appeared normal (Fig. 1A). Radiographic skeletal survey showed no excess number of Wormian bones but there was diffuse osteopenia, healing fractures of the right clavicle and several ribs, compression fractures of several thoracic and all lumbar vertebral bodies, bilateral femoral shortening and bowing, tibial bowing with healing fractures, the left fibula displaced laterally with some bowing, and the right fibula with metaphyseal irregularity from a fracture (Fig. 2A,B). Biochemical studies revealed normal serum levels of calcium (Ca), phosphorus (P), parathyroid hormone (PTH), and 25(OH)vitamin D, but elevated alkaline phosphatase (ALP) activity at 544 IU/L (Nl, 110-320 U/L). Additional bone turnover markers (BMTs) were elevated (Table 2). Collagen studies of a skin biopsy specimen were negative for OI, as was sequencing of genes associated with autosomal recessive (AR) OI (FKBP10, CRTAP, LEPRE1, PPIB, SERPINH1, SP7/OSX, SERPINF1, and PLOD2) (Collagen Diagnostic Laboratory, Seattle, WA).
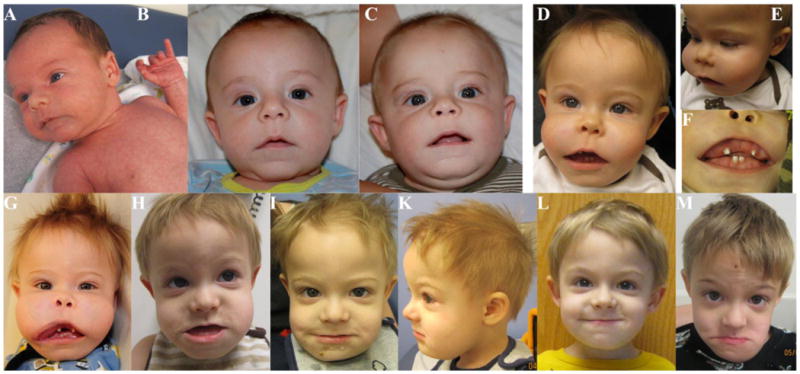
Figure 1. Patient’s Facial Features.
His facial appearance is shown at: (A) 3 weeks, (B) 3 months, (C) 6 months, (D, E, F) 9 months, (G) 12 months (before first debulking), (H) 19 months (after 2 debulking procedures), (I, K) 2 years, (L) 3 years, 4 months, and (M) 5 years of age. Notice the normal appearance at birth but then dramatic increase in size of the maxilla and mandible over 9 months, as well as the changes following debulking procedures.
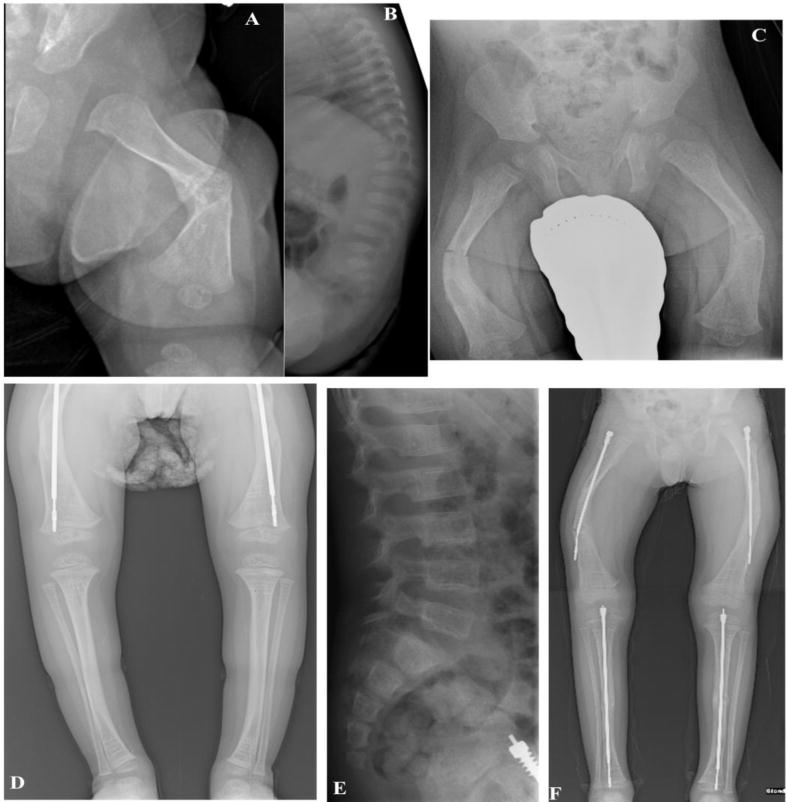
Figure 2. Patient’s Radiographic Findings.
(A) Left femur at age 3 weeks shows the angulation seen in utero, and a mid-shaft healing fracture.
(B) Lateral projection of the spine at age 3 weeks demonstrates osteopenia and compression fractures of all lumbar vertebral bodies and T6 and T12.
(C) Frontal projection of the pelvis and hips at 10 months reveals osteopenia, femoral bowing, and healing fractures in both femoral mid-shafts.
(D) Lower extremities at 3 years-of-age shows healing femoral osteotomies, Fassier-Durval nails, osteopenia, and multiple sclerotic lines from intravenous pamidronate.
(E) Lateral lumbar spine at 4 years demonstrates osteopenia and an L5 pars defect and spondylolisthesis, but some vertebral body compression fracture improvement.
(F) There is incomplete healing of tibial osteotomies, tibial and femoral nailings, and incomplete healing of the femoral osteotomies.
Table 2.
