

Abstract
Based on complete bacterial genome sequence data, we demonstrate a correlation between bacterial chromosome length and the G+C content of the genome, with longer genomes having higher G+C contents. The correlation value decreases at shorter genome sizes, where there is a wider spread of G+C values. However, although significant (P<0.001), the correlation value (Pearson R=0.58) suggests that other factors also have a significant influence. A similar pattern was seen for plasmids; longer plasmids had higher G+C values, although the large number of shorter plasmids had a wide spread of G+C values. There was also a significant (P<0.0001) correlation between the G+C content of plasmids and the G+C content of their bacterial host. Conversely, the G+C content of bacteriophages tended to reduce with larger genome sizes, and although there was a correlation between host genome G+C content and that of the bacteriophage, it was not as strong as that seen between plasmids and their hosts.
Keywords: genome G+C content, genome length, bacteria, plasmids
Data Summary
Jupyter notebooks for the analysis of the data can be found at: https://github.com/atolgrp/Microbial-G-C-Content.
Impact Statement.
Larger genomes provide an opportunity for containing more genes due to the larger amount of DNA. However, the reasons associated with this are still debated and relatively unclear. Using genome sequences accessible from public databases, this paper examines the potential relationship between G+C content and genome length. In addition to studying bacterial genomes, the work also looks at this relationship between G+C content and genome length for both plasmids and bacteriophages. We also compare the G+C content of both plasmid and bacteriophage genomes relative to the G+C content of the organism from which they were isolated. In general, we found that larger bacterial genomes tend to have higher G+C contents, as was the case for plasmids. However, in bacteriophages, the G+C content appeared to reduce with an increase in size. There was a high level of correlation between the G+C content of plasmids and their host organism, a pattern that was seen to some extent between bacteriophages and the organisms they infect, but with a lower correlation level.
Introduction
The redundancy of the genetic code, where as many as six different codons may encode a single amino acid, allows at least some tolerance of the nucleotides used by different organisms. This tolerance, at least in part, means that the bacterial genomic guanine+cytosine (G+C) content may vary enormously, depending on the species. Recently, this range was shown to extend from 17 to 75 mol% [1]. The factors influencing this variation have been debated for at least 50 years [2, 3], including the suggestion that mutational bias acts upon genomes. This bias, together with environmental factors, was thought to exert a selection pressure towards the most adapted genome composition for a given habitat. Subsequent research suggested that this mutational bias generally acts across all bacterial species and promotes a trend towards genomes with higher adenine+thymine (A+T) content [4–6]. Other research revealed that the G+C content of individual bacterial species is correlated to a number of factors. These factors are not mutually exclusive and have included variables such as the organism’s living environment [7], the ability or inability to fix atmospheric nitrogen [8], an organism’s preference for aerobic or anaerobic conditions [8, 9], and normal optimal temperature range [10, 11]. The interconnection of these intrinsic and extrinsic factors means that no single condition is likely to be responsible for the G+C content of an organism, but rather this is due to multiple factors, which in turn makes identification of the relationships between them difficult to analyse.
Various approaches have been adopted to analyse the factors that might influence the G+C content, including traditional (laboratory-based) microbiology and in silico analyses using phylogenetic studies in an attempt to identify similarities between organisms with particular G+C contents. One of the simplest hypothesized relationships was that of a potential correlation between the genomic G+C content of an organism and genome size. This was first proposed by Sueoka [3] and has been studied further by others since (e.g. [12–15]). Initial investigations relying on examining the genome size posed problems due to shearing of DNA during the extraction process, thereby potentially leading to under-estimations of the correct size. Even with the advent of pulsed-field gel electrophoresis [16], which greatly overcame the potential problem of DNA fragmentation, this issue was not fully resolved. However, with the improvements to DNA sequencing methods, particularly the increased use of next-generation sequencing to determine complete genome sequences, accurate values for both genome size and G+C content are becoming increasingly available.
The present study makes use of data from genome sequences and is, to our knowledge, the largest investigation undertaken to date to assess the potential relationship between genome size and G+C content. Furthermore, it also includes plasmids in the analysis and compares their G+C content to that of their host organism.
