

Summary
Essential processes in biology are carried out by large macromolecular assemblies, whose structures are often difficult to determine by traditional methods. Increasingly, researchers combine measured data and computed information from several complementary methods to obtain “hybrid” or “integrative” structural models of macromolecules and their assemblies. These integrative/hybrid (I/H) models are not archived in the Protein Data Bank because of the absence of standard data representations and processing mechanisms. Here, we present the development of data standards and a prototype system for archiving I/H models. The data standards provide the definitions required for representing I/H models that span multiple spatiotemporal scales and conformational states as well as spatial restraints derived from different experimental techniques. Based on these data definitions, we have built a prototype system called PDB-Dev, which provides the infrastructure necessary to archive I/H structural models. PDB-Dev is now accepting structures and is open to the community for new submissions.
Keywords: Integrative/Hybrid modeling, PDBx/mmCIF, PDB, PDB-Dev, data standards, structural biology, data archiving, IHM dictionary, multi-scale models, spatial restraints, Structural Biology
eTOC
Vallat et al., describe the data representation and prototype system for archiving structural models of biological macromolecules computed by integrative/hybrid modeling. The PDB-Dev prototype system enables the deposition, archiving and dissemination of integrative structural models in a standard form.

1 Introduction
The Protein Data Bank (PDB) archives experimentally derived structures of biological macromolecules (Berman et al., 2000). The PDB currently holds over 135,000 structures that were determined primarily by X-ray crystallography (X-ray), Nuclear Magnetic Resonance (NMR) spectroscopy and three-dimensional Electron Microscopy (3DEM). An outcome of a workshop held in 2005 was a new policy that the PDB should only contain models of structures determined from experimental measurements on physical samples, and that purely computational models should be held in a separate repository (Berman et al., 2006). Subsequently, the Model Archive was built to archive computational models obtained through comparative and ab initio modeling methods (Haas et al., 2013; Haas and Schwede, 2013).
Integrative/hybrid (I/H) methods of structure determination (Alber et al., 2007a; Alber et al., 2008; Ward et al., 2013; Sali et al., 2015) use spatial restraints derived from a variety of experimental techniques to build the structures of biological macromolecules (Figure 1). These I/H methods are especially useful to model structures that are not amenable to primary methods of structure determination, such as X-ray, NMR and 3DEM. The different types of experiments that contribute spatial restraints used in I/H modeling include Chemical Crosslinking (CX), Mass Spectrometry (MS), Small Angle Scattering (SAS), Electron Tomography (ET), Fluorescence Resonance Energy Transfer (FRET), Electron Paramagnetic Resonance (EPR) spectroscopy, Atomic Force Microscopy (AFM) and various Proteomics methods (Alber et al., 2007a; Alber et al., 2008; Ward et al., 2013; Sali et al., 2015). The experimentally derived spatial restraints are supplemented by spatial restraints obtained from statistical analyses and physical principles to assemble a given set of structural components (experimental or computational) into the complete structure of an assembly. Several modeling applications such as Integrative Modeling Platform (IMP) (Russel et al., 2012), ROSETTA (Leaver-Fay et al., 2011), HADDOCK (Dominguez et al., 2003), BCL (Woetzel et al., 2011; Karakas et al., 2012; Weiner et al., 2014) and others have been developed and/or extended to handle I/H modeling. Structures of many macromolecular complexes have been determined using I/H methods (Sali et al., 2015), including the nuclear pore complex (Alber et al., 2007a; Alber et al., 2007b) and its sub-complexes (Kim et al., 2014; Shi et al., 2014; Fernandez-Martinez et al., 2016; Upla et al., 2017), the type III secretion system needle (Loquet et al., 2012), the proteosomal lid sub-complex (Politis et al., 2014), the ESCRT-I complex (Boura et al., 2011) and an RNA ribosome-binding element from the turnip crinkle virus genome (Gong et al., 2015). Although these structures have been described in the scientific literature, they have not been archived in the PDB because there were no standard mechanisms to represent, validate, process, archive and disseminate these models.
Figure 1. Illustration of integrative modeling.
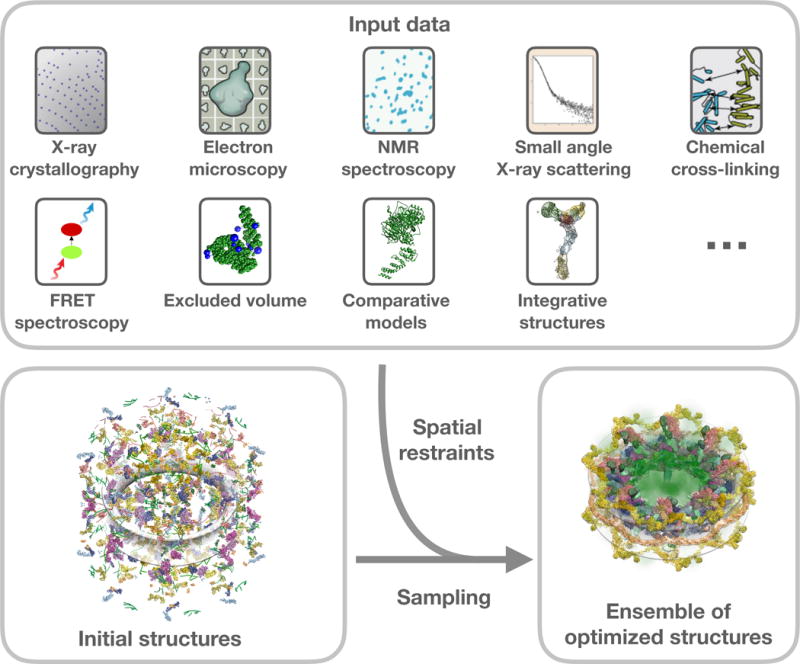
Examples of experimental and computational methods that can provide spatial restraints for integrative modeling (top). Atomistic and coarse-grained starting structural models of components of a macromolecular assembly are shown in various representations (bottom left). Extensive conformational and/or configurational sampling is carried out to yield the optimized assembly models that satisfy the input spatial restraints (bottom right).
In a wwPDB I/H Methods Task Force workshop held in 2014, a set of recommendations was put forward to enable the archiving of I/H models (Sali et al., 2015). A key recommendation proposed the development of a flexible model representation that allows for ensembles of multi-scale models (with atomistic and coarse-grained, non-atomistic representations), multi-state models (allowing for simultaneous multiple conformations), and ordered models (models related by time or other order). In addition, the recommendations proposed the creation of a federated system of model and data archives that interoperate with each other and the development of methods to estimate uncertainties of I/H models and the experimental data on which they are based.
In this paper, we describe the development of a flexible data representation to encode I/H structural models and the creation of a prototype system for archiving I/H models, called PDB-Dev. Together, these provide the foundation for building a robust data pipeline for validation, curation and dissemination of I/H models.
2 Results and Discussion
We have chosen to create the new I/H methods data representation as an extension of the existing PDBx/mmCIF data representation (Fitzgerald et al., 2005; Westbrook, 2013) used by the wwPDB to archive structures of biological macromolecules. The fundamental concepts used in creating data standards, the structure and contents of the new I/H methods data representation and the development of the PDB-Dev prototype system are discussed below. We use the all-inclusive term “data” to broadly refer to the data contents of the PDB-Dev prototype system, which includes the I/H structural models, associated spatial restraints, modeling protocols, and supporting metadata.
