

Abstract

Mycobacterium tuberculosis (MTb) possesses two nonproton pumping type II NADH dehydrogenase (NDH-2) enzymes which are predicted to be jointly essential for respiratory metabolism. Furthermore, the structure of a closely related bacterial NDH-2 has been reported recently, allowing for the structure-based design of small-molecule inhibitors. Herein, we disclose MTb whole-cell structure–activity relationships (SARs) for a series of 2-mercapto-quinazolinones which target the ndh encoded NDH-2 with nanomolar potencies. The compounds were inactivated by glutathione-dependent adduct formation as well as quinazolinone oxidation in microsomes. Pharmacokinetic studies demonstrated modest bioavailability and compound exposures. Resistance to the compounds in MTb was conferred by promoter mutations in the alternative nonessential NDH-2 encoded by ndhA in MTb. Bioenergetic analyses revealed a decrease in oxygen consumption rates in response to inhibitor in cells in which membrane potential was uncoupled from ATP production, while inverted membrane vesicles showed mercapto-quinazolinone-dependent inhibition of ATP production when NADH was the electron donor to the respiratory chain. Enzyme kinetic studies further demonstrated noncompetitive inhibition, suggesting binding of this scaffold to an allosteric site. In summary, while the initial MTb SAR showed limited improvement in potency, these results, combined with structural information on the bacterial protein, will aid in the future discovery of new and improved NDH-2 inhibitors.
Keywords: Mycobacterium tuberculosis, mercapto-quinazolinones, structure−activity relationship, type II NADH dehydrogenase, small molecule NDH-2 inhibitors, respiration
Tuberculosis (TB) is a major global health problem, resulting in significant morbidity each year. Although mortality has fallen dramatically since 1990, TB now ranks alongside HIV as a leading cause of death worldwide. While HIV-related deaths have been declining largely as a result of improved access to, and availability of, better HIV treatments, this has not been the case for TB,1 the treatment of which requires 6 months of chemotherapy with a combination of four agents (isoniazid, rifampicin, pyrazinamide, and ethambutol) to achieve durable cure of drug-sensitive TB.2 The large number of TB patients, coupled with the chemotherapeutic burden, often leads to poor patient adherence and suboptimal treatment outcomes in the developing world, as well as the emergence of multidrug resistant TB (MDR-TB, defined as resistance to isoniazid (INH) and rifampicin (RIF)) and extensively drug-resistant tuberculosis (XDR-TB, defined as MDR-TB plus resistance to any fluoroquinolone and one of three second-line injectable drugs, capreomycin, kanamycin (Kan), and amikacin). To address this global TB health problem, an improved treatment regimen is needed which will reduce treatment duration and prevent relapse and the development of TB drug resistance.3−8 However, achieving this goal will require discovery of multiple novel and mechanistically distinct antimycobacterial agents possessing reduced liabilities for investigation of new drug regimens that might shorten the duration of treatment and simplify management of the disease by improving adherence and reducing costs.9−12
Identification of novel drug targets that will lead to treatment shortening is challenging. Targets of drugs currently in use or phase 3 clinical evaluation for TB chemotherapy include cell wall biosynthesis, translation, transcription, folate synthesis, ATP generation, and maintenance of DNA topology as broad categories, although the nitroimidazoles delaminid and pretomanid generate reactive nitrogen intermediates that inhibit several essential processes.13 Mechanistically novel drugs would conceptually target distinct processes from the above, and to date, successes in the field have all emerged from target identification of hits discovered in Mycobacterium tuberculosis (MTb) whole cell screens.13
In this work, we describe the identification of a 2-mercapto-quinazoline scaffold identified from a MTb whole cell screen, which had been previously reported to inhibit the mycobacterial type II NADH dehydrogenase,14 providing further evidence for its inhibition of the MTb type II NADH dehydrogenase (NDH-2). MTb encodes two NDH-2 genes of which the one encoded by ndh plays a critical role for growth both in vitro and in vivo.15−17 NDH-2 catalyzes the transfer of electrons from NADH into the mycobacterial respiratory pathway and has been proposed to be targeted by a number of early stage inhibitors.16,18−20 In contrast, the proton pumping type I NADH dehydrogenase can be deleted without apparent effects on growth both in vitro or in vivo.15−17 As the current series was considered to have a promising drug-like profile (Table 1), a focused optimization program was initiated.
Table 1. Profiling of Confirmed Hits 1, 2, and 3.