Methods
Data were downloaded from the National Center for Biotechnology Information (NCBI) database, on 12 June 2017. For that purpose, Linux shell commands were used (awk for address parsing and wget for downloading), wrapped in a python script. At the time of downloading, the database contained 14 774 genome entries. The downloaded dataset included a number of draft and incomplete sequences. Only entries containing the text string ‘complete’ in their Fasta definition line (defline) were selected. The same criterion was applied for the separation of plasmids and phages, namely the existence of the text strings ‘plasmid’ and ‘phage’. The rest were assigned as bacterial genomic sequences. The majority of bacteriophage genomes were downloaded from a separate directory in NCBI, but some sequences were also included in the main dataset for microbial genomes. These two datasets were merged after cleaning and any duplicates were removed computationally. Further entries described in their defline as ‘putative’ or ‘endosymbiont’ were also removed. This subset was comparatively small and lacked clear annotation.
All data manipulation and statistical analysis was performed using python 2.7 (implemented in anaconda 2, v4.4.0) (Python Software Foundation, https://www.python.org), in a Linux 64-bit environment. Standard python libraries were used for data cleaning and subsequent analysis, such as pandas, scipy and numpy.
Ordinary least squares (OLS) was applied for linear regression, using python with statsmodels.OLS. This method still provides an unbiased regression estimation in the presence of unequal variance across the data (heteroskedasticity) [17], as the latter were evident across all datasets. One drawback, however, is that when heteroskedasticity is present, OLS has no predictive power, as the error margins and P values can be too small or too large, and cannot be trusted. To mitigate this effect, OLS was used with the HCCM (heteroskedasticity consistent covariance matrix) method [17], which in python statsmodels is implemented with the cov_type=‘HC0’ option.
Plots were produced using matplotlib (v2.0.1) [18] and seaborn (v0.7.1) (M. Waskom, O. Botvinnik, D. O'Kane, P. Hobson, D. C. Gemperline et al., 2016). To enable researchers to easily re-apply our analysis protocols, we have made all code used to generate plots and tables available as jupyter notebooks at https://github.com/atolgrp/Microbial-G-C-Content.
Results
After cleaning, the dataset comprised 12 424 complete genome sequences from bacterial sources; 6671 from bacterial chromosomes, 5744 from plasmids and 4580 from phages. Inevitably, extensively studied microbial species, such as Escherichia coli or Bacillus spp., were represented by more than one strain.
The G+C content ranged from 13.5 mol% (Zinderia insecticola CARI) to 87.5 mol% (Streptomyces autolyticus strain CGMCC0516 plasmid), with a mean value of 48.4 mol%. In distributions with heavy skew, the median is a better estimate of a representative value. For the whole dataset, this was slightly higher than the mean, at 48.5 mol%. Lengths varied from 744 bp (Tremplaya phenacola PAVE plasmid) to 16 Mb (Minicystis rosea strain DSM 24000). Mean and median lengths were 2.08 and 1.64 Mb, respectively.
Bacterial genomes
Bacterial genomic sequence length ranged from 112 kb (Nasuia deltocephalinicola strain PUNC) to 16 Mb (M. rosea strain DSM 24000), with a mean length of 3.66 Mb and a median of 3.78 Mb (Table 1). The lowest G+C content was that of Z. insecticola CARI at 13.5 mol% and the highest that of Anaeromyxobacter dehalogenans 2CP-C, at 74.9 mol% (Table 2). The mean G+C content was 48.8 mol% and the median was 49.3 mol%.
Table 1. Microbes, plasmids and phages with extreme values of length.
The five longest and shortest values are shown in each case. G+C values have been rounded to one decimal place.