Biochemical Study of our Patient with GDD
| Age (months) | 8 | 12 | 14 | 18* | 21* | 25* | 31* | 36* | 50* | 62* | Nl: (RC Lab)* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Serum | |||||||||||
| Calcium | 10.3 | 9.2 (8.0 - 10.9 mg/dL) | 9.5 (8.9 - 9.9 mg/dL) | 9.2 (8.9 - 9.9 mg/dL) | 9.6 (8.9 - 9.9 mg/dL) | 9.7 | 9.1 | 9.3 (8.9 - 9.9 mg/dL) | 8.8 (9.0 - 10.1 mg/dL) | 9.2 (9.0 - mg/dL) 10.1 | 8.6-11 mg/dL |
| Calcium Ionized | 4.5 | 4.9 | 5.0 | 5.1 | N/A | 4.7 | N/A | (4.5-5.3 mg/dL) | |||
| Phosphorus | 4.2 | 4.2 (ref. rng N/A) | 4.2 (4.3-5.4 mg/dL) | 4.7 (4.3-5.4 mg/dL) | 4.6 (4.3-5.4 mg/dL) | 4.4 | 4.1 | 4.7 (4.3-5.4 mg/dL) | 4.9 (3.7-5.4 mg/dL) | 5.4 (3.7-5.4 mg/dL) | 3-6 mg/dL |
| %CK-BB | 2.5 | 2.5 | 0.6 | 4.9 | 6.7 | 2 | 7 | ||||
| Alkaline Phosphatase | 544 | 643 (101-394 U/L) | 650 (185-383 U/L) | 530 (185-383 U/L) | 606 (185-383 U/L) | 528 | 339 | 454 (172 - 405 U/L) | 445 (172 - 405 U/L) | 403 (172 - 405 U/L) | 110-320 U/L |
| Bone AlkPhos | 433.7 (26.0 - 259.0 U/L) | 512.5 (26.0 - 259.0 U/L) | 338.5 (26.0 - 259.0 U/L) | 454.9 (26.0 - 259.0 U/L) | 286.8 (0.0 - 208.0 U/L) | 270.7 (0.0 - 208.0 U/L) | 218.7 (0.0 - 208.0 U/L) | ||||
| PTH | 37 | 23 (10.0 - 69.0 pg/mL) | 41 (10.0 - 69.0 pg/mL) | 27 (10.0 - 69.0 pg/mL) | 26 (10.0 - 69.0 pg/mL) | 12 (10.0 - 69.0 pg/mL) | 39 (10.0 - 69.0 pg/mL) | 39 (15.0 65.0 pg/mL) - | 10-69 pg/mL | ||
| 25 OH vitamin D | 30 | 55.9 (19.1 - 57.6 ng/mL) | ND | 33.6 (30.0 - 100.0 ng/mL) | 26.5 (30.0 - 100.0 ng/mL) | 28.3 (30.0 - 100.0 ng/mL) | 25.9 (30.0 - 100.0 ng/mL) | 34.8 (30.0 - 100.0 ng/mL) | 30-100 ng/mL | ||
| Creatine Kinase | 56 (55 - 370 units/L) | 117 (ref. rng N/A) | 104 (ref. rng N/A) | 110 (25 - 172 units/L) | 96 - (31 152 units/L) | 192 (24 - 204 units/L) | 192 (24 - 204 units/L) | ||||
| Urine | |||||||||||
| Ca/Cre Ratio | 377 | 134 | 65 | 186 | 157 | 385 | 75-250 | ||||
| U Calcium | 109 | 32 | 41 | 153 | 36 | 289.1 | |||||
| Phos/Creat Ratio | 0.6 | 2.3 | 2.2 | 1.3 | 1.9 | 1.6 | 0.8-1.8 | ||||
| Hours Collected | 24 | 6 | 22.5 | 15 | 14 | 27 | |||||
| Bone Turnover Markers | |||||||||||
| Serum | |||||||||||
| TRAP 5b | 33.0 | 29.6 | 25.8 | 30.1 | 27.8 | Research Value | Research Value | 6.3-26.7 U/L | |||
| Urine | |||||||||||
| Deoxypyrid/Cr | 66.8 | 77.8 | 30.2 | 36.8 | 13.7-41.0 nmoles/mmoles |
During courses of PMD Therapy
Despite knowledgeable care from his parents, he suffered three fractures during the first year-of-life. At 5 weeks-of-age, he re-fractured his left femur. At 8 and 10 months-of-age, he fractured the left and right femoral shaft, respectively; both treated by closed reduction and casting. At age 8 months, a second skeletal survey showed persistent osteopenia and cystic enlargements of the maxilla and mandible. At that time, all thoracic and lumbar vertebral bodies were compressed. There were unhealed femoral mid-shaft fractures whereas tibial fractures had healed with residual bowing.
At birth his face appeared normal, but by 2 months-of-age progressive enlargement and expansion of the maxilla and mandible was noticed (Fig. 1B). Then, gingival hypertrophy caused malpositioned teeth and a thickened palate. Maxillofacial CT at age 8 months demonstrated large, expansile, and partially calcified masses involving his maxilla and mandible bilaterally (Fig. 3A&B). Cherubism, fibrous dysplasia (FD), juvenile Paget’s disease, and GDD were considered diagnostic possibilities. However, sequencing of SH3BP2 (GeneDx, Gaithersburg, MD) associated with cherubism, and TNFRSF11A and TNFRSF11B (Prevention Genetics, Marshfield, WI) associated with juvenile Paget’s disease, was negative.
Figure 3. CT Findings.
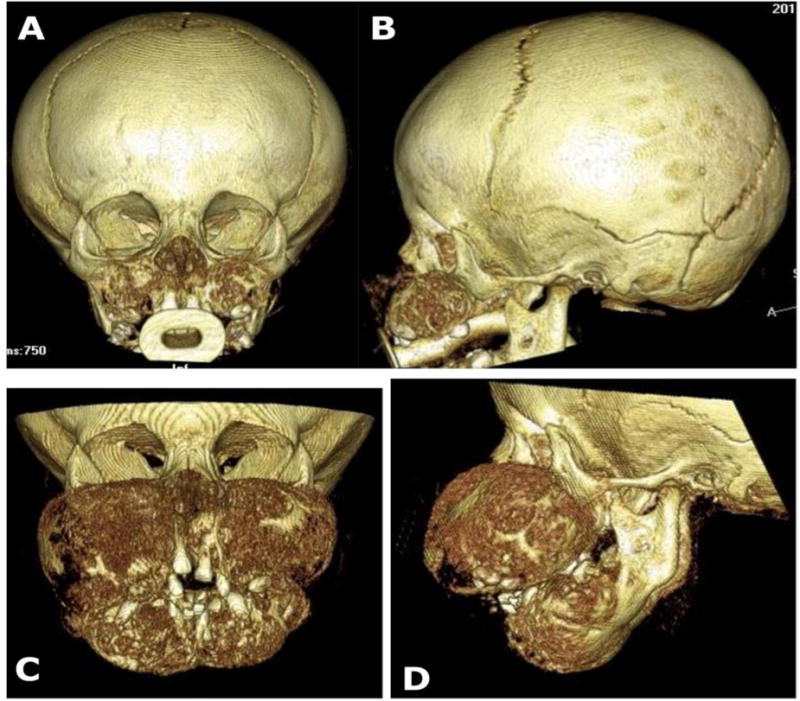
Three-dimensional reconstructions of computed tomography scans document the rapid growth of the patient’s maxilla and mandible. AP and lateral views are shown at age 8 months (A, B) and 13 months (C, D).
