

Abstract
About 7,000 rare, or orphan, diseases affect more than 350 million people worldwide. Although these conditions collectively pose significant health care problems, drug companies seldom develop drugs for orphan diseases due to extremely limited individual markets. Consequently, developing new treatments for often life-threatening orphan diseases is primarily contingent on financial incentives from governments, special research grants, and private philanthropy. Computer-aided drug repositioning is a cheaper and faster alternative to traditional drug discovery offering a promising venue for orphan drug research. Here, we present eRepo-ORP, a comprehensive resource constructed by a large-scale repositioning of existing drugs to orphan diseases with a collection of structural bioinformatics tools, including eThread, eFindSite and eMatchSite. Specifically, a systematic exploration of 320,856 possible links between known drugs in DrugBank and orphan proteins obtained from Orphanet reveals as many as 18,145 candidates for repurposing. In order to illustrate how potential therapeutics for rare diseases can be identified with eRepo-ORP, we discuss the repositioning of a kinase inhibitor for Ras-associated autoimmune leukoproliferative disease. The eRepo-ORP dataset is available through the Open Science Framework at https://osf.io/qdjup/.
Keywords: drug repositioning, drug repurposing, orphan diseases, rare diseases, DrugBank, Orphanet, eMatchSite, eFindSite, pocket alignment, drug-binding site alignment
Graphical abstract
Introduction
Rare diseases are conditions afflicting a small subset of people in a population, where “small” is uniquely defined by each country. For example, the United States denotes disorders affecting fewer than 200,000 patients as rare diseases, also referred to as orphan diseases. Although each of approximately 7,000 orphan conditions has a tiny number of patients, they amount to 30 million patients in the U.S., 30 million in Europe, and around 350 million globally [1]. Because pharmaceutical companies seldom develop drugs for orphan diseases due to the lack of consumers, special attention needs to be placed on treating these conditions. After the success of the Orphan Drug Act signed into law in the U.S. by President Reagan in 1983, other governments adopted similar mechanisms to facilitate orphan drug development, mostly by granting market exclusivity and reducing research and development costs [2]. These actions allow for not only sufficient financial incentives for pharmaceutical companies, but also manageable costs for non-profits. Fewer financial difficulties, various governmental inducements, increasing public awareness, together with advances in research techniques have stimulated a global interest in orphan drug development and rare disease research [3].
Certainly, without the support of quality datasets and resources, the progress in orphan drug research might not be as consistent as it has been. For instance, Orphanet, the de facto rare disease reference source, contributes quality, robust data on rare diseases as well as reliable clinical practice guidelines [4]. Most importantly, Orphanet enables researchers to share common language and information to undergo controlled scientific analysis and, ultimately, orphan drug discovery. Similar to Orphanet, the Genetic and Rare Diseases Information Center (GARD) at the National Institutes of Health provides comprehensive information regarding rare diseases and orphan drugs [5]. Last but not least, the Developing Products for Rare Diseases & Conditions section of the U.S. Food and Drug Administration (FDA) website hosts freely accessible official legal documentation regarding orphan drug development and regulations [6]. These rich resources on orphan diseases available to researchers worldwide facilitate the development of new treatments for rare conditions. For instance, a systems-level approach to find connections between existing drug products and orphan diseases, known as drug repositioning, holds a significant promise to greatly expand the repertoire of orphan drugs.
As an alternative strategy to drug discovery, compound repositioning finds new indications for existing drugs. This approach can dramatically improve the success rates by shortening the time of drug development to about 3–12 years at the reduced safety and pharmacokinetic uncertainty [7]. Repurposing of already-approved drugs would most likely bypass initial clinical trials, especially if the corresponding dosage does not exceed the maximum approved by a regulatory agency. Although efficacy tests for the new treatment are still required, an existing drug is likely to have well characterized long-term toxicity and off-target effects. Further, the magnitude of side effects may be an important determinant to repurpose a drug. For example, a drug with a high risk of significant side effects might not be appropriate when the primary goal is to maintain the quality of life of a patient, however, repurposing the same drug to treat a life-threatening disease may be acceptable.