| Genome | Length (bp) | G+C (mol%) | |
|---|---|---|---|
| Bacterial genomes | |||
| Longest bacterial genomes | |||
| 1 | Minicystis rosea strain DSM 24000 (CP016211.1) | 16 040 666 | 69.1 |
| 2 | Sorangium cellulosum So0157-2 (CP003969.1) | 14 782 125 | 72.1 |
| 3 | Nonomuraea sp. ATCC 55076 (CP017717.1) | 13 047 416 | 71.8 |
| 4 | Sorangium cellulosum ‘So ce 56' (AM746676.1) | 13 033 779 | 71.4 |
| 5 | Archangium gephyra strain DSM 2261 (CP011509.1) | 12 489 432 | 69.4 |
| Shortest bacterial genomes | |||
| 1 | Candidatus Nasuia deltocephalinicola strain PUNC (CP013211.1) | 112 031 | 16.6 |
| 2 | Candidatus Nasuia deltocephalinicola str. NAS-ALF (CP006059.1) | 112 091 | 17.1 |
| 3 | Candidatus Hodgkinia cicadicola isolate TETUND1 (CP007232.1) | 133 698 | 46.8 |
| 4 | Candidatus Tremblaya princeps PCIT (CP002244.1) | 138 927 | 58.8 |
| 5 | Candidatus Tremblaya princeps PCVAL (CP002918.1) | 138 931 | 58.8 |
| Plasmid genomes | |||
| Longest plasmids | |||
| 1 | Cupriavidus metallidurans CH34 megaplasmid (CP000353.2) | 2 580 084 | 63.6 |
| 2 | Burkholderia caribensis MBA4 plasmid (CP012748.1) | 2 555 069 | 62.4 |
| 3 | Rhizobium gallicum bv. gallicum R602 plasmid pRgalR602c (CP006880.1) | 2 466 951 | 59.4 |
| 4 | Sinorhizobium fredii NGR234 plasmid pNGR234b (CP000874.1) | 2 430 033 | 62.3 |
| 5 | Rhizobium gallicum strain IE4872 plasmid pRgalIE4872d (CP017105.1) | 2 388 366 | 59.2 |
| Shortest plasmids | |||
| 1 | Candidatus Tremblaya phenacola PAVE plasmid (CP003983.1) | 744 | 42.2 |
| 2 | Lactococcus lactis subsp. lactis KLDS 4.0325 plasmid 2 (CP007042.1) | 870 | 32.6 |
| 3 | Enterococcus faecium strain ISMMS_VRE_1 plasmid ISMMS_VRE_p5 (CP012433.1) | 886 | 31.3 |
| 4 | Borreliella garinii strain CIP 103362 plasmid cp32 (CP018755.1) | 1 085 | 30.4 |
| 5 | Acinetobacter baumannii strain JBA13 plasmid pJBA13_2 (CP020583.1) | 1 109 | 59.1 |
| Phage genomes | |||
| Longest phages | |||
| 1 | Agrobacterium phage Atu_ph07 (MF403008.1) | 490 380 | 37.1 |
| 2 | Salicola phage SCTP-2 (MF360958.1) | 440 001 | 30.0 |
| 3 | Pectobacterium phage CBB (KU574722.1) | 378 379 | 35.9 |
| 4 | Aureococcus anophagefferens phage BtV-01 (NC_024697.1) | 370 920 | 28.7 |
| 5 | Cronobacter phage vB_CsaM_GAP32 (JN882285.1) | 358 663 | 35.6 |
| Shortest phages | |||
| 1 | Leuconostoc phage L5 (L06183.1) | 2 435 | 33.3 |
| 2 | Enterobacteria phage M (JX625144.1) | 3 405 | 48.0 |
| 3 | Enterobacterio phage KU1 (AF227250.1) | 3 486 | 46.5 |
| 4 | Enterobacteria phage C-1 INW-2012 (JX045649.1) | 3 523 | 48.4 |
| 5 | Enterobacterio phage MS2 isolate DL52 (JQ966307.1) | 3 525 | 51.0 |
Table 2. Microbes, plasmids and phages with extreme values of G+C content.
Only the five highest and lowest values are shown in each case. G+C values have been rounded to one decimal place.