2.1 Overview of structural biology data standards
Data standards provide an essential foundation for building an archive. Data standards are technical specifications describing the semantics, logical organization, and physical encoding of the data and metadata to be archived. These specifications can be represented as a dictionary of terminology. Each definition in the dictionary establishes a unique name for a data item and includes precise definitions and examples. Definitions also include metadata used for assessing and maintaining data consistency, such as data type, controlled vocabularies, boundary conditions and parent-child relationships with other data items. Every data file archived in a repository follows the data standards specified in the dictionary. To maximize their utility, data standards are encoded in a software-accessible form, which can be leveraged by the operations of an automated data pipeline supporting repository deposition, validation, curation, archiving and dissemination of standardized data.
Developing community data standards requires a deep understanding of the underlying scientific domain, and how applications produce and consume the data. The latter considerations are critically important in developing data standards to support software automation. In the following sections, we describe current data standards used in structural biology and the extensions that we have developed to describe I/H investigations.
The PDB format
The PDB format provides a standard representation for macromolecular structure data derived from structural biology studies (Westbrook and Fitzgerald, 2009). This representation was created in the 1970’s and a large amount of software has been developed to work with this format. The PDB data file contains information regarding the atomic coordinates provided in a table of fixed-width columns representing particular data terms. The PDB file also contains semi-structured remark records containing metadata describing the molecular systems, experimental methods, authors, citations, etc. While the PDB format has endured for decades due to its simplicity and convenience, the use of fixed-width columns in the atomic coordinate tables limits its ability to handle large structures. This limitation among others led to the development of the mmCIF data framework for X-ray crystallography (Fitzgerald et al., 2005).
The PDBx/mmCIF framework
The data and publication standard of the International Union of Crystallography (IUCr) for diffraction experiments on small molecules is the Crystallographic Information Framework (CIF) (Hall et al., 1991). This data representation was revised to describe the process and results of macromolecular structure determinations. By design, the Macromolecular Crystallographic Information Framework (mmCIF) is extensible (Fitzgerald et al., 2005). Over time, the wwPDB worked with the NMR, 3DEM and SAS communities to extend the metadata framework, allowing for representing and exchanging the data required to archive and validate model structures obtained from these experimental methods. PDBx/mmCIF has evolved while serving as the metadata and archiving data standard for the PDB archive (Westbrook et al., 2005; Westbrook and Fitzgerald, 2009; Westbrook, 2013). It was officially adopted as the Master format for the PDB in 2011.
2.2 Data standards for I/H methods
Data standards describing I/H modeling methods have been developed as an extension of the PDBx/mmCIF framework (section 2.1). The advantages of building on an existing data standard are multi-fold:
Interoperability: The extension facilitates seamless interoperation with the PDB archive, which is based on the PDBx/mmCIF data standard.
Reusability: Carefully crafted descriptions of biological macromolecules, their polymeric sequences, atomic structures and associated ligands can be reused.
Extensibility: The PDBx/mmCIF standard and related extensions are themselves highly extensible and provide the flexibility and scalability required for adapting to future needs as the field evolves.
Automation: Software tools have been developed to handle the dictionaries and data files that follow the PDBx/mmCIF specifications. These existing tools can be adapted to manage the new I/H methods data dictionary (referred to as the IHM dictionary) and associated data files (referred to as IHM data files).
These advantages make the PDBx/mmCIF extension a convenient choice for creating the data representation for structures determined using I/H methods.
We have used specific examples of I/H models (described in the STAR methods section) as use cases to guide the development of the IHM dictionary. The dictionary and associated documentation are freely available from a public GitHub repository (https://github.com/ihmwg/IHM-dictionary) (Vallat et al., 2016a; Vallat et al., 2016b). The dictionary contains definitions and examples for over 300 new data items that collectively establish the data standards for I/H methods. Figure 2 summarizes the data contents of the IHM dictionary, which includes definitions for input sequence and structural data, restraints obtained from experimental sources as well as descriptions of multi-scale, multi-state and ordered ensembles of macromolecular assemblies. The important concepts and contents of the IHM dictionary are discussed below.
Figure 2. Illustration of the data contents captured in the IHM dictionary.
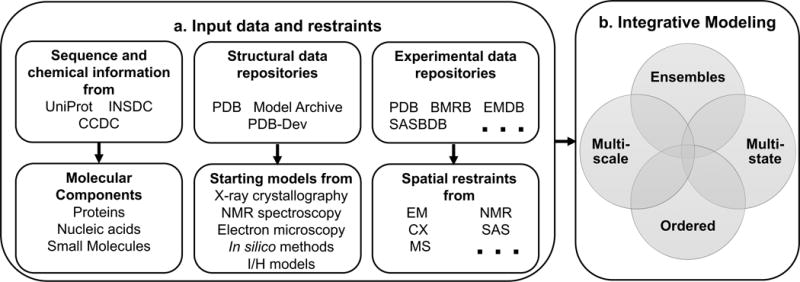
(a) The top row shows existing external resources that provide information regarding macromolecular sequence (UniProt and INSDC), small molecule data (CCDC), macromolecular structures (PDB and Model Archive), and various types of experimental data (PDB, BMRB, EMDB, SASBDB etc.). The second row shows the information derived from the external repositories, which is described in the IHM dictionary. This information includes details of the molecular components (reused from PDBx/mmCIF dictionary), the starting structural models of individual molecular components, and the spatial restraints derived from experimental methods (Electron Microscopy (EM), NMR, CX, MS, SAS, etc.). It is important to note that not all types of experimental information used in I/H modeling are currently archived in an experimental data repository. For instance, FRET and CX communities are beginning to address the requirements for building their own data archives. (b) The details of the integrative modeling algorithm that can produce an ensemble of multi-scale, multi-state and ordered models are described in the dictionary.
Extending existing definitions from PDBx/mmCIF
In developing the IHM dictionary, we have extended the data definitions in the current PDBx/mmCIF dictionary. Figure 3a shows some of the existing data definitions in the PDBx/mmCIF dictionary that pertain to the representation of small molecules (ligands), polymeric macromolecules and biomolecular complexes. These definitions include generic descriptions of the macromolecular components, representations of ligands, amino acid residues, nucleotides, and polymer sequences, and atomic coordinates and related structural features. Other definitions that contain information about software used in modeling, bibliographic citations and authors of the structure are also included in the PDBx/mmCIF dictionary. The descriptions of complex structural assemblies and representations of multi-scale models are provided as extensions in the IHM dictionary (Figure 3b).
Figure 3. Extensions to the definitions of the molecular system.
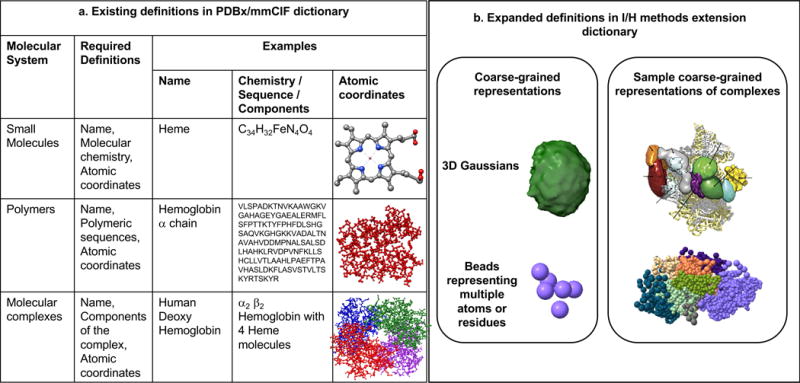
(a) Selected definitions in the PDBx/mmCIF dictionary (Westbrook, 2013) that relate to the descriptions of small molecules (e.g., Heme), polymers (e.g., Hemoglobin α chain) and molecular complexes (e.g., human deoxy Hemoglobin) are shown. The definitions necessary to represent these molecular systems are provided in column 2. Specific examples that illustrate these definitions are provided in columns 3-5. (b) Examples of selected extensions in the IHM dictionary that describe coarse-grained representations such as spheres and 3D Gaussians (eg: segmentation of a 3DEM map) are shown in the left box. Examples of structural assemblies comprised of multi-scale representations including coarse-grained spheres and 3D Gaussians are shown in the right box. The different components of the assembly are shown in different colors.
