

Abstract
Premise of the Study
The tropical tree family Lecythidaceae has enormous ecological and economic importance in the Amazon basin. Lecythidaceae species can be difficult to identify without molecular data, however, and phylogenetic relationships within and among the most diverse genera are poorly resolved.
Methods
To develop informative genetic markers for Lecythidaceae, we used genome skimming to de novo assemble the full plastome of the Brazil nut tree (Bertholletia excelsa) and 23 other Lecythidaceae species. Indices of nucleotide diversity and phylogenetic signal were used to identify regions suitable for genetic marker development.
Results
The B. excelsa plastome contained 160,472 bp and was arranged in a quadripartite structure. Using the 24 plastome alignments, we developed primers for 10 coding and non‐coding DNA regions containing exceptional nucleotide diversity and phylogenetic signal. We also developed 19 chloroplast simple sequence repeats for population‐level studies.
Discussion
The coding region ycf1 and the spacer rpl16‐rps3 outperformed plastid DNA markers previously used for barcoding and phylogenetics. Used in a phylogenetic analysis, the matrix of 24 plastomes showed with 100% bootstrap support that Lecythis and Eschweilera are polyphyletic. The plastomes and primers presented in this study will facilitate a broad array of ecological and evolutionary studies in Lecythidaceae.
Keywords: Amazonian trees, Bertholletia excelsa, DNA barcoding, genetic markers, Lecythidaceae, plastome
Lecythidaceae (sensu lato) is a pantropical family of trees with three subfamilies: Foetidioideae, which is restricted to Madagascar; Barringtonioideae, found in the tropical forests of Asia and Africa; and the Neotropical clade Lecythidoideae, which contains approximately 234 of the approximately 278 known species in the broader family (Mori et al., 2007, 2017; Huang et al., 2015; Mori, 2017). Neotropical Lecythidaceae are understory, canopy, or emergent trees with distinctive floral morphology and woody fruit capsules. Among Lecythidaceae species are the iconic Brazil nut tree, Bertholletia excelsa Bonpl.; the oldest documented angiosperm tree, Cariniana micrantha Ducke (dated at >1400 years old in Manaus, Brazil; Chambers et al., 1998); the cauliflorous cannonball tree commonly grown in botanical gardens, Couroupita guianensis Aubl.; and important timber species (e.g., Cariniana legalis (Mart.) Kuntze). Lecythidaceae is the third most abundant family of trees in the Amazon forest, following Fabaceae and Sapotaceae (ter Steege et al., 2013). The most species‐rich genus, Eschweilera Mart. ex DC., with approximately 99 species (Mori, 2017), is also the most abundant tree genus in the Amazon basin (ter Steege et al., 2013), and E. coriacea (DC.) S. A. Mori is the most common tree species in much of Amazonia (ter Steege et al., 2013). Lecythidaceae provide important ecological services such as carbon sequestration and are food resources for pollinators (bats and large bees) and seed dispersers (monkeys and agouties) (Prance and Mori, 1979; Mori and Prance, 1990).
Tools for species‐level identification and phylogenetic analyses of Lecythidaceae could significantly advance research on Amazon tree diversity. However, despite their ease of identification at the family level, species‐level identification of many Lecythidaceae (especially Eschweilera) is notoriously difficult when based on sterile (i.e., without fruit or floral material) herbarium specimens, and flowering of individual trees often occurs only at multi‐year intervals (Mori and Prance, 1987). As a complement to other approaches, DNA barcoding (Dick and Kress, 2009; Dexter et al., 2010) may help to identify species or clades of Lecythidaceae.
A combination of two protein‐coding plastid regions (matK and rbcL) has been proposed as a core plant DNA barcode (Hollingsworth et al., 2009), although other coding and non‐coding plastome regions (psbA‐trnH, rpoB, rpoC1, trnL, and ycf5) and the ITS of nuclear ribosomal genes have been recommended as supplemental barcodes for vascular plants (Kress et al., 2005; Lahaye et al., 2008; Li et al., 2011). However, an evaluation of a subset of these markers (ITS, psbA‐trnH, matK, rbcL, rpoB, rpoC1, and trnL) on Lecythidaceae in French Guiana (Gonzalez et al., 2009) showed poor performance for species identification. Furthermore, the use of traditional markers (plastid ndhF, trnL‐F, and trnH‐psbA, and nuclear ITS) for phylogenetic analysis has produced weakly supported trees (Mori et al., 2007; Huang et al., 2015), indicating a need to develop more informative markers and/or increase molecular sampling.
The main objectives of this study were to (1) assemble, annotate, and characterize the first complete plastome sequence of Lecythidaceae from the iconic Brazil nut tree, B. excelsa; (2) obtain a robust backbone phylogeny for the Neotropical clade using newly assembled draft plastome sequences for an additional 23 species; and (3) develop a novel set of informative molecular markers for DNA barcoding and broader evolutionary studies.
METHODS
Plant material and DNA library preparation
We performed genomic skimming on 24 Lecythidaceae species, including 23 Lecythidoideae and one outgroup species (Barringtonia edulis Seem.) from the Barringtonioideae. The sampling included all 10 Lecythidoideae genera (Appendix 1). Silica‐dried leaf tissue from herbarium‐vouchered collections was collected by Scott Mori and colleagues and loaned by the New York Botanical Garden. Total genomic DNA was extracted from 20 mg of dried leaf tissue using the NucleoSpin Plant II extraction kit (Machery‐Nagel, Bethlehem, Pennsylvania, USA) with SDS lysis buffer. Prior to DNA library preparation, 5 μg of total DNA was fragmented using a Covaris S‐series sonicator (Covaris Inc., Woburn, Massachusetts, USA) following the manufacturer's protocol to obtain approximately 300‐bp insert sizes. We prepared the sequencing library using the NEBNext DNA library Prep Master Mix and Multiplex Oligos for Illumina Sets (New England BioLabs Inc., Ipswich, Massachusetts, USA) according to the manufacturer's protocol. Size selection was carried out prior to PCR using Pippin Prep (Sage Science, Beverly, Massachusetts, USA). Molecular mass of the finished paired‐end library was quantified using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, California, USA) and by quantitative PCR using an ABI PRISM 7900HT (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at the University of Michigan DNA Sequencing Core (Ann Arbor, Michigan, USA). We sequenced the libraries on one lane of the Illumina HiSeq 2000 (Illumina Inc., San Diego, California, USA) with a paired‐read length of 100 bp.
Plastome assembly
Illumina adapters and barcodes were excised from raw reads using Cutadapt version 1.4.2 (Martin, 2011). Reads were then quality filtered using Prinseq version 0.20.4 (Schmieder and Edwards, 2011), which trimmed 5ʹ and 3ʹ sequence ends with a Phred quality score <20 and removed all trimmed sequences <50 bp in length, with >5% ambiguous bases, or with a mean Phred quality score <20. A combination of de novo and reference‐guided approaches was used to assemble the plastomes. First, chloroplast reads were separated from the raw read pool by BLAST‐searching all raw reads against a database consisting of all complete angiosperm plastome sequences available on GenBank (accessed in 2014). Any aligned reads with an E‐value <1–5 were retained for subsequent analysis. The filtered chloroplast reads were de novo assembled using Velvet version 7.0.4 (Zerbino and Birney, 2008) with k‐mer values of 71, 81, and 91 using a low‐coverage cutoff of 5 and a minimum contig length of 300. The assembled contigs were then mapped to a reference genome (see below) using Geneious version R8 (Kearse et al., 2012) to determine their order and direction using the reference‐guided assembly tool with medium sensitivity and iterative fine‐tuning options. Finally, raw reads were iteratively mapped onto the draft genome assembly to extend contigs and fill gaps using the low‐sensitivity, reference‐guided assembly in Geneious. We first assembled the draft genome of B. excelsa; the plastomes of the remaining 23 species were assembled subsequently using the plastome of B. excelsa as a reference. The B. excelsa plastome was annotated using DOGMA (Wyman et al., 2004) with the default settings for chloroplast genomes. Codon start and stop positions were determined using the open reading frame finder in Geneious and by comparison with the plastome sequence of Camellia sinensis (L.) Kuntze var. pubilimba Hung T. Chang (GenBank ID: KJ806280). A circular representation of the B. excelsa plastome was made using OGDraw V1.2 (Lohse et al., 2007). The complete annotated plastome of B. excelsa and the draft plastomes of the remaining 23 Lecythidaceae species sampled were deposited into GenBank (Appendix 1).
