

Abstract
Leukemic stem cells (LSCs) are an emerging target of curative anti-leukemia therapy. In acute lymphoblastic leukemia (ALL), LSCs frequently express CD34 and often lack CD38. However, little is known about markers and targets expressed in ALL LSCs. We have examined marker- and target expression profiles in CD34+/CD38− LSCs in patients with Ph+ ALL (n = 22) and Ph− ALL (n = 27) by multi-color flow cytometry and qPCR. ALL LSCs expressed CD19 (B4), CD44 (Pgp-1), CD123 (IL-3RA), and CD184 (CXCR4) in all patients tested. Moreover, in various subgroups of patients, LSCs also displayed CD20 (MS4A1) (10/41 = 24%), CD22 (12/20 = 60%), CD33 (Siglec-3) (20/48 = 42%), CD52 (CAMPATH-1) (17/40 = 43%), IL-1RAP (13/29 = 45%), and/or CD135 (FLT3) (4/20 = 20%). CD25 (IL-2RA) and CD26 (DPPIV) were expressed on LSCs in Ph+ ALL exhibiting BCR/ABL1p210, whereas in Ph+ ALL with BCR/ABL1p190, LSCs variably expressed CD25 but did not express CD26. In Ph− ALL, CD34+/CD38− LSCs expressed IL-1RAP in 6/18 patients (33%), but did not express CD25 or CD26. Normal stem cells stained negative for CD25, CD26 and IL-1RAP, and expressed only low amounts of CD52. In xenotransplantation experiments, CD34+/CD38− and CD34+/CD38+ cells engrafted NSG mice after 12–20 weeks, and targeting with antibodies against CD33 and CD52 resulted in reduced engraftment. Together, LSCs in Ph+ and Ph− ALL display unique marker- and target expression profiles. In Ph+ ALL with BCR/ABL1p210, the LSC-phenotype closely resembles the marker-profile of CD34+/CD38− LSCs in chronic myeloid leukemia, confirming the close biologic relationship of these neoplasms. Targeting of LSCs with specific antibodies or related immunotherapies may facilitate LSC eradication in ALL.
Abbreviations: ALL, acute lymphoblastic leukemia; BM, bone marrow; CML, chronic myeloid leukemia; GO, gemtuzumab-ozogamicin; LSC, leukemic stem cell; MNC, mononuclear cell; NSG, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; OS, overall survival; PB, peripheral blood; Ph, Philadelphia chromosome; SCT, stem cell transplantation; TKI, tyrosine kinase inhibitor
Introduction
Acute lymphoblastic leukemia (ALL) is a life-threatening malignancy defined by leukemic expansion and accumulation of lymphoid blast cells in hematopoietic tissues, including the bone marrow (BM), spleen, and lymph nodes [1], [2]. The clinical course and prognosis vary among patients, depending on age, comorbidities, the variant of ALL, and cytogenetic and molecular features. In approximately one third of all adult patients, ALL cells display the Philadelphia chromosome (Ph) and the associated BCR/ABL1 oncogene [1], [2], [3], [4], [5]. In most cases, leukemic cells display the p190-form of BCR/ABL1, whereas in a smaller group of patients, BCR/ABL1p210 is found.
Before BCR/ABL1 blockers had been introduced in clinical practice, patients with Ph+ ALL had a quite unfavorable prognosis [3], [4], [5]. However, since the advent of imatinib and other more effective BCR/ABL1-targeting tyrosine kinase inhibitors (TKIs), the prognosis of Ph+ ALL has improved substantially [3], [6], [7], [8], [9], [10], [11], [12], [13], [14]. Nevertheless, not all patients respond to chemotherapy or/and to targeted drugs [8], [9], [10], [11], [12], [14]. Depending on age, co-morbidities and donor-availability, stem cell transplantation (SCT) is recommended for high-risk patients [15], [16], [17], [18], [19], [20]. The overall treatment plan may include chemotherapy with subsequent SCT as well as BCR/ABL1-targeting drugs [16], [18], [19]. However, despite SCT and other treatment options, not all patients with ALL can be cured. Therefore, current research is attempting to identify new drug-targets and novel treatment approaches, including immunotherapies and other targeted therapies, with the hope to improve treatment outcome and prognosis.
An emerging new target of therapy in clinical hematology is the leukemic stem cell (LSC). The concept of LSCs has been established with the intention to explain cellular hierarchies in leukemic clones, and to improve drug therapy through the elimination of disease-initiating cells [21], [22], [23], [24], [25], [26], [27]. The LSC-hypothesis is based on the assumption that leukemias are organized hierarchically, with more mature cells programmed to undergo apoptosis after a limited number of cell divisions, and LSCs which have self-renewal and thus unlimited disease-propagating ability [21], [23], [24], [25]. In Ph+ chronic myeloid leukemia (CML), LSCs are considered to reside within a CD34+/CD38− fraction of the clone [22], [23], [28], [29]. In ALL, the phenotype of LSCs is less well defined. In adult patients with Ph+ ALL, NOD/SCID-repopulating LSCs supposedly reside within a CD34+/CD38− compartment [30], [31], [32]. However, in other (childhood) variants of ALL, NOD/SCID-repopulating LSCs may also be detectable in other CD34+ sub-fractions or even in CD34− populations [31], [32], [33]. Overall, little is known about markers and target expression profiles in ALL LSCs.
The aim of the current study was to establish the phenotype and target expression profile of LSCs in Ph+ and Ph− ALL in adults. Our data show that depending on the type of ALL, LSCs exhibit unique phenotypes and variable combinations of aberrantly expressed surface targets which may assist in LSC purification and the development of LSC-eradicating treatment strategies.
Material and Methods
Patients and Cell Lines
Peripheral blood (PB) and/or BM samples were collected in 49 patients with ALL and 10 with Ph+ CML. The patients´ characteristics are shown in Supplementary Table S1. All patients gave written informed consent before blood or BM was obtained. The study was approved by the ethics committee of the Medical University of Vienna. The following cell lines were used: the Ph+ cell lines Z-119 (RRID: CVCL_IU88), BV-173 (RRID: CVCL_0181), TOM-1 (RRID: CVCL_1895) and NALM-1 (RRID: CVCL_0091), the Ph− cell lines RAJI (RRID: CVCL_0511), RAMOS (RRID: CVCL_0597), REH (RRID: CVCL_1650) and BL-41 (RRID: CVCL_1087), the CML cell line CML T1 (RRID: CVCL_1126), and the myeloid cell line M-07e (RRID:CVCL_2106) expressing or lacking BCR/ABL1. A detailed description is provided in the Supplement.
Monoclonal Antibodies (mAb) and Other Reagents
A detailed description of reagents used in this study is provided in the Supplement. A list of mAb employed is shown in Supplementary Table S2.