At 9 months-of-age, myringotomy tubes were placed for recurrent otitis media. His maxilla was biopsied showing histopathological findings consistent with a benign fibro-osseous lesion characterized by fibroblastic stroma with distinct loci of stromal condensation, but the psammomatoid bodies characteristic of GDD were absent(1). The histological findings were consistent with benign fibro-osseous lesions analogous to those observed in cemento-ossifying fibroma (see pathology below).
At almost 1 year-of-age, the patient was admitted to the Center for Metabolic Bone Disease and Molecular Research (Research Center) at Shriners Hospital for Children, St. Louis, Missouri, USA. He received his ad libitum calcium intake of 220 mg/day estimated from a 7-day food record (2011 US RDA 260 mg). Microcytic anemia was attributed to almost exclusive breast-feeding. Baseline biochemical assessment (serum and urine) of mineral homeostasis (Table 2) and radiographs were obtained in anticipation of bisphosphonate therapy. The spine radiographs showed osteopenia and reduced height of essentially all thoracic and lumbar vertebrae. Hand and knee radiographs revealed osteopenia with possible femoral modeling abnormalities and healed fractures.
At age 13 months, CT showed progressive expansion of the mandibular and maxillary tumors (Fig. 3C&D), which were then surgically debulked because they interfered with feeding and speech. At age 15 months, intravenous infusions of pamidronate (PMD) began (0.5 mg/kg as a single intravenous infusion). At 13 days prior to his first infusion, serum and urine biochemistries included BTMs (Table 2). Then, allowing for healing of multiple surgical procedures of his bones and jaw (see below), PMD was administered for a total of 7 doses at ages 15, 18, 21, 24, 31, 34, and 39 months (Fig. 4). He was re-evaluated at the Research Center at ages 18, 21, and 36 months and ages 4 and 5 years.
Figure 4. Clinical Timeline.

The patient’s fractures, debulking operations, and pamidronate infusions are depicted monthly. Severity of facial swelling: 0 = no swelling, 4 = severe swelling. Fractures: 1 = one site involvement, 2 = 2 sites simultaneously.
Because of rapid and continuous growth of the mandibular and maxillary masses (Fig. 1), two additional debulking procedures were performed at ages 16 and 23 months. Histopathological findings remained similar. Later, the growth of the masses stopped, allowing a surgical hiatus until debulking was required again at age 39 months (Fig. 4) with facial improvement (Fig. 1L). Five additional re-fractures included both femora, one left wrist buckle fracture, and buckle fractures of fingers.
At 29 months-of-age, significant femoral bowing (~50° bilaterally) led to femoral osteotomies with insertion of telescopic Fassier-Duval intramedullary nails (Pega Medical, Quebec, Canada) for stabilization (Fig. 2D).
At 39 months-of-age, a depressed skull fracture occurred without known head trauma. Because of progressive tibial bowing and recurrent fractures, he underwent bilateral segmental tibial osteotomies and telescopic intramedullary fixation using Fassier-Duval intramedullary nails at age 4 years, 9 months (Fig. 2F).
At age 5 years, significant osteopenia with incomplete but stable healing of the osteotomy sites persisted in the tibias and femurs. Radiographs also showed multiple horizontal sclerotic lines in metaphyses from his PMD treatment.
Motor delay was attributed to his bone disease and inability to perform weight-bearing activities. At age 2 years, he could crawl and started to pull up to stand. Language and fine motor skills were acquired on time. At 5 years-of-age, he was partially ambulatory, active, and consumed a soft diet with high-caloric nutritional supplementation. Subsequent imaging characterized his cemento-ossifying fibroma and its progression.
His radiographic findings up to age 10 months were reported by Herman et al. in 2014 (17). In 2017, Jin et al (10) confirmed our molecular findings including our identification of his unique, heterozygous, ANO5 missense mutation (see below).
B) Histopathology
Surgical resection of bone tissue with tumor from the maxilla and mandible at ages 9 and 16 months-of-age provided specimens that were fixed in 4% neutral buffered formalin overnight. The bone/tumor tissue was decalcified using 4% EDTA in phosphate buffer, pH 7. Teeth after fixation were acid decalcified using 10% hydrochloric acid. Specimens were then paraffin embedded, sectioned at 5μm thickness, and stained with hematoxylin and eosin. Jamshidi needle biopsy of his left iliac crest was performed during a debulking procedure at 16 months-of-age. This specimen was fixed in 70% ethanol, embedded in methylmethacrylate, and then stained with Goldner trichrome.
C) Biochemical Analysis
Fasting serum and timed urine collections acquired at the Research Center underwent biochemical profiling (Dade Behring Dimension Xpand instrument: Siemens Health Care Diagnostics, Inc., Los Angeles, CA, USA). Key parameters of bone and mineral homeostasis were compared also to our values obtained in 2006 and 2007 from 34 healthy children (18). To supplement our initial BTM data (Table 2), skeletal remodeling was further evaluated by measuring serum osteocalcin (OCN: Kit #LKON1; Siemens Health Care Diagnostics), bone-specific ALP (BAP) by ELISA (Quidel Comp., San Diego, CA, USA), and urine free deoxypyridinoline (DPD: Immulite 1000 Pyrilinks–D Kit; Siemens Medical Solutions Diagnostics Ltd., Lianberis, Gwynedd, UK). Also, to assess the impact of his PMD therapy on osteoclast (OC) numbers and perhaps integrity, serum levels of the brain isoenzyme of creatine kinase (CK-BB) (Kit #K20; Sebia, Norcross, GA, USA)(19) and tartrate-resistant acid phosphatase (TRAP-5b) (Kit #8033, Quidel; Los Angeles, CA, USA)(18) were assayed. Serum 25(OH) vitamin D levels were quantitated using Immunodiagnostic Systems (25-hydroxyvitamin D EIA; Gaithersburg, MD, USA), and then for 1.5 years to 4 years-of-age using LabCorp (Dublin, OH, USA).