Identification of molecular markers
Chloroplast simple sequence repeats (cpSSRs) in B. excelsa were identified using the Phobos Tandem Repeat Finder version 3.3.12 (Mayer, 2010) by searching for uninterrupted repeats of nucleotide units of 1 to 6 bp in length, with thresholds of ≥12 mononucleotide, ≥6 dinucleotide, and ≥4 trinucleotide repeats, and ≥3 tetra‐, penta‐, and hexanucleotide repeats (Sablok et al., 2015). We developed primers to amplify the cpSSRs using Primer3 version 2.3.4 (Untergasser et al., 2012) with the default options and setting the PCR product size range between 100 and 300 bp.
The 24 plastomes were aligned with MAFFT version 7.017 (Katoh et al., 2002) and then scanned for regions of high nucleotide diversity (π; Nei, 1987) using a sliding window analysis implemented in DNAsp version 5.10.1 (Librado and Rozas, 2009) with a window and a step size of 600 bp. Levels of nucleotide diversity were plotted using the native R function “plot” (R Core Team, 2017), and windows with values over the 95th percentile were considered of high π.
Taking into account that DNA barcodes can also be used in phylogenetic analyses and because regions with high π do not necessarily have high phylogenetic signal (e.g., unalignable hypervariable regions), we employed a log‐likelihood approach modified from Walker et al. (2017) to identify phylogenetically influential regions. First, we inferred a phylogenetic tree with the plastome alignment (including only one inverted repeat) by performing 100 independent maximum likelihood (ML) searches using a GTRGAMMA model with RAxML version 8.2.9 (Stamatakis, 2014). Those searches resulted in the same topology that was subsequently annotated with the summary from 100 bootstraps using “sumtrees.py” version 4.10 (Sukumaran and Holder, 2010). We then calculated site‐specific log‐likelihoods in the alignment over the plastome phylogeny and calculated their differences site‐wise to the averaged log‐likelihood per site of 1000 randomly permuted trees (tips were randomly shuffled). Log‐likelihood scores were calculated with RAxML using a GTRGAMMA model. The site‐wise log‐likelihood differences (LD) were calculated using 600‐bp non‐overlapping windows with a custom R script (see below). We interpreted greater LD as an indication of greater phylogenetic signal, and windows with an LD above the 95th percentile were considered to have exceptional phylogenetic signal.
Primers flanking the top 10 regions with high π were designed using Primer3 with default program options. We employed a maximum product size of 1300 bp because lower cutoff values (i.e., 600 bp) made the primer design extremely challenging due to the lack of conserved regions. Primers were designed to amplify across all 23 Neotropical species without the use of degenerate bases. However, primers with a small number of degenerate bases were permitted for some regions where primer development otherwise would not have been possible due to high sequence variability in the priming sites. We investigated the potential of our markers to produce robust phylogenies by calculating individual gene trees in RAxML version 8.2.9 in an ML search with 100 rapid bootstraps (option “‐f a”) using the GTRGAMMA model. To evaluate the number of markers needed to obtain a resolved tree with an average of ~90 bootstrap support (BS), we first concatenated the two markers with the highest π and inferred a tree; subsequently, we added the marker with the next highest π score. We iterated this process until we obtained a matrix with each of the 10 markers developed. For every tree obtained, we calculated its average BS and its Robinson–Foulds distance (RF; Robinson and Foulds, 1981) from the plastome phylogeny using a custom R script employing the packages APE (Paradis et al., 2004) and Phangorn (Schliep, 2011). The scripts and alignments used in this study can be found at https://bitbucket.org/oscarvargash/lecythidaceae_plastomes.
RESULTS
Lecythidaceae plastome features
The sequenced plastome of B. excelsa contained 160,472 bp and 115 genes, of which four were rRNAs and 30 were tRNAs (Fig. 1, Table 1). The arrangement of the B. excelsa plastome had a typical angiosperm quadripartite structure with a single‐copy region of 85,830 bp, a small single‐copy region of 16,670 bp, and two inverted repeat regions of 27,481 bp each. Relative to C. sinensis var. pubilimba, we found no gene gain/losses in B. excelsa. The only structural difference we found is that B. excelsa contains the sequential genes trnH‐GUG, rps3, rpl22, and rps19 in the inverted repeat region, whereas C. sinensis var. pubilimba contains these genes in the large single‐copy region. Similarly, no gene gain/losses were found when B. excelsa was compared to other Neotropical Lecythidaceae plastomes assembled herein (Table 2). In addition to B. excelsa, the plastome of Eschweilera alata A. C. Sm. was also completely assembled; the coverage for the remaining plastomes ranged between 85% and 99.60% (Appendix 1).
Figure 1.
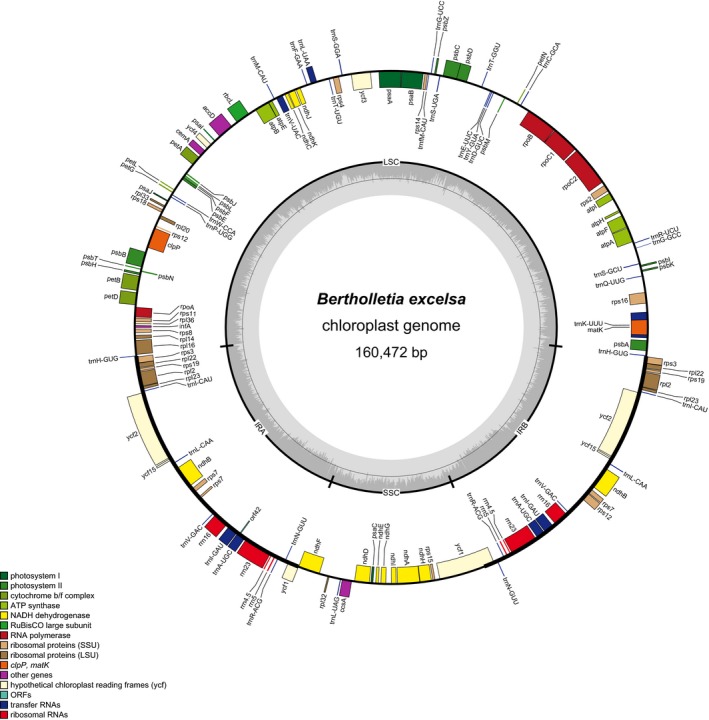
Plastome map of the Brazil nut tree, Bertholletia excelsa. Genes outside of the circle are transcribed clockwise; genes inside of the circle are transcribed counterclockwise. Gray bars in the inner ring show the GC content percentage.
Table 1.