Flow Cytometry and Cell Sorting
Flow cytometry was performed on heparinized BM or PB cells or MNCs to characterize the phenotype of CD34+/CD38− and CD34+/CD38+ cells as described [29], [34], [35]. The gating-strategy is shown in Supplementary Figure S1 and the antibody-combinations applied in Supplementary Table S3. In selected patients with Ph+ ALL (n = 6), Ph− ALL (n = 6), and CML (n = 3), CD34+/CD38− and CD34+/CD38+ cells were purified to homogeneity from MNCs by cell sorting on a FACSAria (BD Biosciences, San José, CA, USA) as described [29], [34], [35].
Quantitative PCR (qPCR)
qPCR was performed as reported [35], [36] using primers shown in Supplementary Table S4 and RNA from cell lines, unfractionated MNCs and sorted CD34+/CD38− and CD34+/CD38+ cells. mRNA levels were quantified on a QuantStudio-3 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using iTaq SYBR Green Supermix with ROX from Bio-Rad (Hercules, CA, USA). ABL1 served as reference gene.
Evaluation of InVitro Effects of Targeted Drugs on ALL Cells
Primary ALL cells, CD34+ sub-fractions (CD34+/CD38− and CD34+/CD38+) and cell lines were incubated in control medium or various concentrations of gemtuzumab-ozogamicin (GO) (48 hours), rituximab (1 hour), or alemtuzumab (1 hour) at 37 °C. After incubation, the percentage of viable cells was determined by AnnexinV and/or PI staining essentially as described [37]. In a separate set of experiments, proliferation of drug-exposed cells was examined by measuring 3H-thymidine uptake. Drug combination effects were determined as reported [29], [35], [36]. A detailed description of drug incubation experiments is provided in the Supplement.
Xenotransplantation of ALL Cells
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG; RRID: IMSR_ARC:NSG) were used to study in vivo engraftment of untreated or drug-pre-incubated ALL cells (total MNCs depleted of CD3+ T cells or sorted CD34-subfractions) or CD52+ BL-41 cells. Twenty four hours prior to injection, mice were irradiated in flat (sterile) irradiation-cages (2.4 Gy). Cells were injected into the lateral tail vein of NSG mice (1 x 106 cells per mouse, 5 mice/group). Mice were inspected daily and sacrificed as soon as they developed disease symptoms or after 20 weeks. Mice were then sacrificed and BM cells recovered. ALL engraftment was measured by flow cytometry. A detailed description of mouse experiments is provided in the Supplement.
Statistical Analysis
Statistical evaluations are described in the Supplement.
Results
CD34+/CD38− ALL Stem Cells Express a Unique Profile of Cell Surface Antigens
As assessed by flow cytometry, CD34+/CD38− ALL LSCs invariably expressed the stem cell-homing receptors CD44 and CXCR4 (CD184) (Table 1 and Supplementary Table S5). CD19 was identified on LSCs in patients with B-lineage ALL, but was not detected on LSCs in patients with T ALL or CML. Interestingly, LSCs displayed CALLA (CD10) in patients with Ph+ ALL but not in patients with Ph− ALL (Table 1). IL-1RAP, a LSC marker in CML, was found to be expressed on ALL LSCs in a subset of patients (13/29 = 45%), including most cases (7/11; 64%) with Ph+ ALL (Supplementary Table S5). CD25 was detected on CD34+/CD38− ALL LSCs in 12/41 patients (29%), including 11/20 (55%) with Ph+ ALL (Supplementary Table S5). CD26 was expressed on ALL LSCs in 5/38 patients (13%), including 5/19 (26%) with Ph+ ALL. In Ph− ALL, CD34+/CD38− LSCs expressed IL-1RAP in a subset of cases (6/18 = 33%), but displayed CD25 in only 1/21 patients (5%), and did not exhibit CD26 (0/20) (Table 1; Supplementary Table S5). Normal CD34+/CD38− BM stem cells did not express CD25, CD26 or IL-1RAP. Since NSG-engrafting LSC also reside in the CD34+/CD38+ fractions of ALL, we also examined this cell-subset. In patients with Ph+ ALL, these cells expressed CD10, CD44, CD184 and IL-1RAP (Supplementary Tables S6 and S7). Overall, the phenotype of CD34+/CD38+ stem/progenitor cells in our ALL patients was similar compared to that of CD34+/CD38− LSCs (Supplementary Tables S6 and S7). Again, CD26 was only expressed on CD34+/CD38+ cells in a subset of patients with Ph+ ALL. In patients with Ph-negative ALL, CD34+/CD38+ cells were found to lack CD25, CD26 and IL-1RAP.
Table 1.
Expression of Lineage-related Markers, Stem Cell Markers, and Niche-related Antigens on ALL LSCs, CML LSCs, and Normal CD34+/CD38− Bone Marrow (BM) Stem Cells
| Expression of Marker/Antigen on CD34+/CD38− Cells in |
|||||||
|---|---|---|---|---|---|---|---|
| BCR/ABL1p210 | BCR/ABL1p190 | ||||||
| CD | Marker/Antigen | T ALL | Ph− ALL | Ph+ ALL | Ph+ ALL | CML | Normal BM |
| CD10 | CALLA | n.t. | − | + | + | n.t. | − |
| CD19 | B4 | − | + | + | + | +/− | − |
| CD22 | Siglec-2 | n.t. | +/− | + | + | n.t. | − |
| CD25 | IL-2RA | − | − | + | +/− | + | − |
| CD26 | DPPIV | − | − | + | − | + | − |
| CD43 | Leukosialin | n.t. | + | + | + | + | + |
| CD44 | Pgp-1 | n.t. | + | + | + | + | + |
| CD51 | VNRA | n.t. | − | − | − | −/+ | − |
| CD61 | VNRB | n.t. | − | − | − | − | − |
| CD90 | Thy-1 | n.t. | − | +/− | +/− | +/− | +/− |
| CD96 | Tactile | − | − | − | − | − | − |
| CD117 | SCFR/KIT | + | −/+ | − | − | + | + |
| CD133 | Prominin-1 | n.t. | +/− | −/+ | − | + | + |
| CD150 | SLAMF1 | − | − | − | − | − | − |
| CD167a | DDR-1 | n.t. | − | − | − | − | − |
| CD184 | CXCR4 | n.t. | + | + | + | +/− | +/− |
| CD243 | MDR-1 | − | − | − | − | − | − |
| CD371 | CLL-1 | n.t. | − | − | − | − | − |
| n.c. | IL-1RAP | +/− | +/− | + | +/− | +/− | − |
| n.c. | ROBO-4 | n.t. | − | − | − | +/− | n.t. |
| n.c. | HLA-DR | n.t. | + | + | + | + | +/− |
Expression of surface markers on CD34+/CD38− BM cells was determined by multi-color flow cytometry as described.30,35,36 Technical details are provided in the Supplement.
Abbreviations: ALL, acute lymphoblastic leukemia; LSCs, leukemic stem cells; CML, chronic myeloid leukemia; BM, bone marrow; CD, cluster of differentiation; n.c., not yet clustered; n.t., not tested. Score of reactivity: +, LSCs positive (median fluorescence intensity (MFI) >3) in >80% of patients; +/−, LSCs positive in 30–80%; −/+, LSCs positive in 10–29%; −, LSCs positive in <10% of patients. Antibody staining results obtained in each individual ALL patient are shown in Supplemental Table S5.