Referencing data from external resources
There are several existing repositories that archive experimental data. These repositories include the PDB for X-ray structure factors (Berman et al., 2014), the Biological Magnetic Resonance Data Bank (BMRB) for NMR data (Ulrich et al., 2008), the Electron Microscopy Data Bank (EMDB) for 3DEM maps (Lawson et al., 2016; Patwardhan and Lawson, 2016), the Electron Microscopy Public Image Archive (EMPIAR) for EM raw micrographs and two-dimensional EM (2DEM) class averages (Iudin et al., 2016), Small Angle Scattering Biological Data Bank (SASBDB (Valentini et al., 2015)) and BIOISIS (Rambo et al., 2017) for SAS data, and the Proteomics Identifiers database (PRIDE (Vizcaino et al., 2016)) and the PEPTIDE ATLAS project (Desiere et al., 2006) from the ProteomeXchange consortium (Vizcaino et al., 2014) for proteomics data. Other communities, such as those collecting FRET, EPR, and MS data, are beginning to address the requirements for building their own data archives. Similarly, the structural model repositories include the PDB archive for structural models determined predominantly using X-ray, NMR or 3DEM, the Model Archive for computational models (Haas et al., 2013; Haas and Schwede, 2013) and the PDB-Dev prototype system, developed as part of this project, which archives I/H models. Macromolecular sequence information is available through the International Nucleotide Sequence Database Collaboration (INSDC) (Nakamura et al., 2013; Benson et al., 2015) and UniProt (The UniProt Consortium, 2017) and small molecule chemical information is available from the Cambridge Crystallographic Data Center (CCDC) (Groom et al., 2016).
The IHM dictionary provides mechanisms to reference experimental data, structural models and macromolecular sequence information available from external repositories, using appropriate provenance details including database names, accession codes and version numbers (Figure 4a). If the data has not been archived in a public repository, the IHM dictionary provides alternate mechanisms to reference external datasets using Digital Object Identifiers (DOI (The International DOI Foundation, 2006)) or persistent Uniform Resource Identifiers (URI) that may be obtained from a provider (Figure 4a). For example, Zenodo (Nielson, 2017) provides hosting and DOI registration for experimental datasets and GitHub provides hosting services, collaboration tools as well as version controls for data and software.
Figure 4. Descriptions of input data and restraints.
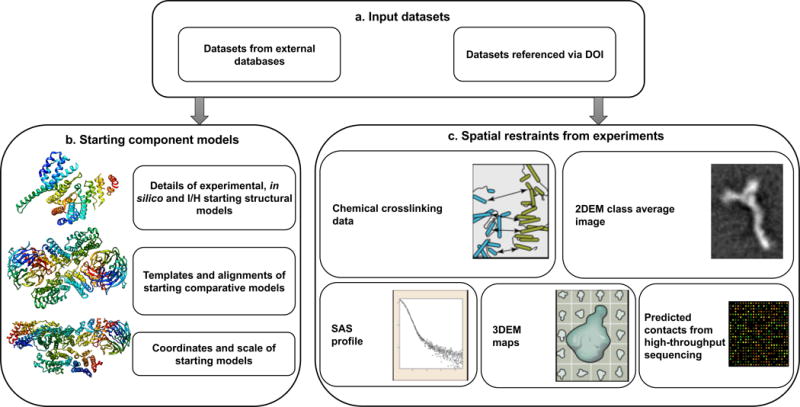
(a) Input datasets that come from external databases or data sets referenced via DOIs. (b) The information captured regarding starting structural models. (c) The different types of spatial restraints derived from experiments.
Structural models of assembly components
Integrative structure modeling often makes use of structural models of assembly components (e.g., domains, proteins and sub-complexes) that may be available in existing structural model repositories, such as the PDB and the Model Archive. The starting structural models of assembly components can be linked to entries in external repositories (Figure 4b). Optionally, the initial sets of coordinates used as input for modeling can be included in the data file. If the starting component model is a comparative model, additional details regarding the structural templates and target-template sequence alignments used to obtain the starting comparative models can also be provided. The dictionary also allows for noting whether the starting models were rigid or flexible during modeling.
Experimentally derived spatial restraints
The primary experimental data underpinning the structural model will be referenced from external resources, such as an experimental data repository or via DOIs as described earlier. The spatial restraints derived from the experimental data will, however, be archived along with the structural models (Figure 4c) to enable analysis, validation and visualization of the structures. Because it is not always possible to decouple method-specific details from the interpretation of the restraints, we have implemented distinct definitions for restraints derived from different kinds of experiments.
For example, the definitions of distance restraints derived from chemical crosslinking experiments address the application of the restraints to the current molecular system as well as the handling of associated experimental ambiguities. Other experimental restraints defined in the IHM dictionary include those derived from two-dimensional electron microscopy class average images (2DEM), 3DEM density maps, data from SAS experiments and predicted contacts from high-throughput sequencing experiments (Figure 4c). The dictionary also contains definitions for generic distance restraints between atoms and residues that may be obtained from different kinds of biophysical and proteomics experiments. These data definitions are based on specific use cases (described in the STAR methods section) and will be expanded as we obtain more modeling examples that utilize different types of experimental data.
Representation of ensembles of multi-scale, multi-state and ordered models
The representation of models in the IHM dictionary extends the scope of the current structural representation of macromolecular data in the PDB archive. Because the existing PDBx/mmCIF dictionary is designed only for single-scale atomistic structures, the IHM dictionary provides the definitions required to represent ensembles of multi-scale, multi-state and ordered collections of macromolecular structures.
Representation of multi-scale models
Experimental techniques do not always provide information at atomic resolution. It is possible for part of a macromolecular complex to have an atomic structure determined by X-ray crystallography, whereas the rest of the complex may not have an experimental or comparative model to begin with. In such cases, it is useful to model the former as atomistic rigid bodies and coarse-grain the latter as flexible single or multi-residue beads leading to a model representation with multiple scales of resolution. Different components or regions in a multi-scale assembly can be represented at different resolutions. Moreover, even the same region of a model can be represented with different granularity, typically to facilitate imposing restraints of varying precision. For example, a chemical cross-link between two residues across a protein interface is conveniently imposed as an upper bound on the distance between the two beads representing the cross-linked residues, whereas an affinity purification observation of interaction between the same two proteins is imposed as a contact between two larger beads representing the interacting proteins. In addition to the descriptions of atomic coordinates provided in the PDBx/mmCIF dictionary, definitions for coarse-grained representations for single and multi-residue spherical beads and three dimensional Gaussian objects characterizing regions of low resolution are provided in the IHM dictionary along with descriptions of how they are applied to the current molecular system (Figure 3b).