Genes contained within the chloroplast genome of Bertholletia excelsa
| Function | Gene group | Gene name |
|---|---|---|
| Self‐replication | Ribosomal proteins (large subunit) | rpl2, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 |
| Ribosomal proteins (small subunit) | rps2, rps3, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19 | |
| RNA polymerase subunits | rpoA, rpoB, rpoC1, rpoC2 | |
| Ribosomal RNAs | rrn4.5, rrn5, rrn16, rrn23 | |
| Transfer RNAs | trnA‐UGC, trnC‐GCA, trnD‐GUC, trnE‐UUC, trnF‐GAA, trnG‐GCC, trnG‐UCC, trnH‐GUG, trnI‐CAU, trnI‐GAU, trnK‐UUU, trnL‐CAA, trnL‐UAA, trnL‐UAG, trnfM‐CAU, trnM‐CAU, trnN‐GUU, trnP‐UGG, trnQ‐UUG, trnR‐AGC, trnR‐UCU, trnS‐GCU, trnS‐GGA, trnS‐UGA, trnT‐GGU, trnT‐UGU, trnV‐GAC, trnV‐UAC, trnW‐CCA, trnY‐GUA | |
| Photosynthesis | Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| NADH dehydrogenase | ndhA, ndhB, ndhC, ndhD, ndhE, ndF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | |
| RuBisCO large subunit | rbcL | |
| Other genes | Subunit of acetyl‐CoA‐carboxylase | accD |
| Envelope membrane protein | cemA | |
| Protease | clpP | |
| c‐type cytochrome synthase | ccsA | |
| Translational initiation factor | infA | |
| Maturase | matK | |
| Unknown function | Hypothetical chloroplast reading frames | ycf1, ycf2, ycf3, ycf4, yc15 |
Table 2.
Comparison for plastome subunits for the samples for which the inverted repeats were completely assembled.a
| Species | LSC length (bp) | SSC length (bp) | IR length (bp) | GC content (%) | Protein‐coding genes | rRNAs | tRNAs |
|---|---|---|---|---|---|---|---|
| Allantoma decandra | 85,269 | 18,738 | 27,618 | 36.9 | 81 | 4 | 30 |
| A. lineata | 85,119 | 18,756 | 27,635 | 36.9 | 81 | 4 | 30 |
| Bertholletia excelsa | 85,840 | 18,950 | 27,841 | 36.4 | 81 | 4 | 30 |
| Corythophora amapaensis | 85,861 | 18,778 | 27,638 | 36.7 | 81 | 4 | 30 |
| C. labriculata | 85,673 | 18,759 | 27,594 | 36.7 | 81 | 4 | 30 |
| Couratari macrosperma | 83,785 | 18,728 | 27,614 | 37.0 | 81 | 4 | 30 |
| C. stellata | 85,547 | 18,491 | 27,576 | 36.9 | 81 | 4 | 30 |
| Eschweilera alata | 85,056 | 18,721 | 27,635 | 36.6 | 81 | 4 | 30 |
| E. caudiculata | 84,713 | 18,759 | 27,638 | 37.0 | 81 | 4 | 30 |
| E. congestiflora | 84,815 | 18,167 | 27,715 | 37.1 | 81 | 4 | 30 |
| E. integrifolia | 84,688 | 18,796 | 27,592 | 36.9 | 81 | 4 | 30 |
| E. micrantha | 85,286 | 18,719 | 27,668 | 36.8 | 81 | 4 | 30 |
| E. wachenheimii | 85,378 | 18,815 | 27,603 | 36.8 | 81 | 4 | 30 |
| Lecythis pneumatophora | 85,506 | 18,845 | 27,622 | 36.7 | 81 | 4 | 30 |
IR = inverted repeat; LSC = large single‐copy region; SSC = small single‐copy region.
Length and GC content of the large single‐copy and small single‐copy regions in partial plastomes are estimates only.
Identification of molecular markers
Within the plastome of B. excelsa we found 23 cpSSRs, 22 of which were in non‐coding regions and one in the ndhD coding region. We designed 19 primer pairs with an acceptable product length, annealing temperature, and GC content for cpSSRs located in non‐coding regions (Table 3). π exceeded the 95th percentile for nine 600‐bp windows (Fig. 2, Tables 4 and 5). Similarly, 13 windows were over the 95th percentile for LD (Fig. 2, Tables 4 and 5), indicating high phylogenetic signal. Although most of the informative windows were in non‐coding regions, two consecutive regions were positioned in the ycf1 gene. Six windows contained both high π and LD. As expected, high π and greater LD largely agreed. Based on the rank of the windows obtained for π, we developed primers for the following regions (ordered from high to low π): ycf1, rpl16‐rps3, psbM‐trnD, ccsA‐ndhD, trnG‐psaB, petD‐rpoA, psbZ‐trnfM, trnE‐trnT, and trnT‐psbD (Table 6).
Table 3.
Primers for the amplification of simple sequence repeats in the plastome of Bertholletia excelsa. All primer pairs amplify non‐coding sequences with the exception of ndhD.a
| Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) | Repeat unit | Location | Region | No. of repeats | Product size (bp) |
|---|---|---|---|---|---|---|
| CCAAAATCATGAACTAACCCCCA | ACCAAGAGGGCGTTATTGCT | A | 396–409 | trnH‐psbA | 14 | 226 |
| TGAAGTCGTGTTGCTGAGATCT | CTGTTGATAAGTTTGCCGAGGT | C | 3686–3702 | trnK intron | 17 | 197 |
| GAGGTTTTCTCCTCGGACGG | ACCACTCATTAAACGAAATGCCT | A | 5680–5691 | rps16 intron | 12 | 244 |
| GTCCACTCAGCCATCTCTCC | AGCCCGGCCATAGGAATAAA | AAAG | 9396–9407 | trnS‐trnG | 3 | 297 |
| TTTATTCCTATGGCCGGGCT | TGCATTGTTTAAGAATCCATAGTTTCA | A | 9769–9780 | trnS‐trnG | 12 | 246 |
| TTTTCCCCACACTTCCCCTC | TGTCCGGTCATTTGATTTGGT | A | 17,925–17,938 | rps2‐rpoC2 | 14 | 192 |
| AAGAGAGGAGAAGTTTTTAGGCA | CCTTACCACTCGGCCATGTC | A | 29,392–29,403 | rpoB‐trnC | 12 | 232 |
| GGGATGCGAGAAAGAGACTT | CAAAAGTATATCTTTCTACGGGTCG | AAAG | 34,775–34,786 | trnT‐psbD | 3 | 250 |
| TACCGGTTTTCAAGACCGGG | TCACAAATGGGCATGCTGGA | AAAAT | 38,160–38,174 | trnS‐psbZ | 3 | 201 |
| ACCCATCAATCATTCGATTCGT | GAAAGATCTTTCCTTGGGGGA | AAAG | 47,627–47,638 | ycf3‐trnS | 3 | 168 |
| No suitable primers found | No suitable primers found | AAAT | 49,610–49,625 | trnT‐trnL | 4 | NA |
| No suitable primers found | No suitable primers found | AATT | 50,016–50,027 | trnT‐trnL | 3 | NA |
| CCACTGAACAAGGGAGAGCC | ACCAAGGCAAACCCATGGAA | AAAATT | 75,475–75,492 | clpP‐psbB | 3 | 128 |
| TGAATCACTGCTTTTCTTTGACTCT | AGGCGGTTCTCGAAAGAAGA | AAAAT | 77,155–77,169 | psbB‐psbT | 3 | 183 |
| TTCAATCTCGGGATTCTTTGAGA | TCGCCTGCGAAAACTTAACT | A | 85,073–85,085 | rpl16‐trnH | 13 | 246 |
| TCGATCAATCCCTTTGCCCT | CGTACTCCTCGCTCAATGAGA | AAAT | 102,172–102,183 | rps12‐trnV | 3 | 248 |
| TGGAGCACCTAACAACGCAT | AGACCTCCGGGAAAAGCATG | A | 106,208–106,219 | trnL intron | 12 | 119 |
| AGAGTAAACACAAGATACAAGGGT | GTGGGTTAGGTCAATCGGGA | AACTT | 117,345–117,359 | rpl32‐trnL | 3 | 194 |
| AGTCAACGTCAAAATTAATGAATGGT | AGGTTGAACGCGAGCGATAT | AT | 117,609–117,622 | rpl32‐trnL | 7 | 177 |
| AAATAACTCCCGCGGTCCAG | GCTTCTCTTGCATTACCGGG | AAAT | 119,729–119,740 | ndhD | 3 | 240 |
| No suitable primers found | No suitable primers found | AAT | 122,820–122,831 | ndhG‐ndhI | 4 | NA |
| No suitable primers found | No suitable primers found | AAT | 122,843–122,854 | ndhG‐ndhI | 4 | NA |
| AACCCGCTTCAAGCCATGAT | AAACGGCTTATAAATTCGCAGT | AATC | 125,271–125,282 | ndhA intron | 3 | 130 |
NA = not applicable.