| Genome | Length (bp) | G+C (mol%) | |
|---|---|---|---|
| Bacterial genomes | |||
| Organisms with highest bacterial genome G+C content | |||
| 1 | Anaeromyxobacter dehalogenans 2CP-C (CP000251.1) | 5 013 479 | 74.9 |
| 2 | Anaeromyxobacter sp. K (NC_011145.1) | 5 061 632 | 74.8 |
| 3 | Streptomyces rubrolavendulae strain MJM4426 (CP017316.1) | 6 543 262 | 74.8 |
| 4 | Corynebacterium sphenisci DSM 44792 (NZ_CP009248.1) | 2 594 799 | 74.7 |
| 5 | Cellulomonas fimi ATCC 484 (NC_015514.1) | 4 266 344 | 74.7 |
| Organisms with lowest bacterial genome G+C content | |||
| 1 | Candidatus Zinderia insecticola CARI (CP002161.1) | 208 564 | 13.5 |
| 2 | Candidatus Carsonella ruddii CE isolate Thao2000 (CP003541.1) | 162 589 | 14.0 |
| 3 | Candidatus Carsonella ruddii HC isolate Thao2000 (CP003543.1) | 166 163 | 14.2 |
| 4 | Candidatus Carsonella ruddii CS isolate Thao2000 (CP003542.1) | 162 504 | 14.2 |
| 5 | Candidatus Carsonella ruddii HT isolate Thao2000 (CP003544.1) | 157 543 | 14.6 |
| Plasmid genomes | |||
| Plasmids with highest G+C content | |||
| 1 | Streptomyces autolyticus CGMCC0516 plasmid unnamed3 (NZ_CP019460.1) | 30 888 | 87.5 |
| 2 | Streptomyces autolyticus CGMCC0516 plasmid unnamed8 (NZ_CP019465.1) | 15 591 | 83.3 |
| 3 | Streptomyces cattleya NRRL 8057 plasmid pSCAT (FQ859184.1) | 1 809 491 | 73.3 |
| 4 | Streptomyces cattleya DSM 46488 plasmid pSCATT (CP003229.1) | 1 812 548 | 73.3 |
| 5 | Streptomyces sp. FR-008 plasmid pSSFR2 (CP009804.1) | 24 272 | 72.9 |
| Plasmids with lowest G+C content | |||
| 1 | Candidatus Baumannia cicadellinicola strain B-GSS plasmid (CP011788.1) | 3 465 | 20.3 |
| 2 | Blattabacterium sp. (Nauphoeta cinerea) plasmid (NC_022551.1) | 3 674 | 20.6 |
| 3 | Borrelia burgdorferi B31 plasmid lp21 (CP009673.1) | 18 777 | 20.6 |
| 4 | Streptobacillus moniliformis DSM 12112 plasmid pSMON01 (CP001780.1) | 10 702 | 20.9 |
| 5 | Brachyspira intermedia PWS/A plasmid pInt (CP002875.1) | 3 260 | 21.0 |
| Phage genomes | |||
| Phage with highest G+C content | |||
| 1 | Streptomyces phage SV1 (NC_018848.1) | 37 612 | 72.7 |
| 2 | Streptomyces phage PapayaSalad (KY092481.1) | 38 411 | 72.6 |
| 3 | Streptomyces phage Picard (KY092480.1) | 39 522 | 72.6 |
| 4 | Streptomyces phage Mojorita (KY092482.1) | 38 496 | 72.5 |
| 5 | Streptomyces phage ToastyFinz (KY676784.1) | 39 693 | 72.5 |
| Phage with lowest G+C content | |||
| 1 | Spiroplasma phage SVTS2 (AF133242.2) | 6 825 | 20.3 |
| 2 | Spiroplasma phage 1-R8A2B (NC_001365.1) | 8 273 | 22.9 |
| 3 | Spiroplasma phage SVGII3 (AJ969242.1) | 7 878 | 23.0 |
| 4 | Spiroplasma phage 1-C74 (NC_003793.1) | 7 768 | 23.2 |
| 5 | Mycoplasma phage phiMFV1 (AY583236.1) | 18 855 | 24.8 |
The data showed a prominent heteroskedasticity. Longer sequences tended to have higher G+C content values, while variation in G+C started high in short genomes and decreased as genomes became longer. In keeping with previous research [13, 14], this creates a data plot of a roughly triangular shape (Fig. 1). There is a positive correlation between genomic G+C content and bacterial genome length, though this is not a simple one: length is associated more with the range of G+C content, rather with its absolute value. As noted above, small sequences accommodate the whole range of G+C content, while as length increases, G+C values tend to occupy the upper part of the range. This is in keeping with the data in Table 1, where the five longest genome sequences all have G+C values of 69 mol% or more, whilst the shortest five examples range from 16.6 to 58.8 mol%.