The IHM dictionary also provides a set of definitions to represent hierarchical structural assemblies comprised of different sub-components of the molecular system (Figure 3b). This hierarchical representation allows for depicting different sub-assemblies and super-assemblies relevant to the integrative modeling study.
Representation of multi-state models
Integrative modeling can involve multiple conformational states (Figure 5a) such as structurally open and closed states, functionally active and inactive states, ligand bound and unbound states, etc. Information regarding conformational diversity may be obtained from single-molecule experiments, where a single molecule changes conformation over time or from other experiments where fractions of molecules in different conformations exist in an equilibrium (Molnar et al., 2014). In case of the latter, structures representing all the states of a multi-state model are required to satisfy the input experimental restraints. We have incorporated descriptions of multi-state models in the dictionary.
Figure 5. Representation of multi-scale, multi-state, and ordered ensembles of models.
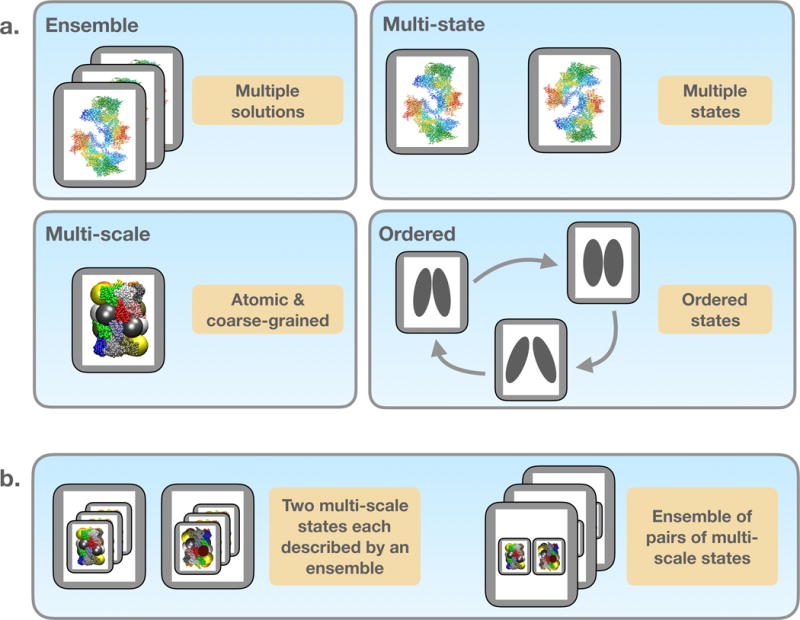
(a) Based on the definitions in the PDBx/mmCIF dictionary, the IHM dictionary allows for ensembles of atomistic models in a single state. In addition, the extension dictionary includes definitions for non-atomistic multi-scale models with coarse-grained representations, models spanning diverse conformational states, and models related by time or other order. Spherical beads of various sizes shown in the structural models represent the multi-scale nature of the I/H models, each grey box depicts a single structural model and a collection of grey boxes represents an ensemble. (b) The four attributes shown in panel (a) can be combined without limitations in an IHM data file. Two examples are shown here. One comprises of two multi-scale states, each described by an ensemble (left). The other example is an ensemble of pairs of multi-scale states (right).
Representation of ordered models
Integrative modeling can lead to time-ordered structures (Figure 5a) or those ordered by other criteria such as the sequence of events in an assembly process. Ordered sets of structural models are defined in the IHM dictionary as directed graphs (Beng-Jensen and Gutin, 2008), where models obtained at a particular time point or event are the nodes and the ordered relationships between them are the directed edges. Linear, branched and cyclic relationships can thus be expressed using the directed graph representation. The graph is stored in the IHM data file as a simple list of edges.
Representation of ensembles of models
The outcome of an integrative modeling study may be an ensemble of models each one of which satisfies the input spatial restraints well (Figure 5a), similar to the model ensemble obtained by satisfying NMR-derived restraints. The IHM dictionary allows for a single data file to include multiple models that belong to an ensemble. Additionally, I/H ensembles can be conformationally diverse as well as compositionally heterogeneous, consisting of models with different assembly components or varying in their multi-scale model representations. The IHM dictionary provides a unique model number identifier for each model, which forms the crucial link between various descriptions of structural assemblies as well as multi-scale, multi-state, and ordered ensembles.
A relatively small number of models (e.g., 100), potentially only a subset of the entire ensemble computed by sampling, can be included in the IHM data file. There is no upper limit currently set for this number and it will evolve over time as PDB-Dev advances. Optionally, the complete set of coordinates from a large collection of sampled models can be provided as external files. It is recommended to provide these coordinates in a binary format, such as the DCD format (Brooks et al., 1983) that allows for inference of the molecular topology from the IHM data file.
Combining multi-state, multi-scale, ordered and ensemble attributes
Importantly, the four attributes of the expanded model representation described above can be combined with each other without limitations (Figure 5b). For example, a model may represent two states of a protein, each described by an ensemble of conformations; alternatively, a model may define an ensemble of pairs of conformations, each of which can be represented in a multi-scale fashion and ordered in time or by some other useful criterion. The flexible model representation in the IHM dictionary is capable of handling the different combinations.
Modeling Workflow
Because different modeling applications can adopt substantially different workflows, it is difficult to describe the specific details of a modeling workflow in a data dictionary; an example is shown in Figure 6 (Shi et al., 2014). Despite these challenges, defining the modeling workflow offers significant advantages. It not only provides richer data content for the end users of the archived data, but also facilitates reproducibility of the results archived in the repository. As a preliminary solution, the IHM dictionary provides a generic representation of the modeling workflow in terms of listing the protocols adopted and the steps followed during modeling and post-modeling processing of the results (Figure 6). These workflow definitions can be used to describe the steps involved, such as conformational sampling, and scoring and clustering of the models. In addition, the software protocols and scripts used as input in the modeling may be provided as external files to facilitate reproducibility of the modeling workflow. These definitions will be extended in the future to describe protocols and workflows yet to be developed by the community.
Figure 6. Description of iterative integrative modeling workflow.