Calculated using StarDrop (http://www.optibrium.com).
Kinetic aqueous solubility was measured using laser nephelometry of compounds in 2.5% DMSO.
Results and Discussion
Screening of a commercial diversity library, details of which will be published elsewhere, afforded a 2-mercapto-quinazolinone cluster of hits (1, 2, and 3), with compounds 1 and 2 showing potent MTb whole-cell minimum inhibitory concentration (MIC) in two distinct growth media. The compounds were assessed in a series of early stage biology profiling assays to understand better the mechanism of action (MoA). These included screening the compounds against a cyd knockout strain which is known to be hyper-susceptible to inhibitors of the cytochrome bc1 complex,21 which suggested that the series was unlikely to target QcrB. Profiling also investigated the extent of upregulation of the promoter of the iniBAC gene cluster, known to be induced by inhibitors of cell wall biosynthesis, such as isoniazid, ethionamide, SQ109, and ethambutol,22,23 which suggested 1 and 2 did not have an effect on cell wall biosynthesis. These biology profiling data were considered promising, especially in conjunction with a recent report14 in which mutations in MTb mutants spontaneously resistant to compound 1 mapped to ndhA, the gene encoding the nonessential type-II NADH dehydrogenase that is involved in NADH reoxidation in the mycobacterial oxidative phosphorylation pathway.14 There are two closely related nonproton pumping type II NADH dehydrogenases in the MTb genome, of which only one (encoded by ndh, Rv1854c) is essential.24−27 Ioerger et al.14 identified promoter mutations in ndhA which resulted in >40-fold upregulation of gene expression, likely compensating for compound 1 inhibiting the essential NDH-2 homologue.
Compound 1 had a promising MTb MIC-derived ligand-lipophilicity efficiency (LLE) drug-likeness profile, suggestive of a quality starting point for medicinal chemistry optimization.28,29 Compound 1 also showed no noticeable cytotoxicity in a mammalian cell line (HepG2). Compounds 1 and 2 also had moderate kinetic solubility and reasonable mouse hepatic microsomal stability, with 1 having excellent human microsomal stability (Table 1). Herein, we report on the development of the structure–activity relationship (SAR) for 1, as well as extended absorption, distribution, metabolism, and excretion (ADME) characterization of key compounds.
Synthetic Chemistry
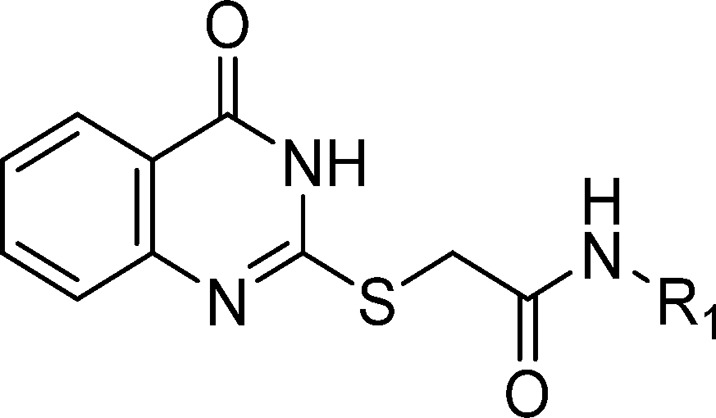
Quinazolinone amides reported herein were synthesized utilizing known procedures, which are detailed in Scheme 1. Commercially available anthranilic acids (28) were cyclized with thiourea, and the resulting 2-mercapto quinazoline-4-diones (29) or commercially available 2-mercapto-4(3H)-quinazolinone was reacted with 2-bromo acetic acid to form the 2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acids (30). Primary and secondary amines (31) were coupled using standard coupling reagents to afford compounds 1, 2, 4–11, and 14.
Scheme 1. General Synthetic Route for Synthesis of Quinazolinone Amides.