Fig. 1.

Scatterplot of G+C content versus sequence length for bacterial chromosomal sequences, showing an approximately triangular shape associated with their relation. Pearson’s R indicates that about 58 mol% of the G+C content variation can be explained by genome length, although there is also apparent heteroskedasticity. G+C content is plotted using values to the nearest percentage point.
Therefore, trying to fit a linear regression model onto this dataset was potentially problematic. Using heteroscedasticity-robust regression, the linear model explained only a small proportion of the variation (Pearson R=0.58, P<0.001). This is equivalent to an r2 of 0.34 and, thus, around 66 mol% of the variation in G+C content cannot be accounted by this model. The heteroskedastic pattern could not be removed by log or root data manipulation, although the Pearson's R value for genomes was raised to 0.61 by log-log transformation (data not shown).
Plasmid genomes
Generally, plasmids were much smaller in size, although a few larger examples existed at >500 kb, e.g. those found in bacteria belonging to the genus Rhizobium. Table 1 shows that the length ranged from 744 bp (T. phenacola PAVE plasmid) to 2.58 Mb (Cupriavidus metallidurans CH34 megaplasmid). The plasmid with the lowest G+C was from Baumannia cicadellinicola strain B-GSS at 20.3 mol%, whilst the two plasmids with the highest G+C content (87.5 and 83.3 mol%, respectively) were from the same organism: S. autolyticus strain CGMCC0516 (Table 2).
Plasmids showed a similar pattern of G+C content variation to that seen in bacterial genomes, namely high variability of G+C in smaller sequences and a tendency for high G+C content as the size increased (Fig. 2). However, given the generally smaller length of these plasmids, the general abundance of shorter sequence lengths generated a rotated L-shape pattern when plotted, rather than the triangular shape seen for bacterial chromosomes.
Fig. 2.

Scatterplot of plasmid G+C content versus plasmid sequence length, showing an approximately rotated L-shape. G+C content is shown to the nearest mol% value.
Correlation between plasmid and host G+C content
A linear relationship (Fig. 3) was evident between plasmid G+C content and the corresponding G+C content of the host organism (Pearson R value=0.74, P<0.0001), although the variance was again not consistent throughout. The linear equation obtained showed approximately a one-to-one relationship between the two variables, with the plasmid G+C content increasing about 0.96 mol% for every 1 mol% increase in host G+C. Nevertheless, about 45 mol% of the variation was not explained by this relationship (r2=0.55, P<0.0001).
Fig. 3.

Comparison of the G+C content of plasmids versus that of their host. G+C content is shown to the nearest mol% value.
Phage genomes
Like plasmids, phages were generally small in size, although a few larger examples existed, the largest being from at almost 500 kb. Table 1 shows that the length ranged from 2435 bp (Leuconostoc phage L5) to 490 kb (Agrobacterium phage Atu_ph07). The phage with the lowest G+C was SVTS2 from Spiroplasma at 20.3 mol%, whilst the phage with the highest five G+C content values (72.7 to 72.5 mol%) were all from S. autolyticus (Table 2).
Phages showed the pattern seen in both bacterial and plasmid genomes, namely high variability of G+C in smaller sequences. However, unlike bacterial and plasmid genomes, those with larger genomes showed a tendency for lower G+C content (Pearson R value=−0.14, P<0.0001) as the size increased (Fig. 4).
Fig. 4.

Scatterplot of phage G+C content versus phage sequence length, showing that longer phages tend to have a lower G+C content. G+C content is shown to the nearest mol% value.
Correlation between phage and host G+C content
A linear relationship (Fig. 5) was evident between phage G+C content and the corresponding G+C content of the host organism (Pearson R value=0.90, P<0.0001). This was the best regression result for the whole dataset. The linear equation obtained approached a one-to-one relationship between the two variables, with the phage G+C content increasing about 0.88 mol% for every 1 mol% increase in host G+C, with about 81 mol% of the variation being explained by this relationship (r2=0.81).
Fig. 5.