Despite the fact that time- and cost-effective rational drug repositioning is expected to play a major role in the development of treatments for rare conditions [8], it is not trivial and poses a number of onerous challenges. It is, therefore, not surprising that most of the repositioned drugs currently on the market are the result of serendipity. Perhaps the most recognizable example is sildenafil; originally intended to treat hypertension and angina pectoris in the 1980s, it was later repurposed to erectile dysfunction as well as pulmonary arterial hypertension [9]. Another instance is memantine [10], synthesized in the 1960s as a potential agent to treat diabetes, although it was found ineffective at lowering blood sugar. Its activity against the N-methyl-D-aspartate (NMDA) receptor was discovered in the 1980s and presently, memantine is used to treat Alzheimer’s disease, vascular dementia and Parkinson’s disease [11]. These examples show that even though drug repositioning is regarded as one of the most promising strategies for translational medicine, many new indications for existing drugs have been found serendipitously. Therefore, there is a clear need to establish rational, preferably computer-guided routines for drug repositioning.
In this communication, we describe eRepo-ORP, a new resource for orphan drug research. eRepo-ORP is a drug repositioning dataset that builds on the results of a large-scale pocket matching between target sites for known drugs and those binding pockets identified in proteins linked to rare diseases. Known drugs and their macromolecular targets are extracted from DrugBank, a unique bioinformatics and cheminformatics resource providing detailed chemical, pharmacological, and structural data on drug-target associations [12], whereas proteins connected to orphan diseases are obtained from Orphanet [4]. Further, we designed a sophisticated protocol incorporating several state-of-the-art algorithms to find potential candidates for repositioning by modeling the high-quality structures of drug targets with eThread [13], comprehensively annotating their binding sites with eFindSite [14, 15], and effectively detecting similar drug-binding pockets with eMatchSite [16, 17]. In general, this approach builds on ligand-binding homology, a technique previously employed in computer-aided drug development to detect binding sites [18] and to discover potential leads through virtual screening [19, 20]. Ras-associated autoimmune leukoproliferative disease is discussed as a representative example illustrating how eRepo-ORP can be used to identify therapeutics for orphan diseases. eRepo-ORP is a large collection of knowledge-based predictions to initiate more extensive basic and clinical research focused on investigating potentially new indications for existing drugs. The complete dataset is freely available to the research community through the Open Science Framework at https://osf.io/qdjup/.
Results and Discussion
Protocol for template-based drug repositioning
eRepo-ORP is constructed based on a large-scale drug repositioning conducted with accurate, template-based techniques according to a protocol presented in Figure 1. The first phase is to generate structural data for FDA-approved drugs and their molecular targets based on information extracted from the DrugBank database (Figure 1A). Structure models of drug targets are constructed by eThread and annotated with drug-binding sites and residues by eFindSite (Figure 1B). Next, for each drug-target pair, we identify in the Protein Data Bank (PDB) [21] a globally similar template binding a ligand that is chemically similar to the DrugBank compound (Figure 1C). This holo-template is structurally superposed onto the DrugBank target (Figure 1D) and then the DrugBank compound is aligned onto the template-bound ligand (Figure 1E). This procedure produces 2,012 atomic models of drug-target complexes involving 348 unique proteins and 715 drugs (Figure 1F). The second phase is to model proteins associated with orphan diseases obtained from the Orphanet database (Figure 1G). Structure models of 922 Orphanet proteins with predicted drug-binding sites and residues (Figure 1H) are generated by a similar protocol to that used for DrugBank targets. The last phase is to identify similar binding sites in DrugBank and Orphanet models in order to reposition existing drugs. This task is accomplished by employing eMatchSite to construct local alignments for 320,856 possible pairs of DrugBank and Orphanet proteins (Figure 1I). For 18,145 pairs producing a statistically significant local alignment, a drug molecule bound to the DrugBank protein is transferred to the Orphanet target and the complex model is subjected to all-atom refinement (Figure 1J). Refined structure models are included in the eRepo-ORP database.
Figure 1.