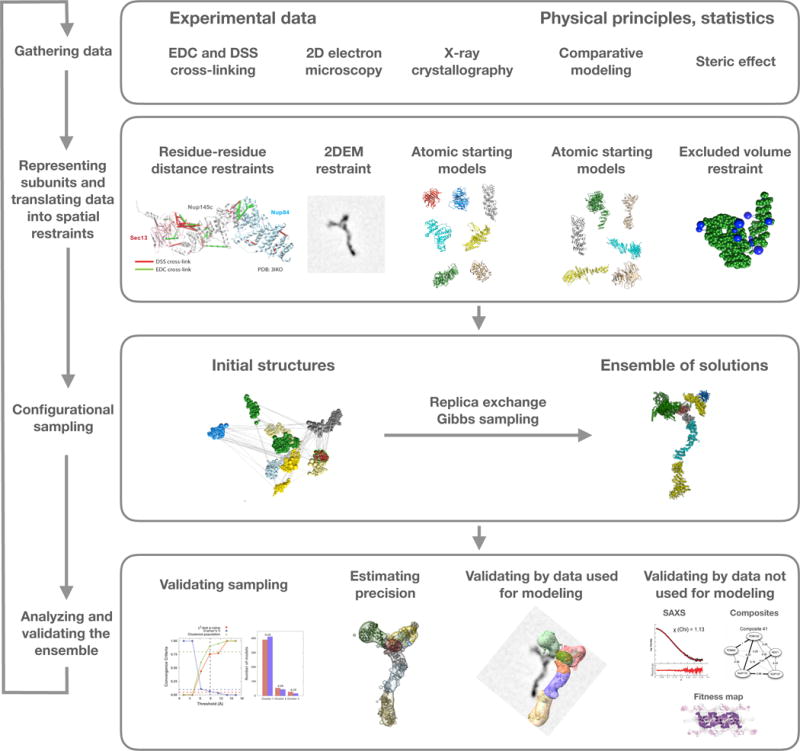
The integrative modeling workflow is illustrated by its application to structure determination of the Nup84 heptamer (Shi et al., 2014). The four stages include: (1) gathering all available experimental data and theoretical information; (2) translating this information into representations of assembly components and a scoring function for ranking alternative assembly structures; (3) sampling and scoring of structural models; and (4) analyzing and assessing the models. In this case, representations of the seven components of the Nup84 complex are based on crystallographic structures and comparative models of their domains. Component representations are coarse-grained by using spherical beads corresponding to multiple amino acid residues, to reflect the lack of information and/or to increase efficiency of sampling. The scoring function consists of spatial restraints that are obtained from CX-MS experiments and 2DEM class average images. The sampling explores both the conformations of the components and/or their configuration, searching for those assembly structures that satisfy the spatial restraints as accurately as possible. In this case, the result is an ensemble of many good-scoring models that satisfy the input data within acceptable thresholds. The sampling is then assessed for convergence, models are clustered, and evaluated by the degree to which they satisfy the data used to construct them as well as omitted data. The protocol can iterate through the four stages, until the models are judged to be satisfactory, most often based on their precision and the degree to which they satisfy the data. Finally, the models are deposited in PDB-Dev (https://pdb-dev.wwpdb.org, section 2.4).
Support for model validation
One of the recommendations from the wwPDB I/H methods task force involves developing procedures for estimating model uncertainty, so that they can be appropriately used by downstream applications. Although development of a complete validation pipeline will require significant participation from the modeling and experimental data communities, some basic definitions that support validation are already provided in the IHM dictionary. The preliminary validation metrics currently defined in the dictionary include information on whether the crosslinking restraints are satisfied or violated by the sampled models.
Localization density is defined as the probability of any volume element being occupied by a given particle (e.g., an atom, residue, or a protein) (Shi et al., 2014). The localization densities provide valuable information regarding the precision (uncertainty) and/or conformational diversity of the sampled ensembles, which is essential for validation and interpretation of the structural model (Shi et al., 2014). The localization densities of sampled ensembles (Figure 6) have been represented as three-dimensional Gaussian objects in the IHM dictionary. The dictionary also allows for localization density files in the standard MRC format (Cheng et al., 2015) to be referenced as external files.
2.3 Software applications that support the IHM dictionary
A key factor that influences the utility of an archive is the availability of software tools that can automatically generate as well as utilize the data files that adhere to the data specifications followed by the archive. Development of such tools demonstrates that the data definitions in the dictionary are software accessible. We have benefited from two software programs to elucidate automated generation and utilization of data files that are compliant with the new IHM dictionary: (1) the IMP software (Russel et al., 2012) has incorporated internal support for the IHM dictionary and can output IHM data files (examples in the STAR methods section); (2) the ChimeraX visualization software (Goddard et al., 2018) can be used to visualize the structural models described in the IHM data file. ChimeraX supports the visualization of multi-scale models comprised of atomistic and non-atomistic representations, input spatial restraints such as distances from chemical crosslinking experiments, 2DEM images and 3DEM maps, preliminary validation metrics regarding satisfaction of input restraints, and the localization densities of sampled ensembles. For example, ChimeraX can display satisfied and violated crosslinks in different colors. Examples of I/H models represented in the IHM data files and visualized using ChimeraX are shown in Figure 7.
Figure 7. Visualization of I/H models in PDB-Dev.
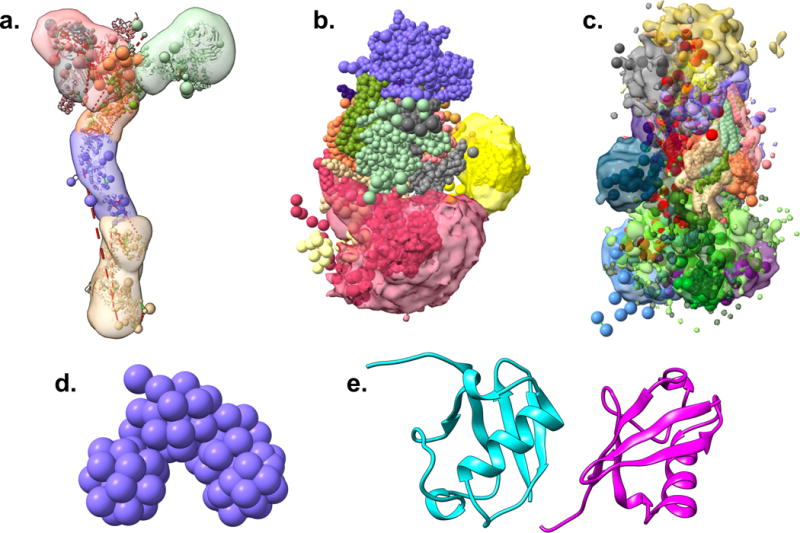
Five I/H model examples from PDB-Dev visualized using ChimeraX (Goddard et al., 2018) are shown. (a) The Nup84 sub-complex of the nuclear pore complex (Shi et al., 2014). (b) The exosome complex (Shi et al., 2015). (c) The mediator complex (Robinson et al., 2015). (d) 3D chromatin model comprising of the first 4.5Mb of Chromosome 2L from the Drosophila melanogaster genome (Trussart et al., 2015). (e) The Diubiquitin model (Liu et al., 2018). In the Nup84, exosome, and mediator structures, the multi-scale coarse-grained models are shown as spheres along with the starting structural models (cartoon), localization densities (transparent contour surfaces) and the distance restraints obtained from chemical crosslinking experiments (dotted lines), where available. The 3D chromatin model is shown using a coarse-grained beaded representation and the Diubiquitin structure is an atomistic model shown using a traditional cartoon representation of the two ubiquitin chains in different colors.
2.4 The PDB-Dev prototype system
Based on the data definitions embodied in the PDBx/mmCIF and the IHM dictionaries, we have built a prototype system for archiving I/H models called PDB-Dev (https://pdb-dev.wwpdb.org) that was jointly announced by the wwPDB leadership and the I/H methods team (Burley et al., 2017). PDB-Dev accepts models that comply with the new IHM dictionary and currently contains nine structural models that exemplify a variety of features of integrative modeling. Three models have been obtained from IMP (Russel et al., 2012; Shi et al., 2014; Robinson et al., 2015; Shi et al., 2015), one from TADbit (Trussart et al., 2015; Serra et al., 2017) with IMP, one from HADDOCK (Dominguez et al., 2003; van Zundert et al., 2015), and four others have been obtained from integrative modeling investigations (Belsom et al., 2016; Liu et al., 2018) that use ROSETTA (Leaver-Fay et al., 2011) and XPLOR-NIH (Schwieters et al., 2018) modeling software (details in the STAR methods section and Figure 7). These structures are currently available for download from PDB-Dev. Six additional structures have been deposited to PDB-Dev and are on hold pending publication. The IMP structures were submitted to PDB-Dev in a dictionary compliant format. The other structures were individually processed by the PDB-Dev team in collaboration with the authors to ensure that they conform to the dictionary. We look forward to working with other I/H modeling investigators to create dictionary-compliant structures for archival in PDB-Dev.