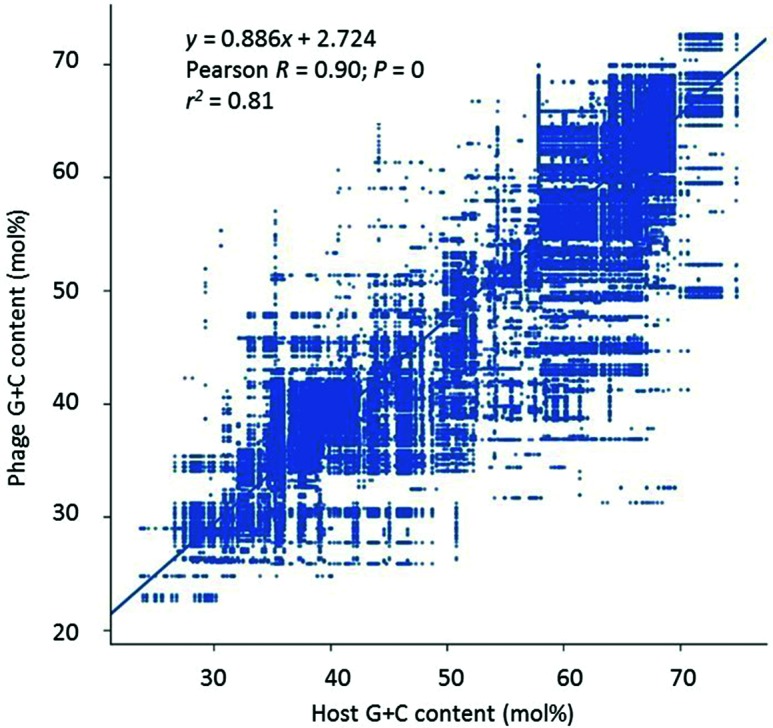
Comparison of the G+C content of phages versus that of their host. G+C content is shown to the nearest mol% value.
Discussion
The data presented here demonstrate that there is a correlation between the length of a bacterial genome and its G+C content, particularly in the case of organisms with longer genome lengths. However, it is also clear that this alone is not enough to explain the complete variation in genome G+C content as evidenced by the results from the linear regression model. Therefore, it is clear that other factors need to be considered to explain the G+C content. Probably the most obvious of these would be the organism’s normal optimal temperature range [10, 11], as the physical property of having a high percentage of triple bonds (G+C rich) is more likely to prevent denaturing of double-stranded DNA than would be the case for those with a high percentage of double bonds (AT rich). However, other environmental factors also need to be considered as well [7–9], together with the physiological capabilities of the organism [8]. Moreover, the heteroskedasticity of the length versus percentage G+C plot suggests that multifactorial variables may be most important in terms of organisms with shorter genome lengths, arguing that the roles played by environmental factors in terms of influencing the G+C content of a bacterial genome will require meta-analytical approaches to elucidate the other key factors. It is also worth noting that to date there has been a bias towards sequencing genomes of organisms that are either medically or agriculturally important. It will be interesting to determine whether the patterns observed continue as more bacterial genome sequences become available from organisms that are not medically important or from those that lack agricultural significance.
In the case of the chromosomal analysis, the G+C content does not go above 75 mol% or below 13 mol%. In part, this may be a reflection of the restrictions of the genetic code, where encoding certain amino acids requires at least some usage of A/T or G/C, e.g. phenylalanine requires TTC or TTT as a codon (with G+C-rich organisms likely to favour TTC) and glycine requires GGN as a codon (with A+T-rich organisms likely to favour either GGA or GGT). In addition to this requirement of compliance to the genetic code, there may also be restrictions imposed whereby unusual or rare codons are incorporated into genes [19], with the possible effect of slowing down the rate of translation to allow correct protein folding to take place. Moreover, there is evidence to suggest that DNA replication in organisms with a higher G+C content is associated with variants in the presence of DNA polymerases present such as polC being used, in addition to the number of and types of variants of the dnaE gene [20], as evidenced by organisms such as Pseudomonas putida [21].