D) Serum Multiplex Biomarker Profiling
Serum multiplex biomarker profiling (SMBP) was performed in 2013 in one comprehensive “batch” assay at Amgen, Inc. (Thousand Oaks, CA, USA) using our published methodology (20–22) and fasting serum acquired from the patient in 2011 and from 20 healthy children (6 boys and 14 girls) obtained in St. Louis, MO, USA in 2012 (Supporting Appendix).
E) Mutation Analyses
Initially, we searched for a somatic mutation at codon 201 in exon 8 of the GNAS1 gene (Center for Genetic Testing at Saint Francis, Tulsa, OK) to exclude fibrous dysplasia (FD) using DNA extracted from a resected mandibular lesion. Then, we ordered ANO5 targeted mutation testing using genomic DNA from the blood of the proband and his parents performed for the full coding exons of ANO5 as well as approximately 50 bases of flanking non-coding DNA (Prevention Genetics, Marshfield, WI).
Subsequently, using an evolutionary action score based on residue importance and substitution odds,(23) we evaluated the patient’s ANO5 missense mutation; its impact as well as a benign polymorphism, p.Leu322Phe (rs7481951). These items are assessed, respectively, with Evolutionary Trace (ET) ranks (24,25) and transition matrices computed over a large set of diverse proteins (PDB database: http://www.rcsb.org/pdb/). The outcome was normalized to become a fractional (percent) score with which every ANO5 missense mutation could be classified. Typically, mutations with action above 60 (higher impact than 60% of mutations) are deleterious, whereas those with action below 30 (lower impact than 70% of mutations) are neutral. We predicted the topology of ANO5 using the TMHMM Server v. 2.0 (26) which suggested that ANO5 protein contains eight transmembrane helices (26).
IV) Results
A) Histological examinations
Grossly, the resected expansive lesions in both the maxilla and mandible consisted of numerous white-pink bony tissue fragments and multiple teeth. After fixation, the tissue fragments were sectioned to show white-tan homogenous cut surfaces (Fig. 5A).
Figure 5. Patient’s Histopathological Findings.

I) Maxilla:
(A) Cut surface of maxillary lesions. Ruler at bottom with 1 mm marks.
(B) Low power image of H&E stained section of maxillary tumor showing a field of bland fibroblasts with intermixed calcified nodules, irregular in shape. Scale bar = 500 μm.
(C) Higher power image of tumor shows lack of cellular atypia or mitotic activity, and the lack of osteoblasts rimming the nodules. Scale bar = 100 μm.
(D) Polarized image of one nodule shows the typical quilted pattern of cementum. Scale bar = 50 μm.
(E) High power image demonstrates perpendicular insertion of collagen fibers into calcified nodule, indicated with white arrowheads. Scale bar = 50 μm.
(F) H&E stained section of tooth root (Rt) with adjacent tumor (Tu). Scale as in B. (G-I) Goldner trichrome stained sections of iliac crest biopsy, scale as in C.
II) Iliac Crest:
(G-I) Goldner trichrome stained sections of iliac crest biopsy, scale as in C.
(G) Tissue near growth plate (GP) shows poorly connected trabecular, numerous osteoclasts off of the bone surface (arrowheads), and focal marrow fibrosis (Fb).
(H) Fragment with only lamellar bone shows peritrabecular fibrosis (Fb), minimal osteoid (Os), and scattered osteoclasts off bone (arrowheads).
(I) Areas with woven bone have an irregular surface without cuboidal osteoblasts or osteoid, and the marrow is exclusively fibrotic (Fb).
The maxilla and the mandible samples demonstrated similar histopathological findings consistent with benign fibro-osseous lesions. Low-power microscopy showed sheets of benign fibroblasts and mature collagen fibers with interspersed bone and cementum-like basophilic, calcified structures, many acellular with a curvilinear configuration (Fig 5B). At higher power, mitotic figures were not seen, and calcified structures were not directly rimmed by cells (Fig. 5C). Polarized light revealed a woven or quilted pattern of cementum (Fig. 5D). The high power H&E stained image of a nodule showed a perpendicular insertion of collagen fibers resembling Sharpey's fibers of the periodontal membrane (Fig 5E). These morphologic findings were analogous to those observed in cemento-ossifying fibroma of the jaw (1).
The teeth appeared to be normally developed, with several showing carries. Sections of the teeth were unremarkable with no apparent developmental anomalies, but did show tumor adjacent to the roots (Fig. 5F).
The iliac crest specimen obtained during jaw surgery (after a single dose of PMD) was processed undecalcified and contained growth plate as well as fragments of cancellous bone with poor trabecular connectivity. Near the growth plate (Fig. 5G), trabeculae consisted of lamellar bone, but with a paucity of osteoid. At ~2 mm from the growth plate, there was fibrotic bone marrow and mostly woven rather than lamellar trabecular bone. Enlarged and/or rounded OCs, predominantly off of the bone surfaces, were abundant and consistent with exposure to PMD (27). Other areas with lamellar trabeculae showed peritrabecular fibrosis (Fig. 5H), and also little osteoid (Os) and more OCs off the bone surface. Additional fragments (Fig. 5I) consisted of woven bone, with irregular scalloped surfaces and few cuboidal osteoblasts (OBs). Marrow fibrosis in these areas was severe. Given the poor trabecular connectivity and history of multiple appendicular and axial fractures, these areas of woven bone may have represented reparative changes associated with fracture.
B) Biochemical Analyses
Biochemical parameters of mineral homeostasis prior to PMD treatment were unremarkable except for elevated serum ALP and BAP (Table 2). TRAP5b was elevated while osteocalcin was low-normal at 14 ng/mL (Nl: 10 – 64). Creatine kinase brain isoemyzme (CK-BB) was 2.5 % of the total level. The 24-hour urine collection revealed an increased Ca/creatinine ratio but low P/creatinine ratio (Table 2). Corrected creatinine clearance was normal at 115 ml/min/1.73 m2. Erythrocyte sedimentation rate was 60 mm/hr (Nl: 0 - 15 mm/hr). A basic metabolic panel was essentially normal.