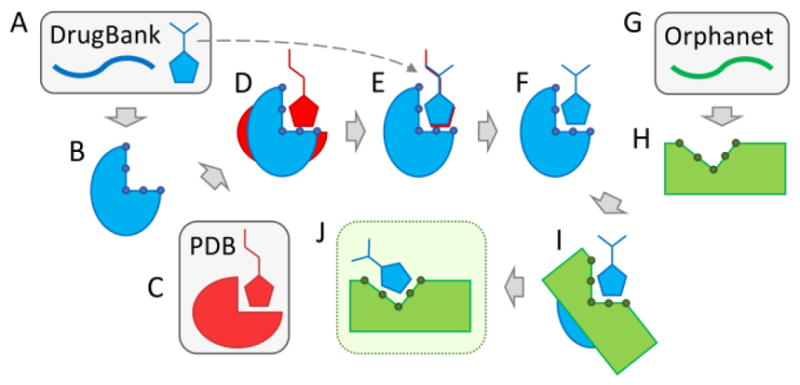
Flowchart of the drug repositioning procedure employed to construct eRepo-ORP. This protocol utilizes data from three sources, DrugBank, Protein Data Bank (PDB), and Orphanet, shown in blue, red, and green, respectively. Databases are indicated by gray boxes. (A) For a given protein sequence from DrugBank, template-based structure modeling is conducted with eThread in order to construct (B) a 3D model subsequently annotated by eFindSite with drug-binding sites and residues represented by little circles. (C) A globally similar template binding a ligand that is chemically similar to the DrugBank compound is selected from the PDB. (D) The template carrying its ligand is structurally aligned onto the DrugBank apo-structure. (E) The DrugBank compound is then aligned onto the template-bound ligand generating (F) a 3D model of the drug-target complex. (G) For a given protein sequence from Orphanet, (H) a 3D model is constructed with eThread and annotated with eFindSite. (I) A local alignment is performed for a pair of binding sites in DrugBank and Orphanet models with eMatchSite. (J) The DrugBank compound is transferred to the Orphanet model when the similarity of binding pockets in DrugBank and Orphanet models is sufficiently high and the resulting complex is refined.
Quality of structural data generated for DrugBank and Orphanet
Structure models are generated for the DrugBank and Orphanet datasets with eThread, a meta-threading approach employing state-of-the-art fold recognition. Initial models constructed by Modeller from eThread alignments are refined with ModRefiner, which performs atomic-level energy minimization in a composite physics- and knowledge-based force field improving side-chain positions and hydrogen-bonding networks. An independent assessment of the quality of protein models is carried out with ModelEvaluator utilizing the predicted secondary structure, relative solvent accessibility, residue contact map, and beta sheet structure. Statistics reported in Supplementary Table S1 show that the template-based modeling protocol employed in this study produces highly confident structure models, whose mean estimated Global Distance Test (GDT)-score [22] values are 0.71 and 0.68 for DrugBank and Orphanet proteins, respectively. In addition, the mean confidence for the top-ranked binding sites predicted in these models by eFindSite is as high as 0.87 for DrugBank and 0.82 for Orphanet targets.
The structure models of DrugBank complexes are constructed by aligning the protein and the drug onto a holo-template selected from the PDB. Supplementary Table S1 reports the mean Tanimoto coefficient (TC) [23] between the DrugBank compound and the template-bound ligand of 0.49 and the mean Template Modeling (TM)-score [24] between receptor proteins of 0.65. Note that both TC and TM-score are even higher when only those cases producing statistically significant pocket alignments are considered. These numbers clearly indicate that globally similar templates binding chemically similar ligands are selected for the majority of drug-protein pairs from DrugBank to produce highly confident complex models. Supplementary Table S1 also provides statistics for DrugBank→Orphanet pairs. Both TM-score and eMS-score values are very low for all data, basically showing that randomly selected pairs of proteins share neither global nor local structure similarity. However, considering the subset of 18,145 pairs producing statistically significant local alignments, the mean eMS-score is as high as 0.91, even though the mean TM-score is still only 0.27. These results demonstrate that the vast majority of similar binding sites included in eRepo-ORP are identified by eMatchSite in DrugBank and Orphanet proteins having unrelated global structures.