Sequences have been deposited to GenBank (BioProject SUB2740669).
Figure 2.
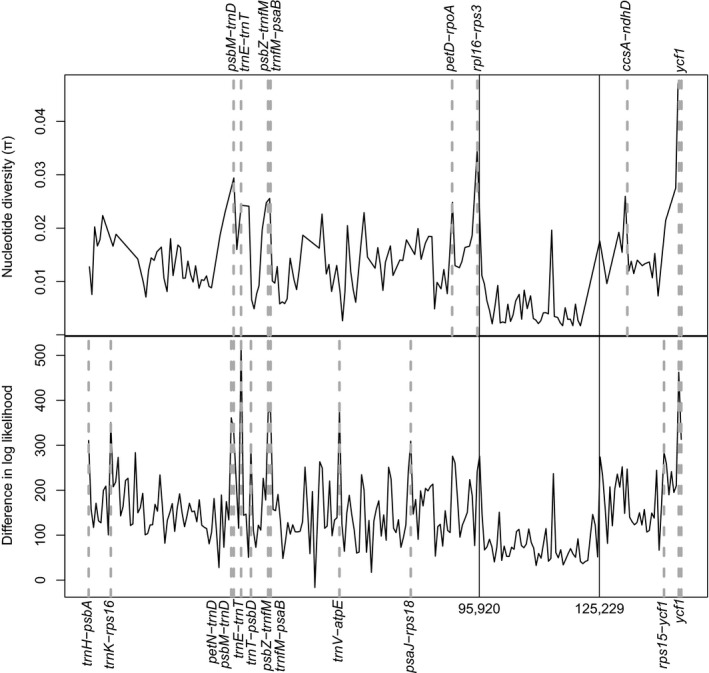
Sliding 600‐site window analyses on the Lecythidaceae plastome alignment of 24 species showing nucleotide diversity (π) (top) and alignment site‐wise differences in log‐likelihood (LD) calculated from the chloroplast topology versus the average scores of 1000 random trees (bottom). Regions with π and LD above the 95th percentiles are indicated with dashed lines. Continuous vertical lines indicate the boundaries, from left to right, among the large single copy, the inverted repeat, and the small single copy.
Table 4.
Regions of the plastome alignment (windows of 600 sites) with significantly high (above the 95th percentile) nucleotide diversity and/or site‐wise log‐likelihood score differences.b
| Location in the alignment | Bertholletia plastome location | Closest flanking expressed region | Region | π | LD | |
|---|---|---|---|---|---|---|
| 5′ | 3′ | |||||
| 1–600 | 1–490 | trnH | psbA | LSC | a | |
| 5401–6000 | 4885–5373 | trnK‐UUU | rps16 | LSC | a | |
| 34,801–35,400 | 30,925–31,450 | petN | trnD‐GUC | LSC | a | |
| 35,401–36,000 | 31,451–31,967 | psbM | trnD‐GUC | LSC | a | a |
| 37,201–37,800 | 33,027–33,573 | trnE‐UUC | trnT‐GGU | LSC | a | a |
| 39,601–40,200 | 34,893–35,433 | trnT‐GGU | psbD | LSC | a | |
| 43,801–44,400 | 38,798–39,254 | psbZ | trnfM‐CAU | LSC | a | a |
| 44,401–45,000 | 39,255–39,744 | trnfM‐CAU | psaB | LSC | a | a |
| 61,201–61,800 | 54,771–55,275 | trnV‐UAC | atpE | LSC | a | |
| 78,601–79,200 | 70,230–70,771 | psaJ | rps18 | LSC | a | |
| 89,401–90,000 | 80,536–81,103 | petD | rpoA | LSC | a | |
| 95,401–96,000 | 85,455–85,906 | rpl16 | rps3 | LSC | a | |
| 131,401–132,000 | 119,237–119,759 | ccsA | ndhD | SSC | a | |
| 140,401–141,000 | 127,827–128,402 | rps15 | ycf1 | SSC | a | |
| 144,001–144,600 | 131,283–131,868 | ycf1 | ycf1 | SSC | a | a |
| 144,601–145,200 | 131,869–132,446 | ycf1 | ycf1 | SSC | a | a |
π = nucleotide diversity (see main text); LD = log‐likelihood score differences; LSC = large single copy; SSC = small single copy.
Signifies regions with high (above the 95th percentile) nucleotide diversity or site‐wise log‐likelihood score differences.
Coding regions are indicated in windows that have the same 5ʹ‐ and 3ʹ‐expressed flanking regions in column 3. Notice that no regions are reported for the inverted repeat (IR). Coordinates are given on the alignment and the Bertholletia excelsa plastome that are assembled with the standard LSC‐SSC‐IR structure.
Table 5.
Nucleotide diversity and differences in log‐likelihood scores of the informative windows identified in this study and of previously proposed barcode markers
| Regiona | πb | LDb |
|---|---|---|
| ccsA‐ndhD | 0.0258 | 247.12 |
| matK | 0.0153 | 136.92 |
| petD‐rpoA | 0.0246 | 260.79 |
| petN‐trnD | 0.0228 | 361.07 |
| psaJ‐rps18 | 0.0176 | 309.10 |
| psbM‐trnD | 0.0292 | 330.41 |
| psbZ‐trnfM | 0.0246 | 373.97 |
| rbcL | 0.0105 | 95.03 |
| rpl16‐rps3 | 0.0345 | 275.89 |
| rpoB | 0.0097 | 120.53 |
| rpoC1 | 0.0103 | 178.60 |
| rps15‐ycf1 | 0.0212 | 284.57 |
| trnE‐trnT | 0.0241 | 522.51 |
| trnfM‐psaB | 0.0254 | 375.76 |
| trnH‐psbA | 0.0126 | 310.47 |
| trnK‐rps16 | 0.0164 | 350.44 |
| trnL | 0.0106 | 192.27 |
| trnT‐psbD | 0.0239 | 291.15 |
| trnV–atpE | 0.0128 | 379.77 |
| ycf1 (1) | 0.0273 | 462.53 |
| ycf1 (2) | 0.0469 | 313.12 |
π = nucleotide diversity; LD = differences in log‐likelihood scores.
Informative windows identified in this study are indicated in bold.
High values (above the 95th percentile) for π and LD are indicated in bold.
Table 6.