Plasmids can be considered as genetic components of the bacterial cell and it is not surprising that their G+C content is correlated to that of their host. This observation has previously been discussed by Campbell and colleagues [22], where a substantial similarity in genomic signatures between prokaryotes and their plasmids was reported, although more recently Rocha and Danchin [23] reported that genetic elements that can be considered as ‘intracellular pathogens’, such as plasmids, phages and insertion sequences, have a tendency to have a lower G+C content than their host organism. However, this conclusion was drawn from a much smaller dataset relative to the current work. Moreover, with the potential benefits associated with some genes on plasmids, it makes sense to see a similarity in terms of G+C content for plasmid-borne genes that rely on the transcriptional and translational factors of the host organism (e.g. the encoding of specific tRNA molecules by the bacterial host). It has also been proposed that similarity in G+C content acts as a way of allowing the bacterial cell to discriminate between compatible and non-compatible DNA [24], although factors such as methylation patterns ensure that this is not as simple a mechanism as relying on the G+C content alone.
Moreover, the increasing number of examples of lateral gene transfer, or horizontal gene transfer, shows that inter-species transfer of genes is more commonplace than first imagined. While there are other means of moving DNA from one organism to another, using plasmids as a vector for this transfer is regarded as one of the most important. This is true for both inter-species conjugation of plasmids or transformational uptake of plasmids that have been released into the ecosystem by an alien species. Therefore, although the plasmids described are known to have been isolated from a particular bacterial species, it is impossible to determine when this plasmid first became part of the bacterial cell, and also what previous organism(s) may have acted as the prior host(s). As above, it will be interesting to put this into context based on both bacterial and plasmid sequence data when sequences from additional organisms become available.
Conversely phages can be regarded as being true parasites of the cells depending on the host organism for expression of their genes, without the potential associated benefit of factors such as antibiotic-resistance genes. However, this in turn also places a dependence on them to maintain a G+C pattern similar to that seen in the organisms they infect. As mentioned above, there have been reports to suggest that intracellular pathogens may have a G+C content lower than their host organism [23], and we also find this to be the case in the current analysis of phages, based on a much larger dataset than was used previously. The evolutionary explanation for this is unclear, although reducing the phage’s metabolic burden via reduced pyrimidine synthesis has been proposed (e.g. [23, 25]).
In terms of phage genome analysis, the site of any incorporation into the bacterial genome (e.g. as part of any lysogenic cycle) could also influence the G+C content of the phage genome. This would be in keeping with reports of heterogeneity of G+C content across bacterial genomes [26], where sliding window analysis identified regions of intragenomic variation of G+C content within a single species.
In conclusion, using a considerably increased dataset relative to previous work, we propose that a simple linear regression between bacterial chromosome length and G+C content accounts for at least some of the relationship. The same relationship is also true for bacterial chromosome G+C content and plasmid G+C content, although phages tend to have a lower G+C content than their hosts. However, in all cases there are other factors involved, although the true extent of each of these factors remains unclear, arguing for additional analyses via techniques such as principal component analysis or multiple regression analysis on data regarding the ecosystems from which organisms have been isolated.
Data bibliography
Almpanis A, Swain M, Gatherer D, McEwan N. Jupyter notebooks, https://github.com/atolgrp/Microbial-G-C-Content (2018).
Funding information
The authors received no specific grant from any funding agency.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
No ethical issues are associated with this work as all data were acquired from publicly accessible databases.
Footnotes
Abbreviation: OLS, ordinary least squares.
We confirm all supporting data, code and protocols have been provided within the article or through supplementary data files.