Matching DrugBank drugs to Orphanet proteins
The results of a large-scale pocket matching between DrugBank and Orphanet proteins are presented as a heat map in Figure 2. Pocket similarity is measured with eMS-score reported by eMatchSite. eMS-score ranges from 0 to 1 with values of ≥0.56 indicating statistically significant local alignments. Further, binding sites predicted by eFindSite in DrugBank and Orphanet proteins are subjected to ligand-based virtual screening employing molecular fingerprints extracted from template-bound ligands. Protein targets in each dataset in Figure 2 are clustered with respect to the chemical similarity of the top-ranked compounds selected by virtual screening. Five distinct groups of proteins marked by rounded boxes bind compounds containing nitrogen bases, carbohydrates, amino acids, fatty acids, and other molecules. Only 5.6% of 320,856 local alignments between DrugBank and Orphanet proteins are statistically significant at an eMS-score of 0.56, indicating that these pairs of pockets bind similar molecules. Although the majority of similar pockets, marked by dark spots in Figure 2, are detected between proteins binding the same type of ligands, e.g. those compounds containing nitrogen bases, similarities are detected between different groups as well. Because some DrugBank proteins bind multiple drugs, more than one drug can be repositioned to the Orphan target based on a single alignment of a pair of pockets. Specifically, eRepo-ORP comprises 31,142 unique putative complexes between DrugBank compounds and Orphanet proteins, modeled from 18,145 pairs of pockets producing statistically significant local alignments. The database can be searched with the disorder name and identification according to Orphanet, as well as the DrugBank identifier. In the following section, we discuss a representative case selected from eRepo-ORP showing a DrugBank compound that can potentially be repositioned to an Orphanet protein associated with a rare disease.
Figure 2.

Heat map visualizing all-against-all binding site matching between DrugBank and Orphanet proteins in eRepo-ORP. Binding pocket similarity is quantified with the eMS-score according to the color scale displayed in the top-left corner. DrugBank and Orphanet proteins are hierarchically clustered by the chemical similarity of their ligands with the resulting dendrograms shown on the left side and at the top of the heat map, respectively. Rounded rectangles identify distinct groups of proteins binding compounds containing nitrogen bases (NB, blue), carbohydrates (CH, red), amino acids (AA, yellow), fatty acids (FA, green), and other molecules (O, gray).
Ras-associated autoimmune leukoproliferative disease and vandetanib
Ras-associated autoimmune leukoproliferative disorder (RALD, ORPHA:268114) is a chronic, non-malignant condition characterized by monocytosis and often associated with leukocytosis, lymphoproliferation, and autoimmune phenomena [25]. RALD is linked to certain mutations in GTPase KRas (KRAS), which plays an important role in the regulation of cell proliferation promoting oncogenic events, thus it is considered a major target in anticancer drug discovery [26]. Specifically, amino acid substitutions in codons 12 and 13 of KRAS in RALD patients cause the constitutive binding of GTP and the activation of the KRAS protein inducing the Raf-MEK-ERK signaling pathway [25]. According to eRepo-ORP, KRAS produces a highly significant local alignment with protein-tyrosine kinase 6 (PTK6) implicated in the regulation of a variety of signaling pathways that control the differentiation and maintenance of normal epithelia, as well as tumor growth [27]. PTK6 is a target for vandetanib, an oral kinase inhibitor of tumor angiogenesis and tumor cell proliferation approved by the FDA to treat non-resectable, locally advanced or metastatic medullary thyroid cancer in adult patients [28].