Primer sequences designed to amplify the 10 most polymorphic Lecythidaceae plastome regions, as sorted by decreasing nucleotide diversity
| Window in the alignment | π | Region | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) | Length (bp)a |
|---|---|---|---|---|---|
| 144,103–145,487 | 0.04691 | ycf1(1) | AGAACCTTTGATTATGTCTCGACG | AGAGACATGCTATAAAAATAGCCCA | 1186 |
| 95,034–95,741 | 0.03446 | rpl16‐rps3 | AGAGTTTCTTCTCATCCAGCTCC | GCTTAGTGTGTGACTCGTTGG | 1014 |
| 35,585–36,413 | 0.02920 | psbM‐trnD | CCGTTCTTTCTTTTCTATAACCTACCC | ACGCTGGTTCAAATCCAGCT | 1093 |
| 143,235–144,102 | 0.02733 | ycf1(2) | TGATTCGAATCTTTTAGCATTAKAACT | KCGTCGAGACATAATCAAAGGT | 1189 |
| 131,180–132,054 | 0.02576 | ccsA‐ndhD | CCGAGTGGTTAATAATGCACGT | GCTTCTCTTGCATTACCGGG | 1180 |
| 44,398–45,132 | 0.02537 | trnG‐psaB | TCGATYCCCGCTATCCGCC | GCCAATTTGATTCGATGGAGAGA | 883 |
| 89,032–89,688 | 0.02464 | petD‐rpoA | TGGGAGTGTGTGACTTGAACT | TGACCCATCCCTTTAGCCAA | 824 |
| 43,412–44,397 | 0.02456 | psbZ‐trnfM | TCCAATTGRCTGTTTTTGCATTAATTG | CCTTGAGGTCACGGGTTCAA | 706 |
| 37,444–38,345 | 0.02409 | trnE‐trnT | AGACGATGGGGGCATACTTG | CCACTTACTTTTTCTTTTGTTTGTTGA | 1324 |
| 38,346–40,085 | 0.02391 | trnT‐psbD | GGCGTAAGTCATCGGTTCAA | CCCAAAGCGAAATAGGCACA | 1717 |
π = nucleotide diversity.
The product size (length) references the Bertholletia excelsa plastome.
Phylogenetics of the plastomes and the developed markers
The ML analysis of the plastome alignment for Lecythidaceae (145,487 sites) yielded a fully resolved phylogeny with high BS for all clades (Fig. 3). Of the genera in which the sampling included multiple species, Eschweilera and Lecythis Loefl. were polyphyletic, whereas Allantoma Miers, Corythophora R. Knuth, Couratari Aubl., and Gustavia L. were monophyletic (Bertholletia is monospecific, and only one species each of Couroupita Aubl., Cariniana Casar., and Grias L. were included in the analysis). The trees obtained from individual markers with high π had an average BS of 73 throughout their nodes, whereas the trees obtained from two or more concatenated regions had an average BS of 89 (Fig. 4, Appendix S1). None of the gene trees, single or combined (Appendix S1), recovered the topology obtained using the complete plastome matrix (none of the gene trees obtained an RF = 0; Fig. 5). In general, matrices with concatenated markers (mean RF = 6) outperformed single markers (mean RF = 13.8; Fig. 5).
Figure 3.
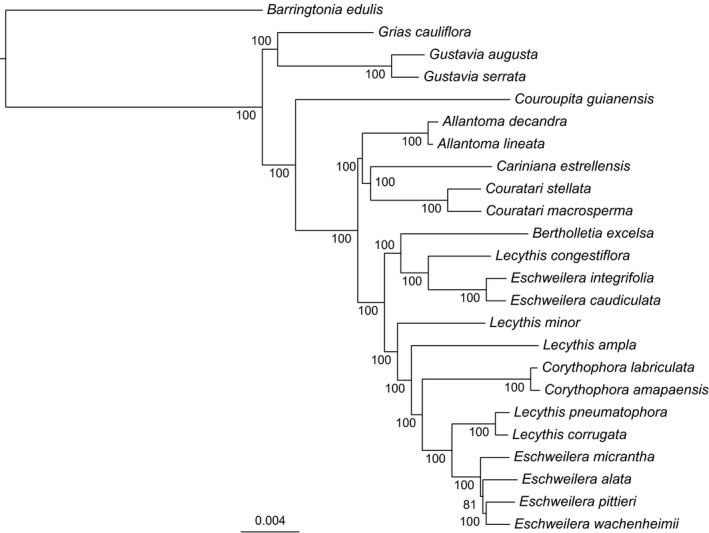
Maximum likelihood phylogeny inferred from plastomes of 23 Neotropical Lecythidaceae. Numbers at nodes indicate bootstrap support.
Figure 4.
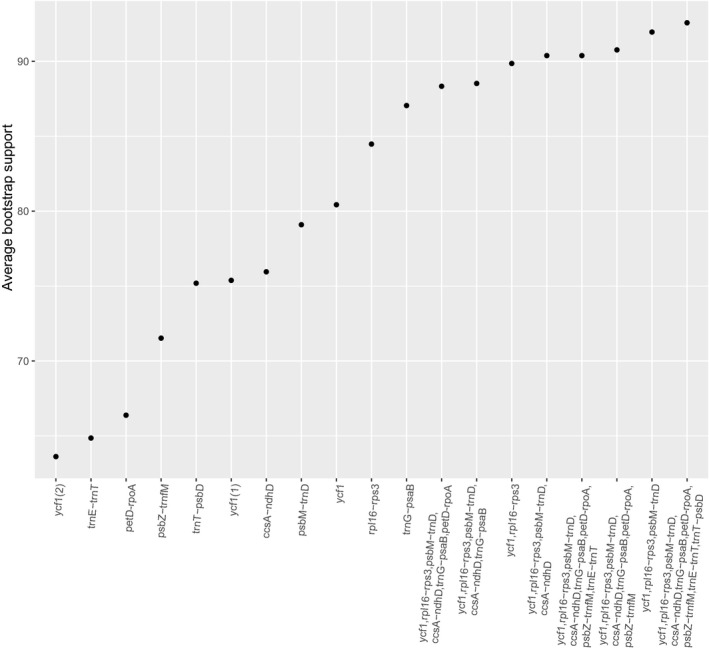
Average bootstrap support for trees inferred from either independent or concatenated regions with high nucleotide diversity, sorted in ascending order.
Figure 5.
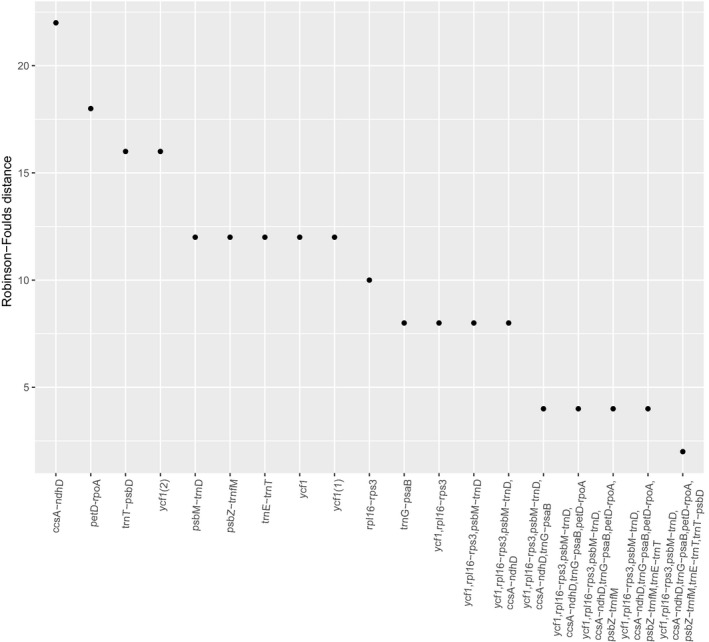
Robinson–Foulds distances (RF) for trees inferred from either independent or concatenated regions with high nucleotide diversity, sorted in descending order. Lower RF distances, which measure the number of different taxa bipartitions from the complete plastome topology, indicate better accuracy.