Correlation Between the Molecular Type of ALL and the Phenotype of LSCs
We have recently shown that CD34+/CD38− LSCs in Ph+ CML co-express IL-1RAP, CD25 and CD26 [29]. In the current study, we found that the phenotype of ALL LSCs correlates with the variant of BCR/ABL1 (p190 vs p210). In fact, in 5/6 ALL patients with BCR/ABL1p210, CD34+/CD38− LSCs expressed CD26 (Figure 1A; Table 1; Supplementary Table S5). By contrast, in ALL patients with BCR/ABL1p190 (n = 12), LSCs failed to express CD26 (Figure 1A, Table 1). However, in most of the BCR/ABL1p190+ patients, CD34+/CD38− LSCs stained positive for CD25 and IL-1RAP. Thus, the LSC-phenotype in BCR/ABL1p210+ ALL closely resembles that of Ph+ CML except for expression of lymphoid antigens in ALL LSCs Expression of surface markers on ALL LSCs was confirmed by qPCR. In particular, CD34+/CD38− LSCs from patients with Ph+ ALL with BCR/ABL1p210 contained transcripts for CD25 and CD26 (Figure 1B). In ALL patients with BCR/ABL1p190, LSCs expressed low amounts of CD26 mRNA but no detectable CD25 (Figure 1B). Based on our engraftment results in NSG mice, we also examined CD34+/CD38+ cells. As shown in Supplementary Tables S6 and S7, these cells expressed CD25, CD44, and CD184 in most patients, independent of the ALL-type. In 5/6 patients with Ph+ ALL exhibiting BCR/ABL1p210, CD34+/CD38+ cells co-expressed CD26. By contrast, in patients with BCR/ABL1p190+ ALL or Ph− ALL, CD34+/CD38+ cells did not express CD26 (Supplementary Table S6 and S7).
Figure 1.

Expression of CD25, CD26, and IL-1RAP on CD34+/CD38− LSCs
A: Bone marrow (BM) cells from patients with Ph+ ALL with BCR/ABL1p210 (upper panel), Ph+ ALL with BCR/ABL1p190 (middle panel) or Ph− ALL (lower panel) were stained with antibodies against CD34, CD38, CD45, CD25, CD26 and IL-1RAP. Expression of CD25, CD26 and IL-1RAP on CD45+/CD34+/CD38− cells was analyzed by multicolor flow cytometry. The black open histograms show the isotype control and the red histograms represent CD25 (left panel), CD26 (middle panel) and IL-1RAP (right panel) expression on CD34+/CD38− cells. B: qPCR analysis of expression of CD25 mRNA (upper panels), CD26 mRNA (middle panels) and IL-1RAP mRNA (lower panels) in CD34+/CD38− cells and CD34+/CD38+ cells obtained from each 3 patients with Ph− ALL (left panels), Ph+ ALL with BCR/ABL1p190 (middle panels) or Ph+ ALL with BCR/ABL1p210 (right panels). mRNA levels are expressed as percent of ABL1.
ALL LSCs Express a Unique Profile of Cytokine Receptors
Growth and function of LSCs are considered to be regulated by various cytokines and their receptors. As assessed by flow cytometry, CD34+/CD38− LSCs were found to display IL-2RA (CD25), IL-3RA (CD123) and IL-1RAP in a majority of patients with Ph+ ALL (Supplementary Table S8A). IL-3RA was also detectable on Ph− ALL LSCs. An unexpected observation was that in patients with B-lineage ALL, LSCs lacked CD117 (KIT). By contrast, KIT was detected on normal CD34+/CD38− stem cells, CML LSCs and LSCs in patients with T ALL. ALL LSCs expressed only low amounts of G-CSFR (CD114) and stained negative for the thrombopoietin (TPO) receptor (CD110) and erythropoietin (EPO) receptor (Supplementary Table S8A). We also examined the expression of cytokine receptors on CD34+/CD38+ cells in our ALL patients. Similar to LSCs, the CD34+/CD38+ stem/progenitor cells expressed IL-2RA, IL-3RA and IL-1RAP in a majority of the patients (Supplementary Table S8B).
Effects of Signal Transduction Blockers on Expression of CD25, CD26 and IL-1RAP
We next investigated whether aberrant expression of CD25, CD26 or IL-1RAP is triggered by BCR/ABL1 or other signaling molecules. To address this question, we incubated ALL cell lines with BCR/ABL1-targeting TKIs (imatinib, nilotinib, ponatinib), the PI3 kinase/mTOR blocker BEZ235, the mTOR blocker RAD001, the MEK inhibitors RDEA119 and PD0325901, and the STAT5-targeting drug pimozide. We found that ponatinib, BEZ235 and RAD001 down-regulate expression of IL-1RAP in the IL-1RAP+ cell lines RAMOS and REH (Supplementary Figure S2A), whereas the other inhibitors induced no significant effects. Interestingly, ponatinib, BEZ235 and RAD001 also upregulated CD25 expression in the CD25+ cell lines BL-41 and RAMOS (Supplementary Figure S2A). By contrast, the other compounds tested did not modulate CD25 expression (data not shown). Unexpectedly, the STAT5-targeting drug pimozide (up to 10 μM) did not inhibit CD25 expression in ALL cells. Furthermore, we tested the effect of BCR/ABL1 on expression of CD25, CD26 and IL-1RAP using M-07e cells expressing or lacking BCR/ABL1. In these experiments, BCR/ABL1 failed to induce the expression of CD25, CD26 or IL-1RAP mRNA (Supplementary Figure S2B).
ALL LSCs Express Several Druggable Cell Surface Targets
Several cell surface antigens serve as molecular targets in clinical hematology. We found that LSCs in B-lineage ALL frequently co-express CD20, CD22, CD33, and CD52. These targets were identified on CD34+/CD38- LSCs in patients with Ph+ and Ph- ALL by flow cytometry and qPCR (Table 2, Figure 2). In addition, we found that in most patients with ALL, CD34+/CD38+ stem/progenitor cells express CD22, CD33 and CD52 (Supplementary Table S7B). We also examined our cell lines for expression of these targets. As shown in Supplementary Table S9, CD20, CD33 and CD52 were expressed variably on these cells, namely Z-119 (CD33+), BV-173 (CD33+), NALM-1 (CD52+), TOM-1 (CD20+), BL-41 (CD20+/CD52+), RAJI (CD20+/CD52+), RAMOS (CD20+), REH (CD52+) and CML-T1 (CD52+).
Table 2.