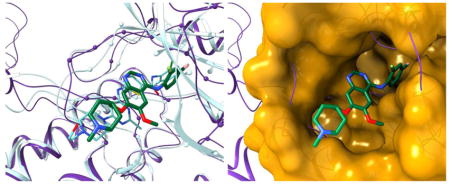
Figure 3 presents structure models of PTK6 (purple) and KRAS (gold). The model of PTK6 constructed with eThread from tyrosine-protein kinase HCK (PDB-ID: 1qcf, chain A, 42.6% sequence identity) [29] is assigned a high estimated GDT-score of 0.74. Further, vandetanib (DrugBank-ID: DB05294) was transferred to PTK6 according to the global structure alignment with cyclin-dependent kinase 6 bound to this inhibitor (PDB-ID: 2ivu, chain A, TM-score of 0.54) [30]. The final model of the vandetanib-PTK6 complex is shown in Figure 3A as solid ribbons and sticks. We selected this particular case because the vandetanib-PTK6 model was generated using the October 2016 version of the PDB and, in January 2017, a crystal structure of PTK6 kinase domain complexed with another inhibitor, dasatinib, was released (PDB-ID: 5h2u, chain A) [31]. This experimental structure superposed onto the vandetanib-PTK6 model is shown in Figure 3A as transparent ribbons and sticks. A TM-score between the PTK6 model and the experimental structure is as high as 0.92 with a Cα-RMSD of 2.3 Å. Further, the root-mean-square deviation (RMSD) calculated over dasatinib-binding residues is only 0.7 Å demonstrating that not only the backbone, but also the binding pocket is modeled with a very high accuracy. Although vandetanib and dasatinib have a low chemical similarity with a TC of only 0.15, both inhibitors have a similar shape and the modeled binding pose of vandetanib resembles the experimental conformation of dasatinib. Moreover, the top-ranked binding site predicted with 99.7% confidence by eFindSite in the PTK6 model substantially overlaps with the dasatinib-binding pocket in the experimental complex structure. The Matthews correlation coefficient (MCC) [32] between predicted and dasatinib-binding residues reported by Ligand-Protein Contacts (LPC) software is 0.62.
Figure 3.

Repositioning of vandetanib from protein-tyrosine kinase 6 (PTK6) to GTPase KRas (KRAS) according to eRepo-ORP. PTK6 and KRAS proteins are colored purple and gold, respectively, whereas ligands are colored by atom type (green/teal – carbon, blue – nitrogen, red – oxygen, yellow – sulfur, citron – chlorine, pink – fluorine, cyan – bromine). (A) Structure model of the complex between PTK6 (purple ribbons) and vandetanib (thick sticks) with predicted binding residues shown as spheres superposed onto the experimental structure of PTK6 (teal ribbons) bound to dasatinib (thin sticks). (B) Structure model of KRAS (gold ribbons) with predicted drug-binding residues shown as spheres superposed onto the experimental structure of KRAS (teal ribbons) bound to ADP (thin sticks). (C) Local superposition of PTK6 (purple ribbons) and KRAS (gold surface) according to the sequence order-independent pocket alignment by eMatchSite. Annotated binding residues in KRAS are solid, whereas the remaining surface is transparent. Vandetanib repositioned to KRAS is represented by thick sticks.
The model of KRAS was constructed from Ras-related protein Rap-1b (PDB-ID: 4m8n, chain G, 58.4% sequence identity) and assigned a high estimated GDT-score of 0.85. Although several inhibitors of KRAS are available, these compounds target the secondary binding site [33]. In Figure 3B, a GDP-bound KRAS (transparent) is superposed onto the model structure (solid). This superposition yields a high TM-score of 0.93 and a low Cα-RMSD of 1.4 Å; furthermore, the RMSD calculated over GDP-binding residues is only 1.1 Å. The top-ranked drug-binding site comprising 27 residues, annotated by eFindSite with 95.7% confidence, has an MCC against GDP-binding residues of 0.61. Despite a very low global sequence identity of 12.9% and a structure similarity with a TM-score of 0.32 between PTK6 and KRAS, eMatchSite reports a significant local similarity of their binding sites with an eMS-score of 0.99. Figure 3C shows the conformation of vandetanib repositioned from PTK6 to KRAS according to the sequence order-independent pocket alignment by eMatchSite, which results in 4.3 Å Cα-RMSD over 25 aligned residues. Repositioned vandetanib fits well into a deep cavity in the KRAS structure forming hydrogen bonds with A18, N116 and K117, aromatic interactions with F28, and hydrophobic contacts with V8 and V9. The interaction energy between vandetanib and KRAS calculated by DFIRE is −441.5, which is only slightly higher than −485.6 obtained for the vandetanib-PTK6 model. Altogether, these results suggest that the nucleotide-binding pocket of KRAS may be a suitable target for vandetanib. If so, we anticipate that the competitive binding of vandetanib to KRAS may subdue its gain-of-function caused by activating mutations, leading to the mitigation of RALD conditions.