Reagents and conditions: General synthetic approach to quinazolinones 1, 2, 4–11, and 14: (a) neat thiourea at 180 °C, 3 h; (b) 2-bromoacetic acid, triethylamine, DMF, 80 °C, 12 h; (c) primary and/or secondary amine 31, EDC·HCl, HOAT, N,N-diisopropylethylamine, DMF/ACN (1:1), room temperature, 12 h or primary and/or secondary amine 31, HATU, N,N-diisopropylethylamine, DCM, room temperature, 12 h.
MTb Whole-Cell SAR
The initial medicinal chemistry plan focused on developing the SAR with the aim of better understanding the pharmacophore. Initial efforts were guided by mouse microsomal metabolite identification (met-ID) studies on 1, which revealed significant oxidation of the cyclohexyl and quinazolin-4(3H)-one rings as well as cleavage of the amide bond (Figure S1). To understand the scope, cycloalkyls 4, 5, and 6 were prepared (Table 2). The large bulky lipophilic cyclohexyl 1 and cycloheptyl 4 were both equally favored, with good whole-cell MIC potency. In contrast, the smaller ring-contracted cyclopentyl 5 and cyclobutyl 6 were slightly less potent (Table 2), and the cyclopropyl and ring deletion NHMe analogues (data not shown) resulted in a complete loss of whole-cell activity, suggesting that a bulky hydrophobic group was required to obtain good whole-cell potency. As mouse met-ID of 1 showed significant oxidation of the cyclohexyl group, the 2-, 3-, and 4-hydroxylated cyclohexyl derivatives were prepared with the aim of improving solubility as well as microsomal stability. Improvements in both kinetic solubility and mouse microsomal stability were indeed achieved, but no whole-cell activity was observed (data not shown). An additional focused set of polar saturated 4-, 5-, and 6-membered oxygen-containing saturated heterocycles was prepared; however, while having good kinetic solubility and mouse microsomal stability, they too lost all whole-cell activity (data not shown). There were also attempts to improve solubility with differentially functionalized 4-substituted piperidines (NAc, NSO2Me, NMe, and NBn), but once again, there was a loss of whole-cell activity (data not shown). As the cyclohexyl group within 1 did not appear to tolerate polar solubilizing groups, we turned our focus to introducing fluorine in an attempt to improve microsomal stability.30,31 We were encouraged to find that the 4,4-difluorocyclohexan 7 retained good whole-cell potency and also had slightly improved mouse microsomal stability versus 1. In comparison to 1, mouse microsomal met-ID on 7 showed a small amount of amide bond cleavage as well as oxidation of the quinazolin-4(3H)-one ring (Figure S2). The 2,2- and 3,3-difluorocyclohexan compounds 8 and 9 had moderate to poor whole-cell potency. Met-ID for both 1 and 7 revealed cleavage of the amide bond, so attempts to hinder the amide through formation of the 1-methyl-, 1-methanol-, and 1-cyano-substituted cyclohexane were explored, all of which resulted in a complete loss of MIC potency as well as increased mouse microsomal instability (data not shown). Addition of a methylene bridge in 1 and 7 to afford 10 and 11 improved MIC potency, further emphasizing the requirement for bulky lipophilic groups.
Table 2. Evaluation R1 (Cyclohexyl) SAR.

Minimum inhibitory concentration (MIC) is the minimum concentration required to inhibit >99% growth of M. tuberculosis in liquid culture. Isoniazid was included as an internal control reference compound (MIC of 0.2 ± 0.1 μM).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes.
Kinetic aqueous solubility was measured using laser nephelometry of compounds in 2.5% DMSO.
There were concerns over the S-linker, based on previous experience from whole-cell screening where confirmed hits with similar S-linker compounds were found to react with glutathione (GSH) both with and without microsomal activation. GSH trapping on 7, with and without human liver microsomes (Figure S3), showed GSH adducts 12 and 13, without microsomal activation. It is presumed that GSH results in cleavage of the sulfur-quinazolinone 7 linker, to afford 12, with GSH coupling to the displaced S-linker to afford 13. Human microsomal oxidation of the quinazolinone ring of 7 was also observed (see Figure 1).
Figure 1.