Most of the I/H structures archived in PDB-Dev have already been published in peer-reviewed journals, while some are on hold pending publication. We expect that more investigators will submit their models to PDB-Dev and that the submission of models to PDB-Dev prior to publication will become routine practice, just as it did for traditional structural biology methods. A critical need is the development of additional software tools that will support the automatic generation of data files that comply with the IHM dictionary. We are working with members of wwPDB I/H methods task force and the developers of I/H modeling software, such as ROSETTA (Leaver-Fay et al., 2011), HADDOCK (Dominguez et al., 2003) and BCL (Woetzel et al., 2011; Karakas et al., 2012; Weiner et al., 2014), to facilitate broader adoption of the new data standards. The creation of an automated deposition and archiving system that can handle models generated by a wide variety of modeling software is the focus of ongoing development. At present, PDB-Dev does not carry out any data curation or model validation activities. These requirements will be addressed in the future with input from the integrative modeling community and the wwPDB I/H methods task force.
3 Conclusions and future directions
The field of structural biology is evolving towards the development of I/H methods that incorporate information derived from a number of experimental and computational sources. To address the needs of the I/H modeling community, we have developed a new set of data standards for archiving I/H models. These data standards are organized in a dictionary of data terms that describe the various features of I/H modeling, including the definitions for spatial restraints derived from different experimental techniques as well as descriptions of models that span multiple spatiotemporal scales and conformational states. The dictionary, consisting of over 300 new data terms, is easily extensible and is capable of handling the growing needs of the I/H methods community. Based on the new data standards, a prototype system for archiving I/H models, called PDB-Dev, has been implemented. PDB-Dev accepts structures that follow the specifications defined in the dictionary and currently contains nine I/H structural models. More structures are requested from the community to facilitate the continued development of software tools that support automated deposition and archiving. Further extension of the prototype system to build a comprehensive data curation and model validation pipeline for I/H models is the focus of future research. These activities will address the implementation of the remaining recommendations from the wwPDB I/H methods task force (Sali et al., 2015).
STAR Methods
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Nup84 sub-complex of the Nuclear Pore complex | Shi et al., 2014 | PDBDEV_00000001 |
| Exosome complex | Shi et al., 2015 | PDBDEV_00000002 |
| Mediator complex | Robinson et al., 2015 | PDBDEV_00000003 |
| Lysine-linked Diubiquitin | Liu et al., 2018 | PDBDEV_00000004 |
| Human serum albumin domains in their native environment | Belsom et al., 2016 | PDBDEV_00000005 PDBDEV_00000006 PDBDEV_00000007 |
| 3D Chromatin model of the first 4.5Mb of Chromosome 2L from the Drosophila Melanogaster genome | Trussart et al., 2015 | PDBDEV_00000008 |
| Ribosomal RNA small subunit methyltransferase A complexed with 16S ribosomal RNA | van Zundert et al., 2015 | PDBDEV_00000014 |
| Software and Algorithms | ||
| IMP | Russel et al., 2012 | https://integrativemodeling.org |
| TADbit | Serra et al., 2017 | http://sgt.cnag.cat/3dg/tadbit/ |
| HADDOCK | Dominguez et al., 2003 | https://haddock.science.uu.nl |
| XPLOR-NIH | Schwieters et al., 2018 | https://nmr.cit.nih.gov/xplor-nih/ |
| ROSETTA | Leaver-Fay et al., 2011 | https://www.rosettacommons.org/software |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| ChimeraX | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| PDBx/mmCIF dictionary |
Fitzgerald et al., 2005 Westbrook, 2013 |
http://mmcif.wwpdb.org |
| IHM dictionary | This work | https://github.com/ihmwg/IHM-dictionary |
| PDB-Dev prototype system | This work | https://pdb-dev.wwpdb.org |
| Python Django Framework | Django Software Foundation | https://www.djangoproject.com |
| Bootstrap Framework | Bootstrap Core Team | https://getbootstrap.com |
| mmCIF dictionary software suite | RCSB PDB | https://sw-tools.rcsb.org/apps/MMCIF-DICT-SUITE/index.html |
| MAXIT software | RCSB PDB | https://sw-tools.rcsb.org/apps/MAXIT/index.html |
Contact for resource sharing
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Brinda Vallat (brinda.vallat@rcsb.org).
Method details
I/H model datasets
Specific examples of I/H models have been used to guide the development of the IHM dictionary. Three of these are multi-scale models obtained from IMP (Russel et al., 2012), which include the heptameric Nup84 sub-complex of the nuclear pore complex (Shi et al., 2014) (Figure 7a), the multi-state exosome complex (Shi et al., 2015) (Figure 7b), and the mediator complex (Robinson et al., 2015) (Figure 7c). These models were used to create the definitions for ensembles of multi-scale, multi-state and ordered models, starting structural models, modeling workflow, validation metrics, localization densities as well as descriptions of experimentally derived spatial restraints from CX-MS, 2DEM and 3DEM data. The multi-scale 3D Chromatin model of the first 4.5Mb of Chromosome 2L from the Drosophila melanogaster genome (Figure 7d) (Trussart et al., 2015) obtained from TADbit (Serra et al., 2017) with IMP, using data from Chromosome Conformation Capture (Hi-C (Dekker et al., 2002)) experiments, was used to develop descriptions of hierarchical structural assemblies as well as to test the multi-scale representations. The atomic model of ribosomal RNA small subunit methyltransferase A complexed with 16S ribosomal RNA (van Zundert et al., 2015) modeled using HADDOCK (Dominguez et al., 2003) with spatial restraints obtained from 3DEM, mutagenesis, and DNA footprinting data was used to create definitions for generic set of distance restraints that may be obtained from different kinds of biophysical and proteomics experiments. Other examples used to develop and validate the dictionary definitions include the multi-state atomistic model of Lysine-linked Diubiquitin (Liu et al., 2018), obtained from XPLOR-NIH (Schwieters et al., 2018) using restraints derived from SAS, single molecule FRET and CX-MS experiments (Figure 7e) as well as atomic structures of human serum albumin domains in their native environment (Belsom et al., 2016) elucidated using ROSETTA (Leaver-Fay et al., 2011) from CX-MS data and predicted residue contacts from high-throughput sequencing experiments. The contents of the IHM dictionary, as described in section 2.2, are based on the inputs, methods and outcomes of these investigations.
The data dictionary
The IHM dictionary is built as an extension to the PDBx/mmCIF dictionary (Fitzgerald et al., 2005; Westbrook, 2013) used by the wwPDB to archive macromolecular structures. Wherever possible, existing definitions in the PDBx/mmCIF dictionary are re-used. New definitions are added to describe the specific features of I/H modeling (Vallat et al., 2016a; Vallat et al., 2016b). These new definitions are mapped back to the molecular system described in the PDBx/mmCIF dictionary.
The IHM dictionary is maintained as a collaborative project on a public GitHub repository (https://github.com/ihmwg/IHM-dictionary). GitHub provides tools for version control and collaborative software development, such as creating branches for working simultaneously with different versions of a repository and reviewing proposed modifications through pull requests. Furthermore, it allows users to create bug reports and request new features that facilitate iterative enhancements. We have used the GitHub platform to freely distribute the IHM dictionary to members of the integrative modeling community, obtain their feedback and incorporate their recommendations.