Conclusions
In this study, we employ a collection of state-of-the-art algorithms to match, at an unprecedented scale, binding sites for known drugs with those pockets identified in proteins associated with rare diseases. Based on these data, we created eRepo-ORP, a new resource for orphan drug research. eRepo-ORP comprises 31,142 putative complexes between DrugBank compounds and Orphanet proteins exposing vast opportunities to reposition existing drugs to rare diseases. In order to illustrate how potential therapeutics for orphan diseases can be identified with eRepo-ORP, we discuss a possibility to repurpose a kinase inhibitor for Ras-associated autoimmune leukoproliferative disease. Freely available through the Open Science Framework at https://osf.io/qdjup/, eRepo-ORP provides a list of pairs of DrugBank and Orphanet proteins sorted by the matching score, structure models of DrugBank and Orphanet proteins with predicted drug-binding sites, sequence and secondary structure profiles, structure models of DrugBank complexes annotated with energy scores, and complex models of DrugBank drugs repositioned to Orphanet proteins with the corresponding energy scores. We expect that eRepo-ORP will prove valuable to orphan disease research by providing a robust, rational drug repositioning component.
Materials and Methods
DrugBank dataset
FDA-approved drugs whose molecular weight is in the range of 150–550 Da and for which at least one target protein is known were selected from DrugBank [12]. Target structures composed of 50–999 amino acids were modeled with eThread, a template-based structure prediction algorithm [13]. eThread employs meta-threading with HH-suite [34], RaptorX [35], and SparksX [36] to select structure templates in the non-redundant and representative subset of the PDB. Comparative structure modeling in eThread is carried out with Modeller [37] based on the top-ranked template and incorporating secondary structure restraints from PSIPRED [38]. Initial models assembled by Modeller were refined with ModRefiner [39]. Finally, each model was assigned an estimated GDT-score by ModelEvaluator [40].
In the next step, drug-binding pockets were predicted by eFindSite [14] in confidently modeled target proteins whose estimated GDT-score is ≥0.4. Pockets assigned by eFindSite a high and moderate confidence were then subjected to fingerprint-based virtual screening [15]. Each target pocket was screened against a library containing drug molecules from DrugBank [12] and a background collection of 244,659 non-redundant compounds selected from the ZINC database [41]. Only those drug-target pairs for which the drug molecule was ranked within the top 10% of the screening library were retained. Further, we devised a two-step alignment protocol to position drug compounds within the predicted binding pockets for each drug-target pair. First, holo-templates selected by eFindSite were structurally aligned onto the target protein with Fr-TM-align [42] and then the drug molecule was superposed onto the template-bound ligand according to the chemical alignment constructed by kcombu [43].
Orphanet dataset
Genes associated with rare disorders were obtained from Orphanet [4] and the sequences of gene products were downloaded from UniProt [44]. Subsequently, for those protein sequences composed of 50–999 amino acids, we employed a protocol described above for the DrugBank dataset to conduct comparative structure modeling with eThread [13] followed by drug-binding pocket prediction by eFindSite [14]. Finally, only protein structures with an estimated GDT-score of ≥0.4 having binding sites predicted with a high and moderate confidence were retained.
Pocket matching with eMatchSite
All-against-all matching of drug-binding pockets in DrugBank and Orphanet proteins was conducted with eMatchSite [16, 17]. eMatchSite constructs sequence order-independent local alignments of pocket residues by solving the assignment problem with machine learning and the Hungarian algorithm [45]. Subsequently, the local alignment is assigned a similarity score, called the eMS-score, calculated based on the overlap of various physicochemical features and evolutionary profiles. eMS-score ranges from 0 for completely dissimilar pockets to 1 for identical pockets, with an optimized threshold of 0.56 accurately distinguishing between pockets binding similar and dissimilar molecules [16]. eMatchSite has been benchmarked against a number of established datasets; a comprehensive recap of its performance is presented in Supplementary Text S1. In addition to calculating the similarity score, eMatchSite superposes two pockets according to the constructed local alignments, so that a drug molecule bound to one pocket can be directly transferred to the other binding site. In this study, we use this feature of eMatchSite to transfer drugs bound to DrugBank target to binding sites in Orphanet proteins. In the last step, the constructed complexes of drugs repositioned to Orphanet proteins are rebuilt with Modeller in order to refine drug-target interactions and eliminate steric clashes. The quality of the final complex models is assessed by a knowledge-based statistical energy function for protein-ligand complexes with the Distance-scaled Finite Ideal-gas REference (DFIRE) potential [46]. Specific interactions between drugs and proteins, such as hydrogen bonds, hydrophobic and aromatic contacts, are identified by LPC [47], LigPlot+ [48] and eAromatic [49].