Expression of Molecular Targets on ALL LSCs, CML LSCs, and Normal BM Stem Cells
| Expression on CD34+/CD38− Cells In |
|||||||
|---|---|---|---|---|---|---|---|
| BCR/ABL1p210 | BCR/ABL1p190 | ||||||
| Marker | Antigen | T ALL | Ph− ALL | Ph+ ALL | Ph+ ALL | CML | Normal BM |
| CD20 | MS4A1 | − | − | +/− | −/+ | − | − |
| CD22 | Siglec-2 | n.t. | +/− | + | + | n.t. | − |
| CD23 | FcεRII | − | − | − | − | − | − |
| CD25 | IL-2RA | − | − | + | +/− | + | − |
| CD30 | Ki-1 | − | − | −/+ | +/− | + | − |
| CD33 | Siglec-3 | +/− | +/− | +/− | −/+ | + | + |
| CD44 | HCAM | n.t. | + | + | + | + | + |
| CD52 | CAMPATH-1 | −/+ | −/+ | +/− | +/− | −/+ | +/− |
| CD117 | KIT/SCFR | + | −/+ | − | − | + | + |
| CD123 | IL-3RA | + | + | + | + | + | + |
| CD135 | FLT-3 | +/− | −/+ | −/+ | − | +/− | −/+ |
| CD300a | CMRF-35H | n.t. | +/− | + | + | + | + |
Expression of markers on CD34+/CD38− bone marrow (BM) cells was determined by multi-color flow cytometry as described.30,35,36 Technical details are provided in the Supplemental Material.
Abbreviations: ALL, acute lymphoblastic leukemia; LSCs, leukemic stem cells; CML, chronic myeloid leukemia; BM, bone marrow; CD, cluster of differentiation; n.t., not tested. Score of reactivity: +, LSCs positive (median fluorescence intensity (MFI) > 3) in >80% of patients; +/−, LSCs positive in 30–80%; −/+, LSCs positive in 10–29%; −, LSCs positive in <10% of patients. Antibody staining results obtained in each individual ALL patient, are shown in Supplemental Table S5.
Figure 2.

Expression of CD20, CD33, and CD52 on ALL LSCs
A: Bone marrow (BM) cells from patients with Ph+ ALL with BCR/ABL1p210 (upper panels), Ph+ ALL with BCR/ABL1p190 (middle panels) or Ph− ALL (lower panels) were stained with antibodies against CD34, CD38, CD45, CD20, CD33 and CD52. Expression of CD20, CD33 and CD52 on CD45+/CD34+/CD38− cells was analyzed by multicolor flow cytometry. The black open histograms show the isotype control and the red histograms represent CD20 (left panel), CD33 (middle panel) and CD52 (right panel) expression on CD34+/CD38− cells. B: qPCR analysis of expression of CD20 mRNA (upper panels), CD33 mRNA (middle panels) and CD52 mRNA (lower panels) in CD34+/CD38− cells and CD34+/CD38+ cells obtained from each 3 patients with Ph− ALL (left panels), Ph+ ALL with BCR/ABL1p190 (middle panels) or Ph+ ALL with BCR/ABL1p210 (right panels). mRNA levels are expressed as percent of ABL1.
Antibody-Based Targeted Drugs Can Kill ALL LSCs In Vitro
Alemtuzumab induced rapid cell lysis in all CD52+ ALL cell lines but not in CD52-negative cell lines (Figure 3, A and B). Cell lysis occurred in complement-containing serum but not in complement-free (heat-inactivated) serum (Figure 3A). In addition, alemtuzumab induced cell lysis in primary LSCs in all patients in whom LSCs expressed CD52, but not in CD52-negative LSCs (Figure 3C). We also examined the effects of rituximab and GO. Both agents were found to induce lysis or/and apoptosis in cell lines expressing the respective target(s), whereas no comparable effects were seen in CD20-negative and/or CD33-negative cells (Supplementary Figure S3). Corresponding results were obtained with primary CD34+/CD38− ALL LSCs and with CD34+/CD38+ stem/progenitor cells (Supplementary Figures S4 and S5). As expected, BCR/ABL1 TKIs suppressed the proliferation of BCR/ABL1+ ALL cell lines and of CML-T1 cells, but did not inhibit growth of Ph-negative cells. In most cell lines, GO also blocked cell proliferation (Supplementary Table S10). Finally, we examined cooperative anti-neoplastic effects in the CD33+/Ph+ cell line Z-119. We found that the drug combinations GO+imatinib, GO+nilotinib and GO+ponatinib produce synergistic growth-inhibitory effects on Z-119 cells (Supplementary Figure S6).
Fig. 3.

Effects of alemtuzumab on growth and survival of neoplastic cells
A: NALM-1 cells (CD52+), BL-41 cells (CD52+), RAJI cells (CD52+) and REH cells (CD52+) and B: BV-173 cells (CD52−) TOM-1 cells (CD52−), Z-119 cells (CD52−) and RAMOS cells (CD52−) were incubated in various concentration of alemtuzumab (10–300 μg/mL) in RPMI-1640 medium with either 30% serum (black bars), or 30% heat-inactivated serum (open bars) at 37 °C for 1 hour. Thereafter, cells were stained with propidium iodide (PI) and analyzed for cell viability on a FACSCalibur. Results represent the mean ± SD from 3 independent experiments. Asterisk (*): P < .05. C: Primary cells from patients with ALL with CD52+ LSCs (#18, #19 and #20; upper panel) or patients with ALL with CD52− LSCs (#44, #46 and #48; lower panel) were incubated in alemtuzumab (10–300 μg/mL) in the presence of 30% human serum at 37 °C (5% CO2) for 1 hour. Then 10 μL calibration beads were added. Cells were washed and then stained with antibodies against CD34, CD45, and CD38 for 15 minutes. Cells were then subjected to DAPI staining to count viable cells on a FACSCanto II. The left panels (black bars) show the effects of alemtuzumab on CD34+/CD38− cells and the right panels (open bars) show the effects of alemtuzumab on CD34+/CD38+ cells. Results represent the mean ± SD from 3 independent experiments. Asterisk (*): P < .05.
Effects of Targeted Antibodies on Engraftment of Ph+ ALL Cells in NSG Mice
In order to confirm that NSG-repopulating Ph+ ALL LSCs indeed reside in a CD34+/CD38− cell fraction, we isolated these cells and injected them into NSG mice. As shown in Figure 4A, control MNCs as well as sorted CD34+/CD38− ALL cells produced leukemic engraftment with comparable engraftment levels in NSG mice after 16–20 weeks. However, surprisingly, CD34+/CD38+ ALL cells were also found to produce leukemic engraftment in NSG mice (Figure 4A). Moreover, xenograft-derived ALL cells obtained from all three primary sample-sources (MNCs, CD34+/CD38− and CD34+/CD38+ cells) engrafted secondary and tertiary recipient mice (Supplementary Table S11). Based on these results, total MNCs (containing both, CD34+/CD38− and CD34+/CD38+ cells) were used in drug incubation experiments. In particular, we pre-incubated primary ALL MNCs with GO (10 μg/mL), alemtuzumab (500 μg/mL), or a combination of both drugs for 1 hour before injecting these cells into NSG mice. Pre-incubation with the drug-combination resulted in reduced engraftment of ALL cells, whereas no substantial effects of single drug-exposures were seen (Fig. 4B). Finally, we injected the CD52+ line BL-41 into NSG mice and treated these mice either with control-buffer or alemtuzumab (250 μg) for 5 weeks. In these experiments, alemtuzumab was found to interfere with leukemic engraftment of BL-41 cells (Fig. 4C) and to counteract leukemia-induced weight loss in NSG mice (Supplementary Fig. S7).