IMP and ChimeraX
The IMP software is a comprehensive suite for integrative modeling of macromolecules and their assemblies (Russel et al., 2012). IMP now provides internal support for the IHM dictionary and can output IHM data files. This support has been implemented as a Python module within the IMP resource and is freely available to the public under the Lesser GPL license. Chimera is a widely-used software for visualizing macromolecules (Pettersen et al., 2004). ChimeraX, which is the next generation version of Chimera (Goddard et al., 2018), can be used to visualize the structural models and associated data, as represented in IHM data files. Because ChimeraX is under active development, it is recommended to use the daily builds of the software for visualization of I/H models.
The prototype archiving system
The PDB-Dev prototype system has been built using the Python Django framework for the backend (Django Software Foundation, 2009) and Bootstrap framework for the frontend (Bootstrap Core Team, 2017). PDB-Dev provides user registration and login functionalities along with a simple interface for uploading data files. Minimal data retrieval capabilities are provided to download the archived data files from PDB-Dev.
Other software tools
The mmCIF dictionary software suite (RCSB Developers, 2013) has been used to validate the syntax and format of the IHM dictionary and compliant data files. The MAXIT software (RCSB Developers, 2017) has been used to convert atomistic I/H models in PDB format to the PDBx/mmCIF format.
Data and software availability
The PDB-Dev website is available at https://pdb-dev.wwpdb.org. The IHM-dictionary is publicly distributed through a GitHub repository, https://github.com/ihmwg/IHM-dictionary.
Highlights.
Integrative structural models of biological macromolecules archived in PDB-Dev
Data standards for archiving integrative structural models
Multi-scale, multi-state, ordered, ensembles of structural models
Spatial restraints derived from various experimental and computational methods
Acknowledgments
We thank the members of the wwPDB I/H methods task force for providing valuable feedback on the dictionary and the PDB-Dev prototype system. In particular, we acknowledge the contributions from members of the Model Validation Subgroup, including Torsten Schwede, Gerhard Hummer, Jens Meiler, Frank Dimaio, Emad Tajkhorshid, and Alexandre Bonvin. We thank Jill Trewhella who co-leads the Federation Subgroup and the wwPDB leadership group, Stephen Burley, Genji Kurisu, John Markley, and Sameer Velankar for their support. We are grateful to the developers of the ChimeraX software, Thomas Ferrin and Thomas Goddard, for enabling the visualization of I/H models archived in PDB-Dev. We thank the authors of the structures in PDB-Dev for their collaboration and Cathy Lawson for her constructive comments on the manuscript. This work was funded by NSF EAGER grant DBI-1519158 and NIH awards R01 GM083960 and P41 GM109824.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, A.S., H.M.B., J.W.; Methodology, B.V., B.W., J.W.; Software, B.V., B.W.; Writing – Original Draft, B.V.; Writing – Review & Editing, B.V., B.W., J.W., A.S., H.M.B.; Supervision, A.S., H.M.B.
Declaration of Interests
The authors declare no competing interests.
References
- Alber F, Dokudovskaya S, Veenhoff L, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait B, Rout M, Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007a;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- Alber F, Dokudovskaya S, Veenhoff L, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait B, Sali A, Rout M. The molecular architecture of the nuclear pore complex. Nature. 2007b;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- Alber F, Forster F, Korkin D, Topf M, Sali A. Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem. 2008;77:443–477. doi: 10.1146/annurev.biochem.77.060407.135530. [DOI] [PubMed] [Google Scholar]
- Belsom A, Schneider M, Fischer L, Brock O, Rappsilber J. Serum Albumin Domain Structures in Human Blood Serum by Mass Spectrometry and Computational Biology. Mol Cell Proteomics. 2016;15:1105–1116. doi: 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beng-Jensen J, Gutin G. Directed graphs: Thoery, Algorithms and Applications. Springer-Verlag; 2008. [Google Scholar]
- Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2015;43:D30–35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Burley SK, Chiu W, Sali A, Adzhubei A, Bourne PE, Bryant SH, Dunbrack RL, Jr, Fidelis K, Frank J, Godzik A, Henrick K, Joachimiak A, Heymann B, Jones D, Markley JL, Moult J, Montelione GT, Orengo C, Rossmann MG, Rost B, Saibil H, Schwede T, Standley DM, Westbrook JD. Outcome of a workshop on archiving structural models of biological macromolecules. Structure. 2006;14:1211–1217. doi: 10.1016/j.str.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Berman HM, Kleywegt GJ, Nakamura H, Markley JL. The Protein Data Bank archive as an open data resource. J Comput Aided Mol Des. 2014;28:1009–1014. doi: 10.1007/s10822-014-9770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootstrap Core Team. Bootstrap. 2017 Retrieved November 7 2017, from http://getbootstrap.com/
- Boura E, Rozycki B, Herrick DZ, Chung HS, Vecer J, Eaton WA, Cafiso DS, Hummer G, Hurley JH. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc Natl Acad Sci USA. 2011;108:9437–9442. doi: 10.1073/pnas.1101763108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Burley SK, Kurisu G, Markley JL, Nakamura H, Velankar S, Berman HM, Sali A, Schwede T, Trewhella J. PDB-Dev: A prototype system for depositing integrative/hybrid structural models. Structure. 2017;25:1317–1318. doi: 10.1016/j.str.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Henderson R, Mastronarde D, Ludtke SJ, Schoenmakers RH, Short J, Marabini R, Dallakyan S, Agard D, Winn M. MRC2014: Extensions to the MRC format header for electron cryo-microscopy and tomography. J Struct Biol. 2015;192:146–150. doi: 10.1016/j.jsb.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–658. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Django Software Foundation. Django: a Python Web framework. 2009 from http://www.djangoproject.com/
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, Williams R, Rice WJ, Stokes DL, Zenklusen D, Chait BT, Sali A, Rout MP. Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell. 2016;167:1215–1228.e1225. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PMD, Westbrook JD, Bourne PE, McMahon B, Watenpaugh KD, Berman HM. 4.5 Macromolecular dictionary (mmCIF) In: Hall SR, McMahon B, editors. International Tables for Crystallography G Definition and exchange of crystallographic data. Springer; 2005. pp. 295–443. [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Schwieters CD, Tang C. Conjoined use of EM and NMR in RNA structure refinement. PLoS One. 2015;10:e0120445. doi: 10.1371/journal.pone.0120445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom CR, Bruno IJ, Lightfoot MP, Ward SC. The Cambridge Structural Database. Acta Crystallogr B. 2016;72:171–179. doi: 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, Schwede T. The Protein Model Portal–a comprehensive resource for protein structure and model information. Database (Oxford) 2013;2013:bat031. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Schwede T. Model Archive. 2013 Retrieved October 12 2016, from http://www.modelarchive.org/
- Hall SR, Allen FH, Brown ID. The Crystallographic Information File (Cif) – a New Standard Archive File for Crystallography. Acta Crystallographica Section A. 1991;47:655–685. [Google Scholar]