Metabolite identification of 7 in a GSH trapping experiment.
While the level of GSH adduct formation for 7 was relatively low and no HepG2 cytotoxicity was observed, this was considered a liability of the series as the reactivity did not require microsomal activation and the ability to predict and quantify the risk of idiosyncratic adverse drug reactions is limited.32,33 We attempted to reduce this liability by modifying the linker. N-Methylation of the amide and/or the methylene linker to afford 14, 15, and 16, was not tolerated. The NH-, O-, and CH2-linkers were readily prepared to afford 17, 18, and 19. However, all resulted in a loss of MIC potency (see Table 3). Oxidation of S-atom in compound 1 was attempted using a variety of conditions and oxidation reagents (for example, 3-chlorobenzoic acid, potassium permanganate, and Oxone). However, all attempts failed to deliver the desired sulfone, possibly as a result of increased reactivity.
Table 3. Evaluation of S-Linker SAR.

Minimum inhibitory concentration (MIC) is the minimum concentration required to inhibit >99% growth of M. tuberculosis in liquid culture. Isoniazid was included as an internal control reference compound (MIC of 0.2 ± 0.1 μM).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes.
Kinetic aqueous solubility was measured using laser nephelometry of compounds in 2.5% DMSO.
Changes to the quinazolinone ring were then explored, starting with N-methylation to afford 20, which was not tolerated. Removal of the carbonyl, by synthesis of quinazoline 21 and quinazolin-4-amine 22, was also not tolerated. Saturation of the quinazolinone core phenyl afforded 23, which retained good MIC potency and also resulted in improved solubility, albeit microsomal stability was poor. The related 5-membered saturated analogue 24 showed similar MIC potency but with improved microsomal stability as well as solubility. However, the dimethyl analogue 25 was less well tolerated, suggesting that a bulky hydrophobic ring was preferred. The above SAR suggested that the pyrimidinone core was a key part of the pharmacophore. As human-metID showed oxidation of the quinazolinone ring of 7, we synthesized the presumably more stable fluorine analogue 26 which, while not as potent as 7, retained good MIC potency. However, the solubility was poor, and the mouse microsomal stability was moderate. In an attempt to improve solubility as well as microsomal stability, the pyridopyrimidinone 27 was prepared; while solubility and microsomal stability were improved, the compound had moderate to weak MIC potency. A summary of the overall whole-cell SAR for 1, as well as the effects on both kinetic solubility and microsomal stability, is shown in Figure 2 and in Tables 2–4.
Figure 2.

Overview of structure–activity and property relationships of the mercapto-quinazolinones.
Table 4. Evaluation of Quinazolinone Core.