C) Serum Multiplex Biomarker Profiling
SMBP used samples drawn before and shortly after onset of PMD therapy. Comparisons were made to the 20 pediatric control samples from the Research Center reflecting ages 6 – 19 years. The most striking abnormalities are shown in Fig. 6 and Supplementary Material. As could be expected, one dose of PMD decreased the BTMs. TRAP5b was elevated in the two patient samples, or near the upper limits of the normal cohort, suggesting increased OC numbers prior to treatment with PMD. Cathepsin K rose more than 5-fold between the time points in keeping with PMD effects (27). BAP and C1CP were higher at 11 months (above normal) but just above normal at 18 months (post initial PMD therapy), suggesting PMD decreased bone turnover. Both pre- and post-PMD sclerostin (SOST) levels were below the lowest value of the control cohort. Osteopontin (OPN) and prostaglandin E2 (PGE2) levels fell ~2.5 times across PMD treatment. Similarly, C2C (Type II collagen cleavage site) levels fell from the lower limit of normal to well below normal for the older controls. Adiponectin (ANC) was the most dramatically elevated analyte before PMD and fell almost 15 fold. ANC levels in the neonate (28) are usually 2 – 3 times normal adult levels, but no data are available for infants and toddlers.
Figure 6. Serum multiplex biomarker profiling.
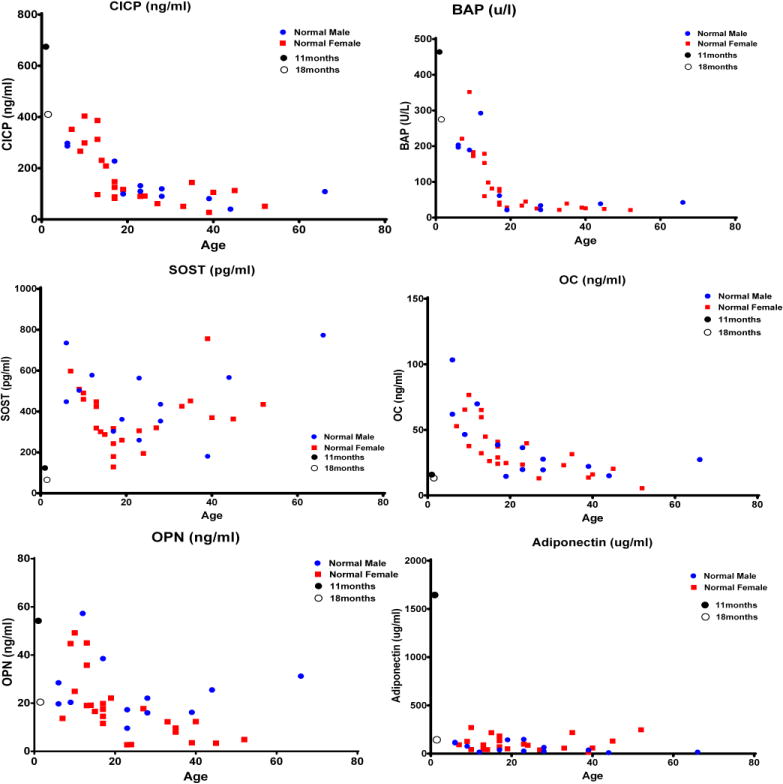
Profiles depict changes in bone turnover markers pre (●) and post (○) initiation of pamidronate treatment in the patient as compared to controls. CICPI- C-terminal propeptide of collagen type I; BAP-bone alkaline phosphatase; SOST-sclerostin; OC-osteocalcin; OPN- osteopontin.
D) Molecular analysis
The lesion resected from the proband’s mandible was negative for the somatic mutation at codon 201 in GNAS1 that causes FD. Sequencing of the patient’s genomic DNA for ANO5 revealed a unique heterozygous missense variant in exon 15 (c.1553G>A) predicted to substitute glycine at position 518 with glutamine (p.Gly518Glu) (Fig. 7). Testing of both parents for this mutation was negative, indicating a de novo event. The ANO5 sequencing also revealed a known polymorphism designated as p.Leu322Phe (rs7481951). The polymorphism p.Leu322Phe and the mutation p.Gly518Glu are predicted to occur within the first and fourth transmembrane helices of the ANO5 protein, respectively (Fig. 7). The p.Leu322Phe polymorphism is predicted to be neutral, with an evolutionary action of 17%, in contrast to the p.Gly518Glu mutation predicted to be deleterious with an evolutionary action of 72% (Fig. 7).
Figure 7. Molecular Data.
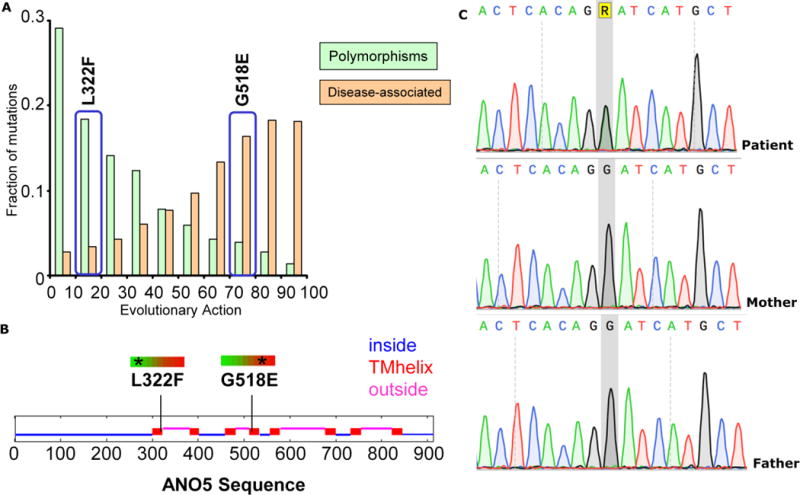
(A) The p.L322F polymorphism is predicted to be neutral with an evolutionary action of 17%, whereas the p.G518E mutation is predicted to be deleterious with an evolutionary action of 72%. The bars represent a reference set of variants found in 218 genes, annotated as polymorphisms or associated to disease in the UniProt database (http://www.uniprot.org/), and analyzed previously (21).
(B) The p.L322F and p.G518E genetic variants of ANO5 are predicted to involve the first and fourth transmembrane helices of the encoded protein.
(C) Electropherogram of the pathogenic variant, c.1553 G > A(R in the figure);p.Gly518Glu, identified in the patient as compared to electropherograms showing the wild type allele in both parents.