DISCUSSION
Genetic markers from the Lecythidaceae plastome
We are publishing the first full plastome for Lecythidaceae, including high‐depth coverage of the Brazil nut tree (B. excelsa) and 23 draft genomes representing all Lecythidoideae genera and a Paleotropical outgroup taxon. We found no significant gene losses or major rearrangements when the plastome of B. excelsa was compared with that of C. sinensis var. pubilimba, a closely related plastome (Theaceae).
We inferred a robust backbone phylogeny for Lecythidoideae using the 24 aligned plastomes. All nodes in our topology had 100% BS except for a node that connects three closely related species of Eschweilera (Fig. 3). The topology agreed with previous but weakly supported (<50% BS) Lecythidaceae phylogenies based on chloroplast and nuclear ITS sequences (Mori et al., 2007; Huang et al., 2015), indicating that Eschweilera and Lecythis are polyphyletic. Although the polyphyly of these two genera is well supported with all available data, some inferred species‐level relationships may change with increased taxonomic sampling and the inclusion of nuclear genomic data.
We measured π and a proxy for phylogenetic signal using an LD modified from Walker et al. (2017). These calculations helped us to evaluate the performance of specific chloroplast regions as potential phylogenetic markers. The core plant DNA barcodes matK and rbcL did not exhibit high π or LD in our analysis (Table 5). Of the secondary plastome barcodes mentioned in the literature (rpoC1, rpoB, trnL, and psbA‐trnH; Kress et al., 2005; Lahaye et al., 2008; Hollingsworth et al., 2009; Li et al., 2011), only psbA‐trnH showed high LD (Table 5), although it did not exhibit exceptionally high values of π. In contrast, the regions ycf1, rpl16‐rps3, psbM‐trnD, ccsA‐ndhD, trnG‐psaB, petD‐rpoA, psbZ‐trnfM, trnE‐trnT, and trnT‐psbD displayed the highest values of π and LD and therefore outperformed all of the previously proposed plant DNA barcodes.
Phylogenetic trees calculated from concatenated marker sets (based on the π rank) outperformed single regions in terms of support (BS) and accuracy (RF; Figs. 4, 5). In fact, tree topologies using single markers deviated from the complete plastome tree (mean RF = 13.8). The most well‐performing concatenated matrix contained all 10 regions for which we developed primers. However, the combination of ycf1 and rpl16‐rps3 produced an average BS of ~90 (Fig. 4) with reasonable accuracy (RF = 4, Fig. 5); we conclude that these two regions, amplified in three PCRs (Table 6), are promising markers for DNA barcoding, phylogeny, and phylogeography in Lecythidaceae. Although barcoding efficiency in species‐rich clades (i.e., Eschweilera/Lecythis) might decline with the addition of more samples, ycf1 and rpl16‐rps3 effectively distinguished between three closely related species within the Eschweilera parvifolia Mart. ex DC. clade (see branch lengths in Appendix S1), suggesting that these markers might effectively distinguish between many other closely related species. Our results and conclusions agree with those of Dong et al. (2015), who proposed ycf1 as a universal barcode for land plants.
The 19 cpSSR markers developed for noncoding portions of the B. excelsa plastome provide a useful resource for population genetic studies. Because of their fast stepwise mutation rate relative to single‐nucleotide polymorphisms, cpSSRs can also be used for finer‐grain phylogeographic analyses (e.g., Lemes et al., 2010; Twyford et al., 2013). This may be especially useful for species that exhibit little geographic structuring across parts of their ranges. Because they are maternally transmitted and can be variable within populations, the cpSSRs may also be used to track the dispersal of seeds and seedlings relative to the maternal source trees.
Because of their high level of polymorphism and phylogenetic signal content, we anticipate using the cpDNA markers presented here to study the phylogeography of widespread Lecythidaceae species such as Couratari guianensis Aubl. and Eschweilera coriacea, which range from the Amazon basin into Central America.
Barcoding of tropical trees
The DNA barcoding of tropical trees has been useful for several applications (Dick and Kress, 2009), including community phylogenetic analyses (Kress et al., 2009), inferring the species identity of the gut content (diet) of herbivores (García‐Robledo et al., 2013), and for species identification of seedlings (Gonzalez et al., 2009). The power of DNA barcodes to discriminate among species should be high if the studied species are distantly related; for example, Kress et al. (2009) were able to discriminate 281 of 296 tree and shrub species from Barro Colorado Island using standard DNA barcodes, but they were not able to discriminate among some congeneric species in the species‐rich genera Inga Mill. (Fabaceae), Ficus L. (Moraceae), and Piper L. (Piperaceae). Gonzalez et al. (2009) encountered similar challenges with Eschweilera species in their study of trees and seedlings in Paracou, French Guiana. The latter study tested a wide range of putative DNA barcode regions (rbcLa, rpoC1, rpoB, matK, trnL, psbA‐trnH, and ITS) but did not include the markers presented in this article.
Limitations of plastome markers for phylogeny and species identification
These newly identified plastome markers are not free of limitations. First, plastome‐based phylogenies should be interpreted with caution, as they can disagree with nuclear markers and species trees as a result of introgression and/or lineage‐sorting issues (Rieseberg and Soltis, 1991; Sun et al., 2015; Vargas et al., 2017). These same processes limit the cpDNA for species identification. For example, cpDNA haplotypes of Nothofagus Blume, Eucalyptus L'Hér., Quercus L., Betula L., and Acer L. were more strongly determined by geographic location than by species identity because of the occurrence of localized introgression within these groups (Petit et al., 1993; Palme et al., 2004; Saeki et al., 2011; Premoli et al., 2012; Nevill et al., 2014; Thomson et al., 2015). To date, the occurrence of haplotype sharing in closely related Lecythidaceae species has not been examined at a large scale and it is therefore not possible to conclude to what extent introgression or incomplete lineage sorting might affect this group. We suggest that future studies utilizing cpDNA barcodes for Neotropical Lecythidaceae test species from several shared geographic localities to examine to what extent haplotypes tend to be shared among species at the same localities. Nuclear DNA markers may also be used to examine phylogenetic incongruence and to identify cases where introgression might have occurred.
DATA ACCESSIBILITY
DNA sequences have been deposited to GenBank (accession no. MF359935–MF359958 and BioProject SUB2740669). Plastome alignment, gene alignments, trees, and R code are available at https://bitbucket.org/oscarvargash/lecythidaceae_plastomes .
Supporting information
ACKNOWLEDGMENTS
The National Science Foundation (grant no. DEB 1240869 and FESD Type I 1338694 to C.W.D.) and the University of Michigan (Associate Professor Award to C.W.D.) provided financial support for this work. The authors thank Scott Mori, Gregory Stull, Caroline Parins‐Fukuchi, Joseph Walker, and three anonymous reviewers for their useful comments, as well as Scott Mori and the New York Botanical Garden for providing access to curated DNA samples of Lecythidaceae.
APPENDIX 1.