Minimum inhibitory concentration (MIC) is the minimum concentration required to inhibit >99% growth of M. tuberculosis in liquid culture. Isoniazid was included as an internal control reference compound (MIC of 0.2 ± 0.1 μM).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes.
Kinetic aqueous solubility was measured using laser nephelometry of compounds in 2.5% DMSO.
Pharmacokinetic studies were initiated in order to assess the potential for in vivo efficacy studies of the 2-mercapto-quinazolinones. Compound 1, when dosed as the free base, had reasonable bioavailability, consistent with its moderate Cli and solubility, and good permeability (Table 5). Compound 7 showed a similar bioavailability and exposure profile to 1 (Table 5).
Table 5. Pharmacokinetic Profiling of Compounds 1, 7, and 11.
| 1 | 7 | 11 | |
|---|---|---|---|
| GAST MIC (μM) | 0.3 (95 ng/mL) | 0.4 (141 ng/mL) | 0.8 (294 ng/mL) |
| 7H9-ADC MIC (μM) | 0.6 (190 ng/mL) | 0.4 (141 ng/mL) | 0.8 (294 ng/mL) |
| HepG2 IC50 (μM) | >50 | >50 | >50 |
| measured CHI-LogD | 1.8 | 1.5 | 1.6 |
| microsomal clearance (mL/min/g) | mouse 2.3 | mouse 1.4 | mouse 4.8 |
| human <0.5 | |||
| MWa | 317 | 353 | 367 |
| cLogPa/cLogDa | 2.3/2.3 | 2.3/2.3 | 2.7/2.7 |
| TPSAa | 75 | 75 | 75 |
| PAMPA (nm/s) | 83 | 64 | 65 |
| kinetic solubility (μM)b | 83 (free base) | 111 (HCl salt) | >250 |
| C57 mouse PK at 3 iv and 10 po (mg/kg) | free base | HCl salt | free base |
| Cmax (ng/mL) | 748 | 399 | 1112 |
| T1/2 (h) | 1.5 | 1.3 | |
| AUC0–8h (ng·min/mL) | 156 800 | 67 085 | 128 309 |
| Clb (mL/min/kg) | 23 | 39 | |
| Vdss (L/kg) | 0.8 | 1 | |
| % F | 46 | 29 | |
| PPB (% unbound) | 14 | 13 | 21 |
Calculated using StarDrop (http://www.optibrium.com).
Kinetic aqueous solubility was measured using laser nephelometry of compounds in 2.5% DMSO.
As compound 7 had comparable MIC potency, mouse microsomal stability, and pharmacokinetic profile to 1, a dose linearity study was conducted to evaluate if exposures above the MIC could be achieved. However, no dose linearity was observed between the 30 and 100 mg/kg doses, determined by the area under the curve (AUC) comparison (Figure S4). This may be a consequence of solubility limited absorption at the highest dose. Compound 11 demonstrated better kinetic solubility and comparable MIC to compounds 1 and 7, yet despite an improved Cmax and Tmax compared to compound 1, likely driven by its improved kinetic solubility, it had a lower exposure (AUC) over time (Figure S5 and Table 5), likely a consequence of it is higher clearance (mouse Cli 4.7 mL/min/g versus 2.3 or 1.4 for compounds 1 and 7, respectively). The modest compound exposures upon oral dosing, combined with the lack of ex vivo intramacrophage efficacy (vide infra), suggested that efficacy studies of these compounds in infected mice were unlikely to validate the target/drug candidate pair in an animal model where the disease is macrophage-based. As such, an efficacy experiment was not performed.
Biological Characterization of the Mercapto-Quinazolinones
Previous work on a mercapto-quinazolinone had suggested that compound 1 might target NDH-2 as evidenced by promoter mutations in the nonessential ndhA gene encoding an orthologue of the type II NADH dehydrogenase.14 We similarly identified promoter mutations for ndhA but were not able to identify polymorphisms in the apparently essential ndh (Rv1854c) (Table S1) suggesting either that mutations were deleterious for enzyme function or that single amino acid mutations alone might not sufficiently decrease affinity of this putative inhibitor. The upregulation of ndhA could serve to compensate for loss of NDH-2 function or could serve to bind excess inhibitor in the cell. It is intriguing that ndh promoter mutations were not identified possibly because this gene is not readily upregulated by single nucleotide substitutions in its promoter. We also identified mutations in Rv0678 (Table S1) encoding a transcriptional repressor that has previously been demonstrated to control expression of the MmpS5-MmpL5 transporter and has been implicated in resistance to bedaquiline and clofazimine.34 In accordance with the predicted essential role of NDH-2 as complex I in oxidative phosphorylation,16 treatment of MTb with compound 1 resulted in depletion of cellular ATP levels, a phenomenon also observed with bedaquiline (TM207), an inhibitor of the ATP synthase, as well as EU306, an inhibitor of the cytochrome bc1 complex35 but not with inhibitors of macromolecular biosynthesis including INH (cell wall), Rif (RNA polymerase), and Kan (protein synthesis) (Figure 3).
Figure 3.