Supplementary Material
Highlights.
Rational drug repositioning is expected to play a major role in the development of treatments for orphan diseases.
eMatchSite is a new computer program to guide structure-based drug repurposing efforts.
State-of-the-art algorithms, eThread, eFindSite and eMatchSite, are employed to construct eRepo-ORP, a new resource for orphan drug research.
eRepo-ORP builds on 320,856 local alignments between target sites for known drugs from DrugBank and proteins associated with rare diseases from Orphanet.
eRepo-ORP exposes a vast number of new opportunities to combat orphan diseases with existing drugs.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119524.
Abbreviations used
- DFIRE
Distance-scaled Finite Ideal-gas REference
- eMS-score
eMatchSite score
- FDA
Food and Drug Administration
- GARD
Genetic and Rare Diseases Information Center
- GDT
Global Distance Test
- KRAS
GTPase KRas
- LPC
Ligand-Protein Contacts
- MCC
Matthews correlation coefficient
- NMDA
N-methyl-D-aspartate
- PDB
Protein Data Bank
- PTK6
protein-tyrosine kinase 6
- RALD
Ras-associated autoimmune leukoproliferative disorder
- RMSD
root-mean-square deviation
- TC
Tanimoto coefficient
- TM-score
Template Modeling score
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rare diseases: Facts and statistics https://globalgenes.org/rare-diseases-facts-statistics/
- 2.Seoane-Vazquez E, Rodriguez-Monguio R, Szeinbach SL, Visaria J. Incentives for orphan drug research and development in the United States. Orphanet J Rare Dis. 2008;3:33. doi: 10.1186/1750-1172-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groft SC. Rare diseases research: expanding collaborative translational research opportunities. Chest. 2013;144:16–23. doi: 10.1378/chest.13-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orphanet: An online database of rare diseases and orphan drugs http://www.orpha.net/
- 5.The Genetic and Rare Diseases Information Center.
- 6.Developing Products for Rare Diseases & Conditions.
- 7.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–83. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 8.Sardana D, Zhu C, Zhang M, Gudivada RC, Yang L, Jegga AG. Drug repositioning for orphan diseases. Brief Bioinform. 2011;12:346–56. doi: 10.1093/bib/bbr021. [DOI] [PubMed] [Google Scholar]
- 9.Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, et al. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8:47–52. [PubMed] [Google Scholar]
- 10.Witt A, Macdonald N, Kirkpatrick P. Memantine hydrochloride. Nat Rev Drug Discov. 2004;3:109–10. doi: 10.1038/nrd1311. [DOI] [PubMed] [Google Scholar]
- 11.Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT, et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr Alzheimer Res. 2012;9:746–58. doi: 10.2174/156720512801322564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brylinski M, Lingam D. eThread: a highly optimized machine learning-based approach to meta-threading and the modeling of protein tertiary structures. PLoS One. 2012;7:e50200. doi: 10.1371/journal.pone.0050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brylinski M, Feinstein WP. eFindSite: improved prediction of ligand binding sites in protein models using meta-threading, machine learning and auxiliary ligands. J Comput Aided Mol Des. 2013;27:551–67. doi: 10.1007/s10822-013-9663-5. [DOI] [PubMed] [Google Scholar]
- 15.Feinstein WP, Brylinski M. eFindSite: Enhanced fingerprint-based virtual screening against predicted ligand binding sites in protein models. Mol Inform. 2014;33:135–50. doi: 10.1002/minf.201300143. [DOI] [PubMed] [Google Scholar]
- 16.Brylinski M. eMatchSite: sequence order-independent structure alignments of ligand binding pockets in protein models. PLoS Comput Biol. 2014;10:e1003829. doi: 10.1371/journal.pcbi.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brylinski M. Local alignment of ligand binding sites in proteins for polypharmacology and drug repositioning. Methods Mol Biol. 2017;1611:109–22. doi: 10.1007/978-1-4939-7015-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci U S A. 2008;105:129–34. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Srinivasan B, Skolnick J. PoLi: A Virtual Screening Pipeline Based on Template Pocket and Ligand Similarity. J Chem Inf Model. 2015;55:1757–70. doi: 10.1021/acs.jcim.5b00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Zhan J, Zhou Y. SPOT-Ligand: Fast and effective structure-based virtual screening by binding homology search according to ligand and receptor similarity. J Comput Chem. 2016;37:1734–9. doi: 10.1002/jcc.24380. [DOI] [PubMed] [Google Scholar]
