
Fig. 6. Pathophysiological immune/inflammatory sequelae triggered by various acute brain injuries.
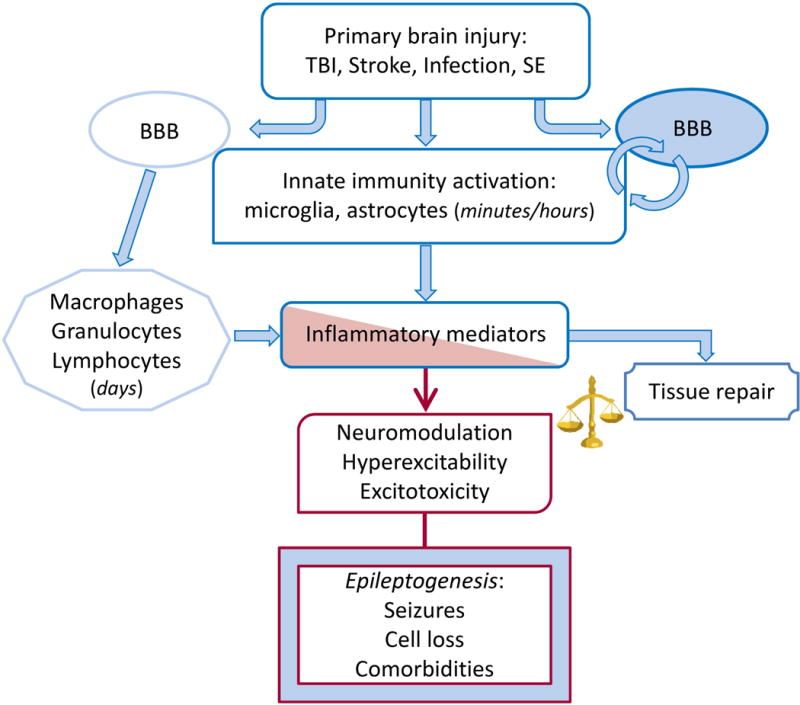
Rapid activation of brain resident innate immunity cells at the site of injury and BBB functional and structural alterations result in the generation of the inflammatory cascade in seizure-prone brain areas. Leukocytes may contribute to perpetuate the inflammatory cascade following interactions with the brain vasculature. The time-locked sequence of these events likely depends on the type of injury. Inflammatory mediators can promote brain damage and dysfunction or contribute to tissue repair depending on their levels and persistence. If the inflammatory milieu exceeds the homeostatic threshold it may lead to hyperexcitability, decrease seizure threshold and promote neuropathology thereby contributing to disease progression. These pathologic events depend on the ability of inflammatory molecules such as cytokines, DAMPs and prostaglandins to alter expression and function of gap junctions, voltage-gated or receptor-coupled ion channels thus contributing to acquired channelopathies, to modify GABA and glutamate release and re-uptake176,177,185 and to alter BBB permeability. BBB dysfunction results in albumin extravasation which compromises homeostatic astrocytic functions and induces excitatory synaptogenesis by activating the TGF-β/ALK5 signaling.310,311,320