
Figure 1. L1000 gene expression platform implementation and validation.
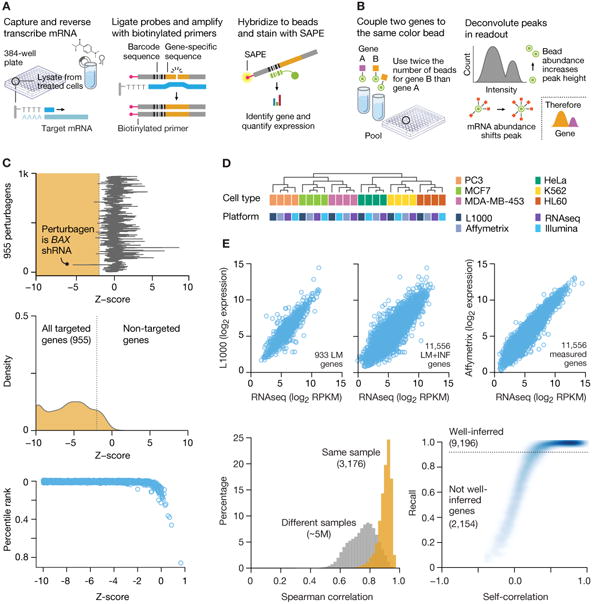
A. Overview of ligation-mediated amplification. Cells are treated in 384-well plates, lysed and mRNA captured on oligo-dT plates. mRNA is reverse-transcribed and oligonucleotide probes designed with transcript-specific, 24-mer unique barcode and universal primer sequences annealed to the cDNA, ligated and PCR-amplified using biotinylated primers. PCR product is hybridized to optically addressed polystyrene microspheres, where each bead is coupled to an oligonucleotide complementary to a landmark gene's barcode. Transcript abundance is quantified by fluorescence using a Luminex FlexMap 3D scanner.
B. Deconvoluting 1,000 landmark genes using 500 bead colors. Each bead is analyzed for its bead color (denoting landmark gene identity) and phycoerythrin intensity (denoting transcript abundance). Aliquots of the same bead color, separately coupled to two different gene barcodes, are combined in a ratio of 2:1. A distribution of fluorescent intensities reveals two peaks (partitioned by k-means clustering), the larger peak designating the landmark for which double number of beads were used.
C. Validation of L1000 probes using shRNA knockdown. MCF7 and PC3 cells transduced with shRNAs targeting 955 landmark genes. Differential expression values (z-scores) were computed for each landmark and the percentile rank of expression z-scores in the experiment in which it was targeted relative to all other experiments was computed. 841/955 genes (88%) rank in the top 1% of all experiments and 907/955 (95%)rank in the top 5%. Top panel: z-score of BAX gene in every experiment. Middle panel: Z-score distribution from all targeted (orange) and non-targeted (white) genes. Distribution from the targeted set is significantly lower than non-targeted (p value <10-16). Bottom panel: Scatter of percentile rank versus expression z-score for 955 targeted genes.
D. Comparison of L1000 with other platforms. Samples of RNA from 6 human cancer cell lines were profiled on L1000, Affymetrix GeneChip HG-U133 Plus 2.0 Array, Illumina Human HT-12 v4 Expression BeadChip Array, and mRNA-seq (Illumina Hi-Seq).
E. Comparison of L1000 with RNA-seq and Affymetrix using patient-derived samples. RNA samples from 3,176 tissue specimens profiled on L1000 and RNA-seq, and a subset on Affymetrix microarrays. Top panels: Scatter plots of L1000 expression versus RNA-seq in landmark (left, Spearman correlation of 0.86) and landmark plus inferred (middle, Spearman correlation of 0.91) expression for a single sample. Bottom left:Spearman correlation distribution for L1000 vs RNA-seq of landmark genes for the same sample (orange) and different samples (gray), across all 3,176 patient samples. Bottom right: All L1000 inferred genes were subject to recall analysis by comparison with their RNA-seq measured equivalents. Scatter plot shows R versus cross-platform correlation for all inferred genes. 9,196 of 11,350 (81%) have an R in the 95th percentile (dotted line).