References
- 1.Brocchieri L. The GC content of bacterial genomes. J Phylogen Evolution Biol. 2014;2:e108. doi: 10.4172/2329-9002.1000e108. [DOI] [Google Scholar]
- 2.Freese E. On the evolution of the base composition of DNA. J Theor Biol. 1962;3:82–101. doi: 10.1016/S0022-5193(62)80005-8. [DOI] [Google Scholar]
- 3.Sueoka N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci USA. 1962;48:582–592. doi: 10.1073/pnas.48.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell A, Graur D. Inferring the pattern of spontaneous mutation from the pattern of substitution in unitary pseudogenes of Mycobacterium leprae and a comparison of mutation patterns among distantly related organisms. J Mol Evol. 2005;61:795–803. doi: 10.1007/s00239-004-0235-0. [DOI] [PubMed] [Google Scholar]
- 5.Sargentini NJ, Smith KC. DNA sequence analysis of γ-radiation (anoxic)-induced and spontaneous lacId mutations in Escherichia coli K-12. Mutat Res. 1994;309:147–163. doi: 10.1016/0027-5107(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 6.Hershberg R, Petrov DA. Evidence of selection upon genomic GC-content in bacteria. PLoS Genetics. 2010;7:e49060. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foerstner KU, von Mering C, Hooper SD, Bork P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005;6:1208–1213. doi: 10.1038/sj.embor.7400538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEwan CE, Gatherer D, McEwan NR. Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas. 1998;128:173–178. doi: 10.1111/j.1601-5223.1998.00173.x. [DOI] [PubMed] [Google Scholar]
- 9.Naya H, Romero H, Zavala A, Alvarez B, Musto H. Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J Mol Evol. 2002;55:260–264. doi: 10.1007/s00239-002-2323-3. [DOI] [PubMed] [Google Scholar]
- 10.Musto H, Naya H, Zavala A, Romero H, Alvarez-Valín F, et al. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 2004;573:73–77. doi: 10.1016/j.febslet.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 11.Musto H, Naya H, Zavala A, Romero H, Alvarez-Valín F, et al. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem Biophys Res Commun. 2006;347:1–3. doi: 10.1016/j.bbrc.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 12.Bohlin J, Skjerve E, Ussery DW. Investigations of oligonucleotide usage variance within and between prokaryotes. PLoS Comput Biol. 2008;4:e1000057. doi: 10.1371/journal.pcbi.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohlin J, Sekse C, Skjerve E, Brynildsrud O. Positive correlations between genomic %AT and genome size within strains of bacterial species: correlation between microbial genome size and % AT. Environ Microbiol Rep. 2014;6:278–286. doi: 10.1111/1758-2229.12145. [DOI] [PubMed] [Google Scholar]
- 14.Guo FB, Lin H, Huang J. A plot of G + C content against sequence length of 640 bacterial chromosomes shows the points are widely scattered in the upper triangular area. Chromosome Res. 2009;17:359–364. doi: 10.1007/s10577-009-9024-3. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell D. GC content and genome length in Chargaff compliant genomes. Biochem Biophys Res Commun. 2007;353:207–210. doi: 10.1016/j.bbrc.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37:67–75. doi: 10.1016/0092-8674(84)90301-5. [DOI] [PubMed] [Google Scholar]
- 17.White H. A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica. 1980;48:817–838. doi: 10.2307/1912934. [DOI] [Google Scholar]
- 18.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 19.Angov E. Codon usage: nature's roadmap to expression and folding of proteins. Biotechnol J. 2011;6:650–659. doi: 10.1002/biot.201000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timinskas K, Balvočiūtė M, Timinskas A, Venclovas Č. Comprehensive analysis of DNA polymerase III α subunits and their homologs in bacterial genomes. Nucleic Acids Res. 2014;42:1393–1413. doi: 10.1093/nar/gkt900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belda E, van Heck RG, José Lopez-Sanchez M, Cruveiller S, Barbe V, et al. The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ Microbiol. 2016;18:3403–3424. doi: 10.1111/1462-2920.13230. [DOI] [PubMed] [Google Scholar]
- 22.Campbell A, Mrázek J, Karlin S. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA. 1999;96:9184–9189. doi: 10.1073/pnas.96.16.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rocha EP, Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18:291–294. doi: 10.1016/S0168-9525(02)02690-2. [DOI] [PubMed] [Google Scholar]
- 24.Forsdyke DR. Different biological species "broadcast" their DNAs at different (G+C)% "wavelengths". J Theor Biol. 1996;178:405–417. doi: 10.1006/jtbi.1996.0038. [DOI] [PubMed] [Google Scholar]
- 25.Agashe D, Shankar N. The evolution of bacterial DNA base composition. J Exp Zool B Mol Dev Evol. 2014;322:517–528. doi: 10.1002/jez.b.22565. [DOI] [PubMed] [Google Scholar]
- 26.Bohlin J, Snipen L, Hardy SP, Kristoffersen AB, Lagesen K, et al. Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genomics. 2010;11:464. doi: 10.1186/1471-2164-11-464. [DOI] [PMC free article] [PubMed] [Google Scholar]