Figure 4.
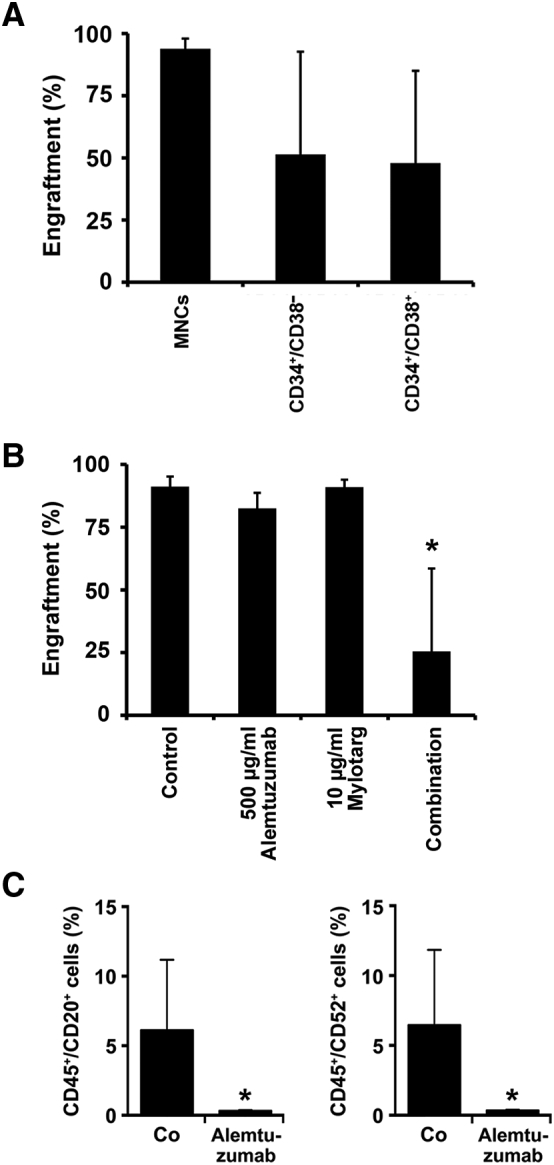
Engraftment of ALL cells in NSG mice
A: Three different fractions of ALL cells were injected intravenously into female NSG mice (5/group): total MNCs depleted of CD3+ T cells (control MNCs), CD34+/CD38− cells and CD34+/CD38+ cells. B: T cell-depleted MNCs were pre-incubated in control medium, alemtuzumab (500 μg/mL), GO (10 μg/mL), or a combination of alemtuzumab (500 μg/mL) and GO (10 μg/mL) at 37 °C for 1 hour prior to injection. A,B: After a maximum observation period of 12–20 weeks, mice were sacrificed and bone marrow (BM) cells (humeri, tibias, femurs) recovered for flow cytometry. Multi-color flow cytometry was performed using antibodies against CD19, CD33, and CD45. TO-PRO-3 was used to exclude non-viable cells. Engraftment was defined as at least 1% human CD45+ cells in mouse BM samples. Asterisk (*): P < .05. C: NSG mice were intravenously injected with BL-41 cells (1 x 105/mouse). At day 7, mice were split into two groups and treated with either alemtuzumab (250 μg intraperitoneally three times a week; 9 mice) or PBS (vehicle control; 5 mice). After 5 weeks, mice were sacrificed. Multicolor flow cytometry was performed on BM cells using antibodies against CD20, CD45 and CD52 to determine engraftment levels.
Prognostic Impact of LSC Markers in Patients With ALL
To define the prognostic significance of expression of various surface markers on ALL LSCs, we correlated overall survival (OS) with expression of CD20, CD25, CD33, and CD52 on LSCs. These markers were differentially expressed on LSCs in patient-subgroups. Accordingly, patients were split into high and low ´LSC-expressers´. We found that higher levels of CD25 and CD33 on LSCs were associated with an increased OS in our ALL patients (Fig. 5). However, the differences did not reach statistical significance. The other markers examined did not correlate with OS.
Figure 5.

Influence of expression of CD20, CD25, CD33 and CD52 on LSCs on survival in ALL patients
The probability of overall survival (OS) in patients with ALL (n = 49) was determined for subgroups of patients in whom CD34+/CD38− stem cells either expressed (pos) or did not express (neg) substantial levels of CD20, CD25, CD33 or CD52. Expressers (pos) were defined as a median fluorescence intensity (MFI) of ≥3 and low expressers (neg) as a MFI of <3. The median follow-up of our ALL patients was 623 days. The probability of OS was calculated by the product limit method of Kaplan and Meier.
Discussion
During the past few years, substantial efforts have been made to identify markers and targets expressed on LSCs in various human leukemias [22], [23], [26], [27], [28], [29], [30], [31], [32], [33], [34]. However, so far, only little is known about the phenotype and function of LSCs in ALL. In patients with Ph+ ALL, the NOD/SCID-repopulating LSCs may reside preferentially in a CD34+/CD38− cell fraction [30], [31], [32]. In the current study, we established the phenotype of CD34+/CD38− LSCs in adult patients with Ph+ ALL and Ph− ALL. We found that CD34+/CD38− LSCs in Ph+ ALL exhibiting BCR/ABL1p210 co-express CD25, CD26 and IL-1RAP in an aberrant manner, thereby resembling the phenotype of CML LSCs [29], [38]. Whereas CD25 and IL-1RAP were also expressed on LSCs in smaller subsets of patients with Ph+ ALL exhibiting BCR/ABL1p190, CD26 was found to be expressed exclusively on ALL LSCs in patients with BCR/ABL1p210, but not in patients with BCR/ABL1p190+ ALL or Ph− ALL. The preferential expression of CD25 on immature progenitor cells in Ph+ ALL compared to Ph− ALL confirms previous studies [39]. We also found that both CD34+/CD38− and CD34+/CD38+ ALL cells engraft NSG mice and that both populations frequently express druggable cell surface targets, including CD20, CD33, and CD52 which may be relevant clinically and may provide a basis for the design of novel LSC-eradicating and thus potentially curative treatment concepts.