- Iudin A, Korir PK, Salavert-Torres J, Kleywegt GJ, Patwardhan A. EMPIAR: a public archive for raw electron microscopy image data. Nature Methods. 2016;13:387–388. doi: 10.1038/nmeth.3806. [DOI] [PubMed] [Google Scholar]
- Karakas M, Woetzel N, Staritzbichler R, Alexander N, Weiner BE, Meiler J. BCL::Fold–de novo prediction of complex and large protein topologies by assembly of secondary structure elements. PLoS One. 2012;7:e49240. doi: 10.1371/journal.pone.0049240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Sampathkumar P, Martel A, Matsui T, Tsuruta H, Weiss TM, Shi Y, Markina-Inarrairaegui A, Bonanno JB, Sauder JM, Burley SK, Chait BT, Almo SC, Rout MP, Sali A. Integrative structure-function mapping of the nucleoporin nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Mol Cell Proteomics. 2014;13:2911–2926. doi: 10.1074/mcp.M114.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CL, Patwardhan A, Baker ML, Hryc C, Garcia ES, Hudson BP, Lagerstedt I, Ludtke SJ, Pintilie G, Sala R, Westbrook JD, Berman HM, Kleywegt GJ, Chiu W. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 2016;44:D396–403. doi: 10.1093/nar/gkv1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gong Z, Cao Y, Ding YH, Dong MQ, Lu YB, Zhang WP, Tang C. Characterizing Protein Dynamics with Integrative Use of Bulk and Single-Molecule Techniques. Biochemistry. 2018;57:305–313. doi: 10.1021/acs.biochem.7b00817. [DOI] [PubMed] [Google Scholar]
- Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. Atomic model of the type III secretion system needle. Nature. 2012;486:276–279. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar KS, Bonomi M, Pellarin R, Clinthorne GD, Gonzalez G, Goldberg SD, Goulian M, Sali A, DeGrado WF. Cys-scanning disulfide crosslinking and bayesian modeling probe the transmembrane signaling mechanism of the histidine kinase, PhoQ. Structure. 2014;22:1239–1251. doi: 10.1016/j.str.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Cochrane G, Karsch-Mizrachi I. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson LH. Sharing your data and software on Zenodo. “Data Management & Open Data” In Life Science; Lausanne, Switzerland: 2017. (Open Science And Reproducibility Series, Workshop III). [Google Scholar]
- Patwardhan A, Lawson CL. Databases and Archiving for CryoEM. Methods Enzymol. 2016;579:393–412. doi: 10.1016/bs.mie.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Politis A, Stengel F, Hall Z, Hernandez H, Leitner A, Walzthoeni T, Robinson CV, Aebersold R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA, Hura GL. BIOISIS. 2017 Retrieved November 7 2017, from http://www.bioisis.net/about.
- Developers RCSB. MMCIF Dictionary Suite. 2013 v2.250 from https://sw-tools.rcsb.org/apps/MMCIF-DICT-SUITE/index.html.
- RCSB Developers. MAXIT Suite. 2017 v10.000 from https://sw-tools.rcsb.org/apps/MAXIT/index.html.
- Robinson PJ, Trnka MJ, Pellarin R, Greenberg CH, Bushnell DA, Davis R, Burlingame AL, Sali A, Kornberg RD. Molecular architecture of the yeast Mediator complex. Elife. 2015;4:e08719. doi: 10.7554/eLife.08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel D, Lasker K, Webb B, Velazquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, Sali A. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012;10:e1001244. doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Berman HM, Schwede T, Trewhella J, Kleywegt G, Burley SK, Markley J, Nakamura H, Adams P, Bonvin AM, Chiu W, Peraro MD, Di Maio F, Ferrin TE, Grunewald K, Gutmanas A, Henderson R, Hummer G, Iwasaki K, Johnson G, Lawson CL, Meiler J, Marti-Renom MA, Montelione GT, Nilges M, Nussinov R, Patwardhan A, Rappsilber J, Read RJ, Saibil H, Schroder GF, Schwieters CD, Seidel CA, Svergun D, Topf M, Ulrich EL, Velankar S, Westbrook JD. Outcome of the First wwPDB Hybrid/Integrative Methods Task Force Workshop. Structure. 2015;23:1156–1167. doi: 10.1016/j.str.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Bermejo GA, Clore GM. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 2018;27:26–40. doi: 10.1002/pro.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra F, Bau D, Goodstadt M, Castillo D, Filion GJ, Marti-Renom MA. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput Biol. 2017;13:e1005665. doi: 10.1371/journal.pcbi.1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT. Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol Cell Proteomics. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Pellarin R, Fridy PC, Fernandez-Martinez J, Thompson MK, Li Y, Wang QJ, Sali A, Rout MP, Chait BT. A strategy for dissecting the architectures of native macromolecular assemblies. Nat Methods. 2015;12:1135–1138. doi: 10.1038/nmeth.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International DOI Foundation. Digital Object Identifier Handbook. 2006 from http://www.doi.org/hb.html.
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussart M, Serra F, Bau D, Junier I, Serrano L, Marti-Renom MA. Assessing the limits of restraint-based 3D modeling of genomes and genomic domains. Nucleic Acids Res. 2015;43:3465–3477. doi: 10.1093/nar/gkv221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upla P, Kim SJ, Sampathkumar P, Dutta K, Cahill SM, Chemmama IE, Williams R, Bonanno JB, Rice WJ, Stokes DL, Cowburn D, Almo SC, Sali A, Rout MP, Fernandez-Martinez J. Molecular Architecture of the Major Membrane Ring Component of the Nuclear Pore Complex. Structure. 2017;25:434–445. doi: 10.1016/j.str.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015;43:D357–363. doi: 10.1093/nar/gku1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat B, Webb BM, Westbrook JD, Sali A, Berman HM. Integrative/Hybrid Methods PDBx/mmCIF dictionary extension. 2016a Retrieved June 9 2016, from https://github.com/ihmwg/IHM-dictionary/blob/master/dictionary/ihm-extension.dic.
- Vallat B, Webb BM, Westbrook JD, Sali A, Berman HM. Integrative/Hybrid Methods PDBx/mmCIF dictionary extension documentation. 2016b Retrieved June 9 2016, from https://github.com/ihmwg/IHM-dictionary/blob/master/dictionary_documentation/documentation.md.
- van Zundert GCP, Melquiond ASJ, Bonvin A. Integrative Modeling of Biomolecular Complexes: HADDOCKing with Cryo-Electron Microscopy Data. Structure. 2015;23:949–960. doi: 10.1016/j.str.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AB, Sali A, Wilson IA. Biochemistry. Integrative structural biology. Science. 2013;339:913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner BE, Alexander N, Akin LR, Woetzel N, Karakas M, Meiler J. BCL::Fold–protein topology determination from limited NMR restraints. Proteins. 2014;82:587–595. doi: 10.1002/prot.24427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook J. PDBx/mmCIF Dictionary Resources. 2013 Retrieved August 25 2015, from http://mmcif.wwpdb.org/
- Westbrook J, Henrick K, Ulrich EL, Berman HM. 3.6.2 The Protein Data Bank exchange data dictionary. In: Hall SR, McMahon B, editors. International Tables for Crystallography. Springer; 2005. pp. 195–198. G. Definition and exchange of crystallographic data. [Google Scholar]
- Westbrook JD, Fitzgerald PMD. Chapter 10 The PDB format, mmCIF formats, and other data formats. In: Bourne PE, Gu J, editors. Structural Bioinformatics. Second. John Wiley & Sons, Inc; 2009. pp. 271–291. [Google Scholar]
- Woetzel N, Lindert S, Stewart PL, Meiler J. BCL::EM-Fit: rigid body fitting of atomic structures into density maps using geometric hashing and real space refinement. J Struct Biol. 2011;175:264–276. doi: 10.1016/j.jsb.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]