V) Discussion
Our patient with GDD appears to be the first documented with prenatal onset. The complications from GDD, age of onset and severity were variable (1–3), but he exhibited an especially severe and atypical postnatal clinical course compared to the ~ 80 reported cases (1–3,6–10) (Table 1). It was the possibility of GDD suggested by our patient’s facial swelling that prompted us to order targeted sequencing of ANO5, typically offered in diagnostic laboratories for AR ANO5-related neuromuscular disorders (29,30).
Diagnosis of GDD can be challenging due to the disorder’s rarity, unusual and severe clinical presentation, rapid progression of its skeletal manifestations, and necessary extensive investigation and complex management involving multidisciplinary teams. Recurrent fractures can suggest OI but in contrast to OI GDD has normal stature, sclerae, and hearing. Progressive overgrowth of our patient’s maxilla and mandible and gingival hypertrophy provided a major clue for diagnosis. Initially, these findings suggested cherubism,(31) but SH3BP2 testing was negative, and the histopathology of the jaw lesions was negative for giant cells (32). In fact, cherubism typically presents later; i.e., between 2-4 years of age, and importantly is not associated with long bone complications (31,33). In FD and McCune-Albright syndrome, skeletal lesions tend to be structurally weak and prone to pathological fractures. FD was considered for our patient, but the radiolucent ground glass pattern characteristics of FD was not present in his radiographs and his molecular testing was negative. FD lesions tend to grow slowly and rarely manifest rapid enlargement and compression of surrounding structures (34). Malignant degeneration occurs in < 1% of FD patients (35). The histopathology of our patient’s excised tumors was inconsistent with FD and instead showed fibro-osseous lesions with interspersed cementum-like calcified structures typically seen in both cemento-ossifying fibroma (36) and in GDD (3,11). Structures referred to as psammomatoid bodies, that are variably observed in GDD, are indistinguishable from the small cementum like spheres seen in our patient. His jaw lesions are similar to the ones described by Jin et al.(10) as “cementum-rich florid osseous dysplasia.” They were also remarkably similar to gigantiform cementoma (OMIM # 137575), as documented in 2016 by Andreeva et al (9) and in the recent 4th edition of WHO classification of head and neck tumors (37).
To date, only 7 missense ANO5 mutations (from 14 families) have been reported in GDD (3,5,7–10,38). Three [p.Cys356Arg (c.1067T>A), p.Cys356Gly (C.1067T>G), and p.Cys356Tyr (C.1067G>A)] are in exon 11 and involve the identical codon for amino acid cysteine at position 356. An additional exon 11 mutation, p.Cys360Tyr, was recently reported. (10) The fifth genetic variant, (c.1538C>T), identified in a large Italian family with GDD, is in exon 15 and substitutes threonine at position 513 with isoleucine (p.Thr513Ile) (3). Jin et al. (10) reported the only exon 7 mutation, p.Arg215Gly, associated with GDD. One report in 2017 describes a 13-year-old boy with GDD harboring a novel mutation in exon 15: c.1499C>T (p.Ser500Phe) (38).
Our patient’s unique heterozygous p.Gly518Glu missense mutation is also located in exon 15 of ANO5. Glycine at position 518 is evolutionarily conserved among human, chimp, cow, and mouse, thus emphasizing its biological significance. His p.Gly518Glu variant has not been observed in 66,672 alleles from European controls in the ExAC database (last accessed 11/14/2017). Interestingly, the chemically related amino acid alanine occurs at this site in opossum, chicken, lizard, frog, and zebrafish and is reported in 5/66,672 alleles from European controls in the ExAc database (http://exac.broadinstitute.org/gene/ENSG00000171714). In silico analysis, predicted this mutation to be deleterious with an evolutionary action of 72% (Fig. 6A).
ANO5 has 22 exons and encodes a protein with eight transmembrane domains (13,15). Its function is not known (14). In contrast, ANO1 and ANO2 encode Ca2+-activated Cl− channels (14), whereas other anoctamins do not conduct Cl− and have roles in phospholipid scrambling (15). ANO5 probably locates intracellularly (39), and has not been found to exhibit either of these activities, suggesting different properties and function (3,39,40). In mice, ANO5 is expressed in the calvarium, femur, and mandible, but in humans it is expressed in the brain, heart, kidney, lung, and skeletal muscle (5). However, there are no data concerning the temporal expression of ANO5 during embryonic life or childhood, perhaps the critical time for its action on skeletal tissue. The protein products of wild type and mutant ANO5 are rapidly degraded via the proteasome pathway, but rescued by inhibition of the PI3K pathway and by the chemical chaperone, sodium butyrate (40).
Our patient with GDD is the first treated with a bisphosphonate. Pre-treatment BTMs revealed elevated serum ALP, BAP, and TRAP 5b, suggesting accelerated bone remodeling. Unfortunately, no apparent improvement followed in the frequency or severity of his fractures, although DEXA spine (L1-L4) BMD Z-score was −2.4 at age 3 years and −1.7 at age 5 years. Importantly, debulking intervals seemed extended (Fig. 4). Perhaps the markedly increased serum cathepsin K level post treatment possibly reflected increased production. The low SOST levels suggested improved to no pathogenetic role for this inhibitor of bone formation in our patient’s osteopenia and bone disease. Prior to PMD treatment, his circulating OPN, PGE2, and ANC levels were relatively high compared to our healthy controls. OPN and PGE2 may have had a role in inhibiting mineralization and accelerating bone remodeling prior to PMD therapy. However, the most remarkable SMBP finding was ANC elevated 8 times the upper limit for our pediatric controls that then fell with PMD therapy to within the normal range. Bone marrow adipose tissue is a significant source of circulating ANC, which has varied inversely with BMD in several studies (41). However, ANC’s role in bone mineral metabolism remains unknown. The markedly elevated ANC level before our patient received PMD suggests ANO5 might modulate its production or degradation.
Bone histology indicated a clear effect of PMD on our patient’s OCs. Of note, the early and rapid progression of his facial tumor slowed significantly as he aged, and the interval between debulking procedures progressively increased. Perhaps the improved course for our patient was because the estimated 85% of growth of the craniofacial skeleton is complete by age 3 years (42). Alternatively, the PMD infusions and/or the debulking operations could have explained this improvement. The surgery could have helped by reducing the numbers of progenitor cells in the periodontal membrane thought to be the source of the cemento-ossifying fibroma in GDD (36). Jaw tumor growth in GDD can slow after multiple debulking procedures (6,9), although not well documented. The PMD could have helped by generating a solid ossification rim surrounding the tumor tissue, preventing or slowing its continuous expansion.