During early lymphopoiesis, B cell precursors co-express CD34 and CD19, whereas later, during maturation, these cells lose CD34 and acquire additional lymphoid markers, including CD19 and CD20 [40], [41]. In our patients with B-lineage ALL, CD34+/CD38− LSCs co-expressed CD19 in all samples tested, thereby confirming the lymphoid nature of these cells. Moreover, in a considerable number of patients, ALL LSCs co-expressed CD10, CD20, and CD22. In addition, we were able to show that purified (sorted) LSCs in Ph+ ALL display BCR/ABL1 mRNA, confirming their clonal origin. In patients with T ALL and CML, LSCs did not express CD19 or CD20. An interesting observation was that LSCs display CALLA (CD10) in patients with Ph+ ALL but not in Ph− ALL.
We and others have recently shown that LSCs in CML express CD25, CD26, and IL-1RAP in an aberrant manner [29], [38], [42]. Therefore, we were interested to learn whether LSCs in our ALL patients express one or more of these LSC-antigens. The results of our study show that LSCs in Ph+ ALL often express CD25 and IL-1RAP, whereas LSCs in most patients with Ph− ALL did not express CD25, and IL-1RAP was only detected on LSCs in a subset of these patients. In a distinct group of patients with Ph+ ALL, namely those where the lymphoblasts exhibited BCR/ABL1p210, LSCs and CD34+/CD38+ stem/progenitor cells also expressed CD26. These data suggest that the phenotype of stem cells in Ph+ ALL exhibiting BCR/ABL1p210 is almost identical to that of CML LSCs [29]. This observation supports the close relationship between these two leukemias. It is noteworthy in this regard, that Ph+ ALL expressing BCR/ABL1p210 is often regarded as primary lymphoid blast phase of CML.
Currently, it remains unknown why stem and progenitor cells in BCR/ABL1p190+ ALL usually lack IL-1RAP and CD26. A possible explanation for this difference is that the two forms of BCR/ABL1 activate different signaling networks [43], [44]. Therefore, we believe that these different signaling pathways and the related oncogenic machineries might be responsible for differential expression of CD26 and other surface molecules on ALL LSCs. For example, it has been described that in Ph+ CML, the STAT5 pathway contributes to aberrant expression of CD25 on LSC [45].
In the current study, we first asked whether BCR/ABL1 could be responsible for aberrant expression of these antigens. However, no effects of imatinib or nilotinib on expression of CD25, CD26 or IL-1RAP in ALL cells were seen. In addition, introduction of BCR/ABL1 into the Ph-negative cell line M-07e did not induce CD25, CD26 or IL-1RAP mRNA expression suggesting that other pathways and molecules are involved. Next, we applied the multi-kinase blocker ponatinib, the PI3 kinase/mTOR inhibitor BEZ235, and RAD001. These drugs were found to down-regulate IL-1RAP expression in the IL-1RAP+ cell lines RAMOS and REH, suggesting that the PI3 kinase/mTOR pathway contributes to expression of IL-1RAP. However, these drugs did not downregulate expression of CD25 in the ALL cell lines tested. Rather, BEZ235 and RAD001 even upregulated expression of CD25 on REH and RAMOS cells. This is of particular interest since a similar effect of these agents has been described in CML LSCs [45].
We also examined the potential prognostic role of aberrantly expressed surface antigens on ALL LSCs. While most LSC markers did not define a distinct prognostic subgroup, expression of CD25 and CD33 on ALL LSCs was found to be associated with improved OS which was an unexpected result. In fact, in patients with AML expression of CD25 and CD52 has been associated with a poor survival [37], [46]. On the other hand, CD25 has been described as a growth-suppressor in CML cells [45]. Whether CD25 can act as a growth-suppressing molecule in ALL LSCs remains unknown.
ALL LSCs were also found to express receptors for various cytokine-ligands, including IL-2RA (CD25), IL-3RA (CD123) and CD135 (FLT3). Interestingly, ALL LSCs in T ALL also expressed KIT (CD117) whereas in B-lineage ALL, CD34+/CD38− LSCs were found to lack KIT, suggesting that KIT does not serve as a major regulator of LSC-function in B-lineage ALL. Correspondingly, the KIT-targeting TKIs imatinib or nilotinib did not show growth-inhibitory effects on Ph-negative ALL cells. The EPO and TPO receptors were not detected on LSCs in Ph+ and Ph− ALL. The G-CSF receptor (CD114) was found to be expressed on ALL LSCs either at low level or was not detected on LSCs. These data may have clinical implications as some of these cytokines are applied in clinical practice during and/or after chemotherapy.
We also identified several druggable surface antigens on ALL LSCs. Among these were CD20, CD33, and CD52. We therefore asked whether antibody-based drugs directed against these molecules would induce cell death in LSCs. The results of our study show that these targeted antibodies are indeed able to induce cell death in ALL LSCs in vitro. However, responses were only seen in patients in whom LSCs expressed these targets, but not in those in whom LSCs did not express these target-antigens. Finally, we tested the hypothesis that these drugs can interfere with leukemic engraftment of LSCs in NSG mice. Since our data showed that both, CD34+/CD38− and CD34+/CD38+ ALL cells produce engraftment, we used T cell-depleted MNCs (bulk of CD34+ cells) in these experiments. However, neither GO alone nor alemtuzumab alone were able to inhibit ALL engraftment in NSG mice, suggesting that some of the LSCs in these cell-fractions were resistant or did not express these targets. On the other hand, a combination of both drugs was found to substantially inhibit engraftment of ALL cells in NSG mice. These data suggest that CD33 and CD52 are variably expressed in sub-fractions of ALL LSCs, and that combined targeting is required to block LSC engraftment. An alternative explanation would be that ALL LSCs can recover rapidly from drug effects in vivo in mice unless multiple drugs are applied. We therefore transplanted the CD52+ ALL cell line BL-41 into NSG mice and asked whether continuous treatment with alemtuzumab is able to interfere with ALL expansion. Indeed, alemtuzumab i.p. injection three times a week led to a significant decrease in engraftment. However, again, no total depletion of ALL cells was achieved. Together, these data suggest that targeting through defined surface antigens may be a promising approach to attack LSCs, but these drugs have to be combined with each other or with other drugs to achieve optimal anti-neoplastic effects.
The application of targeted antibodies in lymphoid malignancies has led to an improvement of the overall outcome and long-term disease-free survival. An impressive example is the use of CD20-targeting antibodies as adjunct to chemotherapy in patients with CD20+ Non Hodgkin lymphomas [47], [48], [49]. In ALL, the application of targeted antibodies has recently also been proposed [50], [51]. In the present study, we show that such ´antibody+TKI´ combinations can produce additive or even synergistic effects on growth and survival of Ph+ ALL cell lines.
In conclusion, our data show that ALL LSCs display a distinct phenotype, including aberrantly expressed markers and druggable target-antigens. Aberrantly expressed surface antigens, such as CD25, IL-1RAP or CD26 may serve as diagnostic LSC markers and may assist in enrichment of LSCs and their separation from normal stem cells. The druggable targets identified may provide a basis for the design of novel LSC-eliminating treatment-strategies, including antibody-based therapies and novel CAR-T cell-based immunotherapies.