Lecythidaceae species sequenced with their voucher, assembly information, and GenBank accession number. All voucher specimens are deposited at the New York Botanical Garden Herbarium (Bronx, New York, USA).
| Species | Voucher | No. of reads | % of ref seqa | Mean coverage | No. of contigs | Average length of assembled contigs (bp) | Minimum contig length (bp) | Maximum contig length (bp) | N50 | GenBank accession no. |
|---|---|---|---|---|---|---|---|---|---|---|
| Allantoma decandra (Ducke) S. A. Mori, Ya Y. Huang & Prance | Mori 25640 | 527,449 | 99.20 | 213 | 9 | 157,957 | 1052 | 42,447 | 22,733 | MF359949 |
| A. lineata (Mart. & O. Berg) Miers | Chevalier 10101 | 697,746 | 99.60 | 295 | 8 | 158,449 | 400 | 34,633 | 32,463 | MF359941 |
| Barringtonia edulis Seem. | Tsou 1552 | 519,377 | 96.10 | 230 | 10 | 152,805 | 2636 | 46,991 | 32,608 | MF359956 |
| Bertholletia excelsa Bonpl. | Mori 25637 | 1,036,874 | 100 | 646 | 14 | 160,472 | 461 | 160,472 | 160,472 | MF359948 |
| Cariniana estrellensis (Raddi) Kuntze | Nee 52828 | 759,042 | 85 | 292 | 70 | 130,037 | 278 | 22,237 | 3803 | MF359938 |
| Corythophora amapaensis Pires ex S. A. Mori & Prance | Mori 24148 | 690,545 | 99.60 | 302 | 4 | 159,222 | 6643 | 75,002 | 42,750 | MF359955 |
| C. labriculata (Eyma) S. A. Mori & Prance | Mori 25518 | 606,728 | 99.60 | 260 | 5 | 158,819 | 6596 | 74,896 | 42,691 | MF359946 |
| Couratari macrosperma A. C. Sm. | Janovec 2506 | 340,696 | 99 | 144 | 9 | 156,981 | 1107 | 45,975 | 42,740 | MF359944 |
| C. stellata A. C. Sm. | Mori 24093 | 493,777 | 99.40 | 211 | 6 | 158,312 | 1374 | 109,344 | 109,344 | MF359936 |
| Couroupita guianensis Aubl. | Mori 25516 | 503,417 | 96.50 | 314 | 12 | 154,792 | 1071 | 47,693 | 18,977 | MF359935 |
| Eschweilera alata A. C. Sm. | Prévost 4607 | 851,683 | 100 | 358 | 2 | 158,981 | 61,051 | 97,930 | 97,929 | MF359940 |
| E. caudiculata R. Knuth | Cornejo 8185 | 273,053 | 98.30 | 116 | 11 | 156,630 | 1117 | 37,154 | 21,598 | MF359957 |
| E. integrifolia (Ruiz & Pav. ex Miers) R. Knuth | Cornejo 8211 | 440,144 | 99 | 187 | 9 | 157,206 | 1105 | 42,497 | 21,591 | MF359942 |
| E. micrantha (O. Berg) Miers | Mori 25410 | 289,775 | 98.90 | 120 | 11 | 157,890 | 1143 | 42,551 | 18,021 | MF359958 |
| E. pittieri R. Knuth | Cornejo 8208 | 160,625 | 97 | 166 | 8 | 154,547 | 551 | 74,876 | 24,636 | MF359954 |
| E. wachenheimii (Benoist) Sandwith | Prévost 4252 | 367,631 | 98.90 | 151 | 11 | 157,757 | 1179 | 37,141 | 21,748 | MF359939 |
| Grias cauliflora L. | Aguilar 7961 | 520,480 | 94.90 | 326 | 41 | 150,768 | 314 | 28,189 | 8102 | MF359952 |
| Gustavia augusta L. | Mori 24255 | 761,640 | 95.50 | 476 | 33 | 152,601 | 358 | 28,353 | 11,657 | MF359943 |
| G. serrata S. A. Mori | Cornejo 8184 | 534,143 | 98.90 | 334 | 10 | 157,746 | 1035 | 35,586 | 22,762 | MF359947 |
| Lecythis ampla Miers | Cornejo 8229 | 606,518 | 94.10 | 241 | 39 | 149,464 | 348 | 28,759 | 6527 | MF359951 |
| L. congestiflora Benoist | Molino 2019 | 1,073,567 | 96.90 | 405 | 27 | 154,882 | 302 | 21,264 | 11,580 | MF359937 |
| L. corrugata Poit. | Mori 24265 | 544,831 | 90.70 | 243 | 50 | 143,859 | 317 | 28,741 | 4496 | MF359950 |
| L. minor Jacq. | Tsou 1542 | 666,355 | 94.80 | 416 | 26 | 151,568 | 354 | 31,188 | 14,768 | MF359945 |
| L. pneumatophora S. A. Mori | Mori 25728 | 690,202 | 99.50 | 301 | 4 | 158,832 | 11,782 | 75,019 | 49,184 | MF359953 |
Percentage of the sequence recovered in relation to Bertholletia excelsa.
Thomson, A. M. , Vargas O. M., and Dick C. W.. 2018. Complete plastome sequences from Bertholletia excelsa and 23 related species yield informative markers for Lecythidaceae. Applications in Plant Sciences 6(5): e1151.
LITERATURE CITED
- Chambers, J. Q. , Higuchi N., and Schimel J. P.. 1998. Ancient trees in Amazonia. Nature 391: 135–136. [Google Scholar]
- Dexter, K. G. , Pennington T. D., and Cunningham C. W.. 2010. Using DNA to assess errors in tropical tree identifications: How often are ecologists wrong and when does it matter? Ecological Monographs 80: 267–286. [Google Scholar]
- Dick, C. W. , and Kress W. J.. 2009. Dissecting tropical plant diversity with forest plots and a molecular toolkit. BioScience 59: 745–755. [Google Scholar]
- Dong, W. , Xu C., Li C., Sun J., Zuo Y., Shi S., Cheng T., et al. 2015. ycf1, the most promising plastid DNA barcode of land plants. Scientific Reports 5: 8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Robledo, C. , Erickson D. L., Staines C. L., Erwin T. L., and Kress W. J.. 2013. Tropical plant‐herbivore networks: Reconstructing species interactions using DNA barcodes. PLoS ONE 8: e52967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, M. A. , Baraloto C., Engel J., Mori S. A., Pétronelli P., Riéra B., Roger A., et al. 2009. Identification of Amazonian trees with DNA barcodes. PLoS ONE 4: e7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth, P. M. , Forrest L. L., Spouge J. L., Hajibabaei M., Ratnasingham S., van der Bank M., Chase M. W., et al. 2009. A DNA barcode for land plants. Proceedings of the National Academy of Sciences, USA 106: 12794–129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. Y. , Mori S. A., and Kelly L. M.. 2015. Toward a phylogenetic‐based generic classification of Neotropical Lecythidaceae–I. Status of Bertholletia, Corythophora , Eschweilera and Lecythis. Phytotaxa 203: 85–121. [Google Scholar]
- Katoh, K. , Misawa K., Kuma K., and Miyata T.. 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse, M. , Moir R., Wilson A., Stones‐Havas S., Cheung M., Sturrock S., Buxton S., et al. 2012. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress, W. J. , Wurdack K. J., Zimmer E. A., Weigt L. A., and Janzen D. H.. 2005. Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences, USA 102: 8369–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress, W. J. , Erickson D. L., Jones F. A., Swenson N. G., Perez R., Sanjur O., and Bermingham E.. 2009. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proceedings of the National Academy of Sciences, USA 106: 18621–18626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye, R. , van der Bank M., Bogarin D., Warner J., Pupulin F., Gigot G., Maurin O., et al. 2008. DNA barcoding the floras of biodiversity hotspots. Proceedings of the National Academy of Sciences, USA 105: 2923–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemes, M. R. , Dick C. W., Navarro C., Lowe A. J., Cavers S., and Gribel R.. 2010. Chloroplast DNA microsatellites reveal contrasting phylogeographic structure in mahogany (Swietenia macrophylla King, Meliaceae) from Amazonia and Central America. Tropical Plant Biology 3: 40–49. [Google Scholar]
- Li, D.‐Z. , Gao L.‐M., Li H.‐T., Wang H., Ge X.‐J., Liu J.‐Q., Chen Z.‐D., et al. 2011. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proceedings of the National Academy of Sciences, USA 108: 19641–19646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , and Rozas J.. 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lohse, M. , Drechsel O., and Bock R.. 2007. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high‐quality custom graphical maps of plastid and mitochondrial genomes. Current Genetics 52: 267–274. [DOI] [PubMed] [Google Scholar]
- Martin, M. 2011. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Mayer, C. 2010. Phobos. Website http://www.rub.de/spezzoo/cm/cm_phobos.htm [accessed 1 February 2017].