In summary, we have detailed the 5-year life-long history of a boy with an atypical presentation and especially severe course of GDD. We discovered that he carries a novel heterozygous missense mutation in ANO5. Growth of his jaw tumor, microscopically analogous to cemento-ossifying fibroma and gigantiform cementoma, slowed with aging but whether PMD treatment and/or tumor debulking explained this remains uncertain. ANO5 function and the mechanism by which our patient’s ANO5 mutation compromised his skeleton remain unknown.
Supplementary Material
Highlights.
Heterozygous mutations of ANO5 cause gnathodiaphyseal dysplasia (GDD)
GDD features fibro-osseous lesions in jawbones and fragility and sclerosis of tubular bones
ANO5 novel de novo mutation p.Gly518Glu causes especially severe atypical GDD
Multiple debulking procedures can control GDD fibro-osseous lesions
Bisphosphonate treatment for GDD requires further assessment
Acknowledgments
We thank the patient’s family for participating in our study. This report was made possible by the skill and dedication of the nurses, radiology technologists, and physical therapists at the Center for Metabolic Bone Disease and Molecular Research, Shriners Hospital for Children, St. Louis, MO, USA.
Supported by Shriners Hospitals for Children, The Clark and Mildred Cox Inherited Metabolic Bone Disease Research Fund at The Barnes-Jewish Hospital Foundation, and the United States Agency for International Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riminucci M, Collins MT, Corsi A, Boyde A, Murphey MD, Wientroub S, Kuznetsov SA, Cherman N, Robey PG, Bianco P. Gnathodiaphyseal dysplasia: a syndrome of fibro-osseous lesions of jawbones, bone fragility, and long bone bowing. J Bone Miner Res. 2001;16:1710–1718. doi: 10.1359/jbmr.2001.16.9.1710. [DOI] [PubMed] [Google Scholar]
- 2.Ahluwalia J, Ly JQ, Norman E, Costello RF, JR, Beall DP. Gnathodiaphyseal dysplasia. Clin Imaging. 2007;31:67–69. doi: 10.1016/j.clinimag.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Marconi C, BrunamontiBinello P, Badiali G, Caci E, Cusano R, Garibaldi J, Pippucci T, Merlini A, Marchetti C, Rhoden KJ, Galietta LJ, Lalatta F, Balbi P, Seri M. A novel missense mutation in ANO5/TMEM16E is causative for gnathodiaphysealdyplasia in a large Italian pedigree. Eur J Hum Genet. 2013;21:613–619. doi: 10.1038/ejhg.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akasaka Y, Nakajima T, Koyama K, Furuya K, Mitsuka Y. Familial cases of a new systemic bone disease, hereditary gnatho-diaphyseal sclerosis. Nippon SeikeigekaGakkaiZasshi. 1969;43:381–394. [PubMed] [Google Scholar]
- 5.Tsutsumi S, Kamata N, Vokes TJ, Maruoka Y, Nakakuki K, Enomoto S, Omura K, Amagasa T, Nagayama M, Saito-Ohara F, Inazawa J, Moritani M, Yamaoka T, Inoue H, Itakura M. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD) Am J Hum Genet. 2004;74:1255–1261. doi: 10.1086/421527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roginsky VV, Ivanov AL, Khonsari RH. Recurring gnathodiaphyseal dysplasia in two Russian brothers. Int J Oral Maxillofac Surg. 2010 Apr;39(4):397–401. doi: 10.1016/j.ijom.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 7.Vengoechea J, Carpenter L. Gnathodiaphyseal dysplasia presenting as polyostotic fibrous dysplasia. Am J Med Genet A. 2015 Jun;167(6):1421–2. doi: 10.1002/ajmg.a.36986. [DOI] [PubMed] [Google Scholar]
- 8.Duong HA, Le KT, Soulema AL, Yueh RH, Scheuner MT, Holick MF, Christensen R, Tajima TL, Leung AM, Mallya SM. Gnathodiaphyseal dysplasia: report of a family with a novel mutation of the ANO5 gene. Oral Surg Oral Med Oral Pathol Oral Radiol. 2016 May;121(5):e123–8. doi: 10.1016/j.oooo.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andreeva TV, Tyazhelova TV, Rykalina VN, Gusev FE, Goltsov AY, Zolotareva OI, Aliseichik MP, Borodina TA, Grigorenko AP, Reshetov DA, Ginter EK, Amelina SS, Zinchenko RA, Rogaev EI. Whole exome sequencing links dental tumor to an autosomal-dominant mutation in ANO5 gene associated with gnathodiaphyseal dysplasia and muscle dystrophies. Sci Rep. 2016 May 24;6:26440. doi: 10.1038/srep26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, Liu Y, Sun F, Collings MT, Blackwell K, Woo AS, Reichenberger EJ, Ying Hu. Three novel ANO5 missense mutations in Caucasian and Chinese families and sporadic cases with gnathodiaphyseal dysplasia. Scientific Reports. 2017;7:40935. doi: 10.1038/srep40935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsutsumi S, Kamata N, Maruoka Y, Ando M, Tezuka O, Enomoto S, Omura K, Nagayama M, Kudo E, Moritani M, Yamaoka T, Itakura M. Autosomal dominant gnathodiaphyseal dysplasia maps to chromosome 11p14.3-15.1. J Bone Miner Res. 2003;18:413–418. doi: 10.1359/jbmr.2003.18.3.413. [DOI] [PubMed] [Google Scholar]
- 12.Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16familymembers are Ca2+-activatedCl− channels. J Physiol. 2009 May 15;587(Pt 10):2127–39. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuta K, Tsutsumi S, Inoue H, Sakamoto Y, Miyatake K, Miyawaki K, Noji S, Kamata N, Itakura M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia gene GDD1. BiochemBiophys Res Commun. 2007;357:126–132. doi: 10.1016/j.bbrc.2007.03.108. [DOI] [PubMed] [Google Scholar]
- 14.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 15.Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins) Physiol Rev. 2014 Apr;94(2):419–59. doi: 10.1152/physrev.00039.2011. [DOI] [PubMed] [Google Scholar]
- 16.van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PG, van Rijn RR, Pals G. Osteogenesis Imperfecta: A Review with Clinical Examples. Mol Syndromol. 2011 Dec;2(1):1–20. doi: 10.1159/000332228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herman TE, Siegel MJ, Sargar K. Gnathodiaphyseal dysplasia. J Perinatol. 2014;34:412–414. doi: 10.1038/jp.2013.178. [DOI] [PubMed] [Google Scholar]