Acknowledgments
Acknowledgements
The authors would like to thank Karin Lind and Tina Bernthaler for excellent technical assistance and Günther Hofbauer and Andreas Spittler (both at the Cell Sorting Core Unit of the Medical University of Vienna) for their support in this study.
Funding sources
This study was supported by a Cancer Stem Cell Grant from the Medical University of Vienna and by the Austrian Science Fund (FWF), grants SFB F4701, F4704, and F4706.
Conflicts of interest
The authors declare no conflicts of interest.
Author contributions
K.B. performed key laboratory experiments (proliferation and apoptosis assays, flow cytometry and mouse experiments) and wrote parts of the manuscript; I.M. and V.S: provided mouse data with BL-41, G.E. performed sorting, flow cytometry and mouse experiments and wrote parts of the manuscript, S.C.-R. and G.S. provided qPCR data; H.H. and I.S. performed flow cytometry experiments, S.H. provided vital logistic support; D.B. performed proliferation assays; A.K. performed mouse experiments; A.H., H.S. and U.J. provided clinical data and patients material; G.H. and C.M. contributed molecular biology data; M.W. and T.R. provided mouse studies; W.R.S. provided clinical data, patients and statistics; J.V.M provided cell lines; P.V. contributed the study design and wrote the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2018.04.004.
Appendix A. Supplementary data
Supplementary material
References
- 1.Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23:6306–6315. doi: 10.1200/JCO.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 2.Faderl S, Jeha S, Kantarjian HM. The biology and therapy of adult acute lymphoblastic leukemia. Cancer. 2003;98:1337–1354. doi: 10.1002/cncr.11664. [DOI] [PubMed] [Google Scholar]
- 3.Crazzolara R, Bendall L. Emerging treatments in acute lymphoblastic leukemia. Curr Cancer Drug Targets. 2009;9:19–31. doi: 10.2174/156800909787314057. [DOI] [PubMed] [Google Scholar]
- 4.Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities in adult acute lymphoblastic leukemia. Blood. 1998;91:3995–4019. [PubMed] [Google Scholar]
- 5.Gleissner B, Gökbuget N, Bartram CR, Janssen B, Rieder H, Janssen JWG, Fonatsch C, Heyll A, Voliotis D, Beck J. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood. 2002;99:1536–1543. doi: 10.1182/blood.v99.5.1536. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 7.Gökbuget N, Hoelzer D. Treatment of adult acute lymphoblastic leukemia. Semin Hematol. 2009;46:64–75. doi: 10.1053/j.seminhematol.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Gruber F, Mustjoki S, Porkka K. Impact of tyrosine kinase inhibitors on patient outcomes in Philadelphia chromosome-positive acute lymphoblastic leukaemia. Br J Haematol. 2009;145:581–597. doi: 10.1111/j.1365-2141.2009.07666.x. [DOI] [PubMed] [Google Scholar]
- 9.Hoelzer D, Gökbuget N, Ottmann OG. Targeted therapies in the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Semin Hematol. 2002;39:32–37. doi: 10.1053/shem.2002.36927. [DOI] [PubMed] [Google Scholar]
- 10.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, Tura S, Fischer T, Deininger MW, Schiffer CA. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100:1965–1971. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 11.Thomas DA, Faderl S, Cortes J, O'Brien S, Giles FJ, Kornblau SM, Garcia-Manero G, Keating MJ, Andreeff M, Jeha S. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood. 2004;103:4396–4407. doi: 10.1182/blood-2003-08-2958. [DOI] [PubMed] [Google Scholar]
- 12.Towatari M, Yanada M, Usui N, Takeuchi J, Sugiura I, Takeuchi M, Yagasaki F, Kawai Y, Miyawaki S, Ohtake S. Combination of intensive chemotherapy and imatinib can rapidly induce high-quality complete remission for a majority of patients with newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia. Blood. 2004;104:3507–3512. doi: 10.1182/blood-2004-04-1389. [DOI] [PubMed] [Google Scholar]
- 13.Vitale A, Guarini A, Chiaretti S, Foà R. The changing scene of adult acute lymphoblastic leukemia. Curr Opin Oncol. 2006;18:652–659. doi: 10.1097/01.cco.0000245317.82391.1b. [DOI] [PubMed] [Google Scholar]
- 14.Yanada M, Takeuchi J, Sugiura I, Akiyama H, Usui N, Yagasaki F, Kobayashi T, Ueda Y, Takeuchi M, Miyawaki S. High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol. 2006;24:460–466. doi: 10.1200/JCO.2005.03.2177. [DOI] [PubMed] [Google Scholar]
- 15.Chao NJ, Blume KG, Forman SJ, Snyder DS. Long-term follow-up of allogeneic bone marrow recipients for Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 1995;85:3353–3354. [PubMed] [Google Scholar]
- 16.Chen H, Liu K, Xu L, Liu D, Chen Y, Zhao X, Han W, Zhang X, Wang Y, Zhang Y. Administration of imatinib after allogeneic hematopoietic stem cell transplantation may improve disease-free survival for patients with Philadelphia chromosome-positive acute lymphobla stic leukemia. J Hematol Oncol. 2012;5:29. doi: 10.1186/1756-8722-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fielding AK, Goldstone AH. Allogeneic haematopoietic stem cell transplant in Philadelphia-positive acute lymphoblastic leukaemia. Bone Marrow Transplant. 2008;41:447–453. doi: 10.1038/sj.bmt.1705904. [DOI] [PubMed] [Google Scholar]
- 18.Lee S, Kim Y-J, Min C-K, Kim H-J, Eom K-S, Kim D-W, Lee J-W, Min W-S, Kim C-C. The effect of first-line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2005;105:3449–3457. doi: 10.1182/blood-2004-09-3785. [DOI] [PubMed] [Google Scholar]
- 19.Oliansky DM, Larson RA, Weisdorf D, Dillon H, Ratko TA, Wall D, McCarthy PL, Hahn T. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of adult acute lymphoblastic leukemia: update of the 2006 evidence-based review. Biol Blood Marrow Transplant. 2012;18:18–36. doi: 10.1016/j.bbmt.2011.07.019. [e6] [DOI] [PubMed] [Google Scholar]
- 20.Stein A, Forman SJ. Allogeneic transplantation for ALL in adults. Bone Marrow Transplant. 2008;41:439–446. doi: 10.1038/bmt.2008.1. [DOI] [PubMed] [Google Scholar]
- 21.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 22.Holyoake TL, Jiang X, Jorgensen HG, Graham S, Alcorn MJ, Laird C, Eaves AC, Eaves CJ. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood. 2001;97:720–728. doi: 10.1182/blood.v97.3.720. [DOI] [PubMed] [Google Scholar]
- 23.Kavalerchik E, Goff D, Jamieson CHM. Chronic myeloid leukemia stem cells. J Clin Oncol. 2008;26:2911–2915. doi: 10.1200/JCO.2008.17.5745. [DOI] [PubMed] [Google Scholar]