- Mori, S. A. 2017. The Lecythidaceae pages. Website http://sweetgum.nybg.org/science/projects/lp/ [accessed 1 February 2017].
- Mori, S. A. , and Prance G. T.. 1987. A guide to collecting Lecythidaceae. Annals of the Missouri Botanical Garden 74: 321–330. [Google Scholar]
- Mori, S. A. , and Prance G. T.. 1990. Lecythidaceae. Part II. The zygomorphic‐flowered New World genera (Couroupita, Corythophora, Bertholletia, Couratari, Eschweilera, & Lecythis) In Kubitzki K. and Renner S. [eds.], Flora Neotropica Monographs, vol. 21, part 2. New York Botanical Garden, Bronx, New York, USA. [Google Scholar]
- Mori, S. A. , Tsou C. H., Wu C. C., Cronholm B., and Anderberg A. A.. 2007. Evolution of Lecythidaceae with an emphasis on the circumscription of Neotropical genera: Information from combined ndhF and trnL‐F sequence data. American Journal of Botany 94: 289–301. [DOI] [PubMed] [Google Scholar]
- Mori, S. A. , Kiernan E. A., Smith N. P., Kelley L. M., Huang Y.‐Y., Prance G. T., and Thiers B.. 2017. Observations on the phytogeography of the Lecythidaceae clade (Brazil nut family). Phytoneuron 30: 1–85. [Google Scholar]
- Nei, M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, New York, USA. [Google Scholar]
- Nevill, P. G. , Després T., Bayly M. J., Bossinger G., and Ades P. K.. 2014. Shared phylogeographic patterns and widespread chloroplast haplotype sharing in Eucalyptus species with different ecological tolerances. Tree Genetics and Genomes 10: 1079–1092. [Google Scholar]
- Palme, A. E. , Su Q., Palsson S., and Lascoux M.. 2004. Extensive sharing of chloroplast haplotypes among European birches indicates hybridization among Betula pendula, B. pubescens and B. nana . Molecular Ecology 13: 167–178. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , Claude J., and Strimmer K.. 2004. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- Petit, R. J. , Kremer A., and Wagner D. B.. 1993. Geographic structure of chloroplast DNA polymorphisms in European oaks. Theoretical and Applied Genetics: International Journal of Plant Breeding Research 87: 122–128. [DOI] [PubMed] [Google Scholar]
- Prance, G. T. , and Mori S. A.. 1979. Lecythidaceae–Part I. The actinomorphic‐flowered New World Lecythidaceae (Asteranthos, Gustavia, Grias, Allantoma, & Cariniana) In Kubitzki K. and Renner S. [eds.], Flora Neotropica Monographs, vol. 21, part 1. New York Botanical Garden, Bronx, New York, USA. [Google Scholar]
- Premoli, A. C. , Mathiasen P., Acosta M. C., and Ramos V. A.. 2012. Phylogeographically concordant chloroplast DNA divergence in sympatric Nothofagus s.s. How deep can it be? New Phytologist 193: 261–275. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2017. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Website https://www.r-project.org/ [accessed 1 March 2017]. [Google Scholar]
- Rieseberg, L. H. , and Soltis D. E.. 1991. Phylogenetic consequences of cytoplasmic gene flow in plants. Evolutionary Trends in Plants 5: 64–84. [Google Scholar]
- Robinson, D. F. , and Foulds L. R.. 1981. Comparison of phylogenetic trees. Mathematical Biosciences 53: 131–147. [Google Scholar]
- Sablok, G. , Padma Raju G. V., Mudunuri S. B., Prabha R., Singh D. P., Baev V., Yahubyan G., et al. 2015. ChloroMitoSSRDB 2.00: More genomes, more repeats, unifying SSRs search patterns and on‐the‐fly repeat detection. Database 2015: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki, I. , Dick C. W., Barnes B. V., and Murakami N.. 2011. Comparative phylogeography of red maple (Acer rubrum L.) and silver maple (Acer saccharinum L.): Impacts of habitat specialization, hybridization and glacial history. Journal of Biogeography 38: 992–1005. [Google Scholar]
- Schliep, K. P. 2011. phangorn: Phylogenetic analysis in R. Bioinformatics 27: 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder, R. , and Edwards R.. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. 2014. RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran, J. , and Holder M. T.. 2010. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26: 1569–1571. [DOI] [PubMed] [Google Scholar]
- Sun, M. , Soltis D. E., Soltis P. S., Zhu X., Burleigh J. G., and Chen Z.. 2015. Deep phylogenetic incongruence in the angiosperm Rosidae clade. Molecular Phylogenetics and Evolution 83: 156–166. [DOI] [PubMed] [Google Scholar]
- ter Steege, H. , Pitman N. C. A., Sabatier D., Baraloto C., Salomão R. P., Guevara J. E., Phillips O. L., et al. 2013. Hyperdominance in the Amazonian tree flora. Science 342: 325–342. [DOI] [PubMed] [Google Scholar]
- Thomson, A. M. , Dick C. W., and Dayanandan S.. 2015. A similar phylogeographical structure among sympatric North American birches (Betula) is better explained by introgression than by shared biogeographical history. Journal of Biogeography 42: 339–350. [Google Scholar]
- Twyford, A. D. , Kidner C. A., Harrison N., and Ennos R. A.. 2013. Population history and seed dispersal in widespread Central American Begonia species (Begoniaceae) inferred from plastome‐derived microsatellite markers. Botanical Journal of the Linnean Society 171: 260–276. [Google Scholar]
- Untergasser, A. , Cutcutache I., Koressaar T., Ye J., Faircloth B. C., Remm M., and Rozen S. G.. 2012. Primer3: New capabilities and interfaces. Nucleic Acids Research 40: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas, O. M. , Ortiz E. M., and Simpson B. B.. 2017. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high‐Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytologist 214: 1736–1750. [DOI] [PubMed] [Google Scholar]
- Walker, J. F. , Brown J. W., and Smith S. A.. 2017. Analyzing contentious relationships and outlier genes in phylogenomics. bioRxiv. https://doi.org/10.1101/115774 [DOI] [PubMed] [Google Scholar]
- Wyman, S. K. , Jansen R. K., and Boore J. L.. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20: 3252–3255. [DOI] [PubMed] [Google Scholar]
- Zerbino, D. R. , and Birney E.. 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences have been deposited to GenBank (accession no. MF359935–MF359958 and BioProject SUB2740669). Plastome alignment, gene alignments, trees, and R code are available at https://bitbucket.org/oscarvargash/lecythidaceae_plastomes .