- 21.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 22.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. Proteins. 1999;(Suppl 3):22–9. doi: 10.1002/(sici)1097-0134(1999)37:3+<22::aid-prot5>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- 23.Tanimoto TT. IBM Internal Report. 1958. An elementary mathematical theory of classification and prediction. [Google Scholar]
- 24.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–10. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 25.Calvo KR, Price S, Braylan RC, Oliveira JB, Lenardo M, Fleisher TA, et al. JMML and RALD (Ras-associated autoimmune leukoproliferative disorder): common genetic etiology yet clinically distinct entities. Blood. 2015;125:2753–8. doi: 10.1182/blood-2014-11-567917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 27.Park SH, Lee KH, Kim H, Lee ST. Assignment of the human PTK6 gene encoding a non-receptor protein tyrosine kinase to 20q13.3 by fluorescence in situ hybridization. Cytogenet Cell Genet. 1997;77:271–2. doi: 10.1159/000134595. [DOI] [PubMed] [Google Scholar]
- 28.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–55. [PubMed] [Google Scholar]
- 29.Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–48. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- 30.Knowles PP, Murray-Rust J, Kjaer S, Scott RP, Hanrahan S, Santoro M, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281:33577–87. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 31.Thakur MK, Birudukota S, Swaminathan S, Battula SK, Vadivelu S, Tyagi R, et al. Co-crystal structures of PTK6: With Dasatinib at 2.24 A, with novel imidazo[1,2-a]pyrazin-8-amine derivative inhibitor at 1.70 A resolution. Biochem Biophys Res Commun. 2017;482:1289–95. doi: 10.1016/j.bbrc.2016.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405:442–51. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 33.Sun Q, Phan J, Friberg AR, Camper DV, Olejniczak ET, Fesik SW. A method for the second-site screening of K-Ras in the presence of a covalently attached first-site ligand. J Biomol NMR. 2014;60:11–4. doi: 10.1007/s10858-014-9849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9:173–5. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 35.Ma J, Wang S, Zhao F, Xu J. Protein threading using context-specific alignment potential. Bioinformatics. 2013;29:i257–65. doi: 10.1093/bioinformatics/btt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Faraggi E, Zhao H, Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–82. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- 38.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 39.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–34. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao R, Cheng J. Protein single-model quality assessment by feature-based probability density functions. Sci Rep. 2016;6:23990. doi: 10.1038/srep23990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irwin JJ, Shoichet BK. ZINC - a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandit SB, Skolnick J. Fr-TM-align: a new protein structural alignment method based on fragment alignments and the TM-score. BMC Bioinformatics. 2008;9:531. doi: 10.1186/1471-2105-9-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawabata T. Build-up algorithm for atomic correspondence between chemical structures. J Chem Inf Model. 2011;51:1775–87. doi: 10.1021/ci2001023. [DOI] [PubMed] [Google Scholar]
- 44.The UniProt C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D69. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn HW. The Hungarian method for the assignment problem. Naval Research Logistics Quarterly. 1955;2:83–97. [Google Scholar]
- 46.Zhang C, Liu S, Zhu Q, Zhou Y. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. J Med Chem. 2005;48:2325–35. doi: 10.1021/jm049314d. [DOI] [PubMed] [Google Scholar]
- 47.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–32. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 48.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 49.Brylinski M. Aromatic interactions at the ligand-protein interface: Implications for the development of docking scoring functions. Chem Biol Drug Des. 2017 doi: 10.1111/cbdd.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.