- 24.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–481. doi: 10.1016/j.molmed.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 26.Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009;23:25–42. doi: 10.1038/leu.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011;11:56–71. doi: 10.2174/156800911793743655. [DOI] [PubMed] [Google Scholar]
- 28.Eisterer W, Jiang X, Christ O, Glimm H, Lee KH, Pang E, Lambie K, Shaw G, Holyoake TL, Petzer AL. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia. 2005;19:435–441. doi: 10.1038/sj.leu.2403649. [DOI] [PubMed] [Google Scholar]
- 29.Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, Hoermann G, Bilban M, Blatt K, Herndlhofer S. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123:3951–3962. doi: 10.1182/blood-2013-10-536078. [DOI] [PubMed] [Google Scholar]
- 30.Cobaleda C, Gutiérrez-Cianca N, Pérez-Losada J, Flores T, García-Sanz R, González M, Sánchez-García I. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000;95:1007–1013. [PubMed] [Google Scholar]
- 31.Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104:2919–2925. doi: 10.1182/blood-2004-03-0901. [DOI] [PubMed] [Google Scholar]
- 32.Kong Y, Yoshida S, Saito Y, Doi T, Nagatoshi Y, Fukata M, Saito N, Yang SM, Iwamoto C, Okamura J. CD34 + CD38 + CD19+ as well as CD34 + CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia. 2008;22:1207–1213. doi: 10.1038/leu.2008.83. [DOI] [PubMed] [Google Scholar]
- 33.le Viseur C, Hotfilder M, Bomken S, Wilson K, Röttgers S, Schrauder A, Rosemann A, Irving J, Stam RW, Shultz LD. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell. 2008;14:47–58. doi: 10.1016/j.ccr.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Florian S, Sonneck K, Hauswirth AW, Krauth M-T, Schernthaner G-H, Sperr WR, Valent P. Detection of molecular targets on the surface of CD34+/CD38-- stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47:207–222. doi: 10.1080/10428190500272507. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann H, Cerny-Reiterer S, Gleixner KV, Blatt K, Herndlhofer S, Rabitsch W, Jäger E, Mitterbauer-Hohendanner G, Streubel B, Selzer E. CD34(+)/CD38(−) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica. 2012;97:219–226. doi: 10.3324/haematol.2010.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerny-Reiterer S, Meyer RA, Herrmann H, Peter B, Gleixner KV, Stefanzl G, Hadzijusufovic E, Pickl WF, Sperr WR, Melo JV. Identification of heat shock protein 32 (Hsp32) as a novel target in acute lymphoblastic leukemia. Oncotarget. 2014;5:1198–1211. doi: 10.18632/oncotarget.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blatt K, Herrmann H, Hoermann G, Willmann M, Cerny-Reiterer S, Sadovnik I, Herndlhofer S, Streubel B, Rabitsch W, Sperr WR. Identification of campath-1 (CD52) as novel drug target in neoplastic stem cells in 5q-patients with MDS and AML. Clin Cancer Res. 2014;20:3589–3602. doi: 10.1158/1078-0432.CCR-13-2811. [DOI] [PubMed] [Google Scholar]
- 38.Valent P, Sadovnik I, Ráčil Z, Herrmann H, Blatt K, Cerny-Reiterer S, Eisenwort G, Lion T, Holyoake T, Mayer J. DPPIV (CD26) as a novel stem cell marker in Ph + chronic myeloid leukaemia. Eur J Clin Investig. 2014;44:1239–1245. doi: 10.1111/eci.12368. [DOI] [PubMed] [Google Scholar]
- 39.Paietta E, Racevskis J, Neuberg D, Rowe JM, Goldstone AH, Wiernik PH. Expression of CD25 (interleukin-2 receptor alpha chain) in adult acute lymphoblastic leukemia predicts for the presence of BCR/ABL fusion transcripts: results of a preliminary laboratory analysis of ECOG/MRC Intergroup Study E2993. Eastern Cooperative Oncology Group/Medical Research Council. Leukemia. 1997;11:1887–1890. doi: 10.1038/sj.leu.2400836. [DOI] [PubMed] [Google Scholar]
- 40.LeBien TW. Fates of human B-cell precursors. Blood. 2000;96:9–23. [PubMed] [Google Scholar]
- 41.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Järås M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, Lassen C, Olofsson T, Bjerrum OW, Richter J. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci U S A. 2010;107:16280–16285. doi: 10.1073/pnas.1004408107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reckel S, Hamelin R, Georgeon S, Armand F, Jolliet Q, Chiappe D, Moniatte M, Hantschel O. Differential signaling networks of Bcr–Abl p210 and p190 kinases in leukemia cells defined by functional proteomics. Leukemia. 2017;31:1502–1512. doi: 10.1038/leu.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cutler JA, Tahir R, Sreenivasamurthy SK, Mitchell C, Renuse S, Nirujogi RS, Patil AH, Heydarian M, Wong X, Wu X. Differential signaling through p190 and p210 BCR-ABL fusion proteins revealed by interactome and phosphoproteome analysis. Leukemia. 2017;31:1513–1524. doi: 10.1038/leu.2017.61. [DOI] [PubMed] [Google Scholar]
- 45.Sadovnik I, Hoelbl-Kovacic A, Herrmann H, Eisenwort G, Cerny-Reiterer S, Warsch W, Hoermann G, Greiner G, Blatt K, Peter B. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph + CML. Clin Cancer Res. 2016;22:2051–2061. doi: 10.1158/1078-0432.CCR-15-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gönen M, Sun Z, Figueroa ME, Patel JP, Abdel-Wahab O, Racevskis J, Ketterling RP, Fernandez H, Rowe JM, Tallman MS. CD25 expression status improves prognostic risk classification in AML independent of established biomarkers: ECOG phase 3 trial, E1900. Blood. 2012;120:2297–2306. doi: 10.1182/blood-2012-02-414425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheson BD. Rituximab: clinical development and future directions. Expert Opin Biol Ther. 2002;2:97–110. doi: 10.1517/14712598.2.1.97. [DOI] [PubMed] [Google Scholar]
- 48.Czuczman MS, Gregory SA. The future of CD20 monoclonal antibody therapy in B-cell malignancies. Leuk Lymphoma. 2010;51:983–994. doi: 10.3109/10428191003717746. [DOI] [PubMed] [Google Scholar]
- 49.Maury S, Chevret S, Thomas X, Heim D, Leguay T, Huguet F, Chevallier P, Hunault M, Boissel N, Escoffre-Barbe M. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med. 2016;375:1044–1053. doi: 10.1056/NEJMoa1605085. [DOI] [PubMed] [Google Scholar]
- 50.Hoelzer D. Targeted therapy with monoclonal antibodies in acute lymphoblastic leukemia. Curr Opin Oncol. 2013;25:701–706. doi: 10.1097/CCO.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 51.Hoelzer D, Gökbuget N. Chemoimmunotherapy in acute lymphoblastic leukemia. Blood Rev. 2012;26:25–32. doi: 10.1016/j.blre.2011.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material