- 18.Whyte MP, Kempa LG, McAlister WH, Zhang F, Mumm S, Wenkert D. Elevated serum lactate dehydrogenase isoenzymes and aspartate transaminase distinguish Albers-Schonberg disease (chloride channel 7 deficiency osteopetrosis) among the sclerosing bone disorders. J Bone Miner Res. 2010 Nov;25(11):2515–26. doi: 10.1002/jbmr.130. [DOI] [PubMed] [Google Scholar]
- 19.Whyte MP, Chines A, Silva DP, Jr, Landt Y, Ladenson JH. Creatine kinase brain isoenzyme (BB-CK) presence in serum distinguishes osteopetroses among the sclerosing bone disorders. J Bone Miner Res. 1996 Oct;11(10):1438–43. doi: 10.1002/jbmr.5650111010. [DOI] [PubMed] [Google Scholar]
- 20.Whyte MP, Wenkert D, Dwyer DC, Lacey DL, Stolina M. Systemic biomarker profiling of metabolic and dysplastic skeletal diseases using multiplex serum protein analyses [Abstract] J Bone Miner Res. 2010;25(Suppl 1):S137. [Google Scholar]
- 21.Whyte MP, Madson KL, Mumm S, McAlister WH, Novack DV, Blair JC, Helliwell TR, Stolina M, Abernethy LJ, Shaw NJ. Rapid skeletal turnover in a radiographic mimic of osteopetrosis. J Bone Miner Res. 2014;29:2601–9. doi: 10.1002/jbmr.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guanabens N, Mumm S, Gifre L, Ruiz-Gaspa S, Demertzis JL, Stolina M, Novack DV, Whyte MP. Idiopathic acquired osteosclerosis in a middle-aged woman with systemic lupus erythematosus. J Bone Miner Res. 2016;31:1774–1782. doi: 10.1002/jbmr.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsonis P, Lichtarge O. A formal perturbation equation between genotype and phenotype determines the evolutionary actin of protein-coding variations on fitness. Genome Res. 2014;24:2050–2058. doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lichtarge O, Sowa ME. Evolutionary predictions of binding surfaces and interactions. Curr Opin Struct Biol. 2002;12:21–27. doi: 10.1016/S0959-440X(02)00284-1. [DOI] [PubMed] [Google Scholar]
- 25.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 26.Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 27.Whyte MP, Wenkert D, Clements KL, McAlister WH, Mumm S. Bisphosphonate-induced osteopetrosis. N Engl J Med. 2003;349:455–61. doi: 10.1056/NEJMoa023110. [DOI] [PubMed] [Google Scholar]
- 28.Scheller EL, Burr AA, MacDougald OA, Cawthorn WP. Inside out: Bone marrow adipose tissue as a source of circulating adiponectin. Adipocyte. 2016;5(3):251–69. doi: 10.1080/21623945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calcium activated chloride channel anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86:213–221. doi: 10.1016/j.ajhg.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahjneh I, Jaiswal J, Lamminen A, et al. A new distal myopathy with mutation in anoctamin 5. NeuromusculDisord. 2010;20:791–795. doi: 10.1016/j.nmd.2010.07.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadaki ME, Lietman SA, Levine MA, Olsen BR, Kaban LB, Reichenberger EJ. Cherubism: best clinical practice. Orphanet journal of rare diseases. 2012;7:1. doi: 10.1186/1750-1172-7-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueki Y, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28(2):125–6. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 33.Von Wowern N. Cherubism: a 36-year long-term follow-up of 2 generations in different families and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(6):765–72. doi: 10.1067/moe.2000.108438. [DOI] [PubMed] [Google Scholar]
- 34.Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7:1. doi: 10.1186/1750-1172-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cholakova R, Kanasirska P, Kanasirski N, ChenchevIv DA. Fibrous dysplasia in the maxillomandibular region–Case report. J IMAB. 2010;16:10–13. doi: 10.5272/jimab.1642010_10-13. [DOI] [Google Scholar]
- 36.Speight PM, Carlos R. Maxillofacial fibro-osseous lesions. Current Diagnostic Pathology. 2006;12:1–10. doi: 10.1016/j.cdip.2005.10.002. [DOI] [Google Scholar]
- 37.El-Mofty SK. Fibro-osseous and osteochonderomatous lesions Familial Gigantiform Cementoma. In: El Naggar AK, Chan KC, Grandis JR, Takata T, Slootweg PJ, editors. WHO Classification of head and neck tumors. 4th. Lyon: IARC Press; 2017. p. 253. [Google Scholar]
- 38.Rolvien T, Koehne T, Kornak U, Lehmann W, Amling M, Schinke T, Oheim R. A novel ANO5 mutation causing gnathodiaphyseal dysplasia with high bone turnover osteosclerosis. J Bone Miner Res. 2017;32:277–284. doi: 10.1002/jbmr.2980. [DOI] [PubMed] [Google Scholar]
- 39.Duran C, Qu Z, Osunkoya AO, Cui Y, Hartzell HC. ANOs 3-7 in the Anoctamin/TMEM16Cl- channel family are intracellular proteins. Am J Physiol. 2012;302:482–493. doi: 10.1152/ajpcell.00140.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran TT, Tobiume K, Hirono C, Fujimoto S, Mizuta K, Kubozono K, Inoue H, Itakura M, Sugita M, Kamata N. TMEM16E (GDD1) exhibits protein instability and distinct characteristics in chloride channel/pore forming ability. J Cell Physiol. 2014;229:181–190. doi: 10.1002/jcp.24431. [DOI] [PubMed] [Google Scholar]
- 41.Johansson H, Oden A, Lerner UH, Jutberger H, Lorentzon M, Barrett-Connor E, Karlsson M, Ljunggren O, Smith U, McCloskey E, Kanis J, Ohlsson C, Mellstrom D. High serum adiponectin predicts incident fractures in elderly men: osteoporotic fractures in men (MrOS) Sweden. J Bone Miner Res. 2012;27:1390–1396. doi: 10.1002/jbmr.1591. [DOI] [PubMed] [Google Scholar]
- 42.Blinkov SM, Glezer II. The Human Brain in Figures and Tables. New York: Plenum Press; 1968. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.