Mercapto-quinazolinones deplete cellular ATP levels in MTb. ATP was measured using the BacTiter Glo assay after 24 h of drug exposure and expressed as a fraction of the drug-free vehicle control levels.
We predicted that inhibition of NDH-2 would result in a more severe phenotype on a mutant lacking the second copy of the type II NADH dehydrogenase. Indeed, while treatment of wild-type MTb H37Rv at MIC concentrations could not fully suppress growth of cells over 7 days of treatment, similar concentrations of compound resulted in an almost 2-log kill of a ΔndhA mutant (Figure 4). Despite the higher vulnerability of the ndhA mutant to killing by compound 1, the MIC to this strain, as well as a nuoG deletion mutant lacking a functional type I NADH dehydrogenase, was indistinguishable from WT cells (results not shown). Inhibition of anaerobic nonreplicating cells with compound 1 did not affect viability (Figure S6) which contrasts with the anaerobic cidal activity of other NDH-2 inhibitors including the phenothiazines16 and a recently described quinolone scaffold,19 possibly due to differences in compound access to the target under anaerobic conditions or to growth-dependent differences in compound modification by MTb. In addition, this compound lacked activity against MTb growing in infected macrophages (Figure S7) possibly due GSH-catalyzed compound inactivation or inability to access the mycobacterial phagosome.
Figure 4.
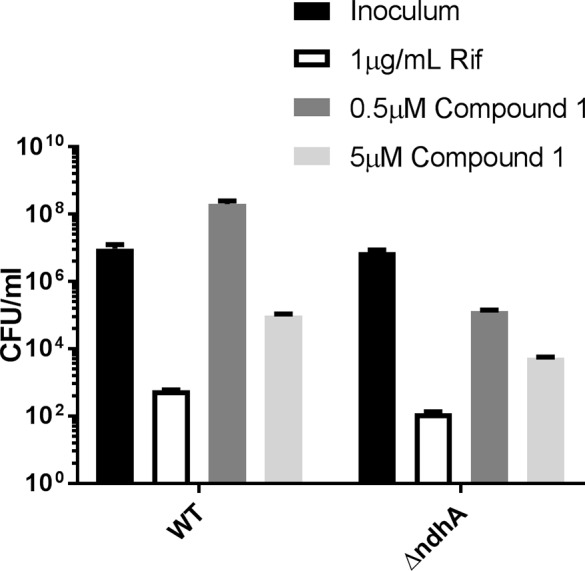
Mtb lacking the alternative type II NADH dehydrogenase encoded by ndhA was more susceptible to compound 1. Colony-forming units (CFU) after 7 days of treatment as compared to CFU at start of drug treatment (inoculum). Unpaired t test comparison between WT and ΔndhA at 0.5 μM compound 1: two-tailed P-value = 0.0023.
The effect of the mercapto-quinazolinones on MTb bioenergetics in whole cells was further demonstrated by analysis of the mycobacterial oxygen consumption rate (OCR) (Figure 5). Addition of compounds 1 and 7 had a minimal effect on basal MTb OCR levels over ∼45 min. The same results were obtained in the presence of glucose or palmitate as carbon sources (Figure 5A,B). Uncoupling of MTb oxidative phosphorylation (OXPHOS) with carbonyl cyanide m-chlorophenyl hydrazone (CCCP) significantly diminished maximal respiration in the presence of the two inhibitors compared to the untreated control. These findings suggest an energy generation pathway common to both fatty acid and glucose oxidation as the target of the compounds. Previous studies21,36 have shown that MTb can rapidly reroute electron flux to overcome inhibition of cytochrome bc1–aa3, cytochrome bd, or ATP synthase, resulting in enhanced respiration. Therefore, we speculated that the target of compounds 1 and 7 was at the point of entry into the electron transport chain (ETC), that is, Complex 1 (NADH dehydrogenase) or Complex II (succinate dehydrogenase). To test this prediction, ATP production was measured in MTb inverted membrane vesicles (IMVs) in the presence of the mercapto-quinazolinones (Figure 5C,D). Notably, ATP production was significantly inhibited when NADH was provided as electron donor but not when succinate was the source of reducing equivalents, suggesting that the site of inhibition was NADH dehydrogenase. The mechanism by which the compounds interfere with NADH dehydrogenase function differs from clofazimine and the quinolinequinones, which have been proposed to interfere with electron transport by activating NDH-2 resulting in production of reactive oxygen species.18,20
Figure 5.

Mercapto-quinazolinones target Complex I of the MTb ETC. At the times indicated by the dotted vertical lines, either glucose (A) or palmitate (B) was added to MTb as carbon source (CS), followed by the mercapto-quinazolinones (Compound) and, last, the uncoupler CCCP to induce maximal respiration. The mercapto-quinazolinones diminished MTb’s uncoupling capacity significantly compared to that of the untreated (UT) control. The oxygen consumption rate (OCR) is reported as a percentage of baseline values. Mercapto-quinazolinones inhibit ATP production in the presence of NADH but not succinate. (C, D) ATP production in inverted membrane vesicles (IMVs) in the presence of NADH or succinate as electron donors after 30 min (C) and over 60 min (D). P-values were calculated by one-way ANOVA using GraphPad Prism 7.02.
To verify that NDH-2 was indeed the target for this class of mercapto-quinazolinones, we expressed MTb NDH-2 (MtNdh) encoded by the predicted essential ndh in E. coli as a recombinant MBP-fusion protein and purified the protein to near homogeneity via amylose-resin affinity chromatography and gel filtration. The recombinant enzyme was highly active with coenzyme Q2 (ubiquinone Q-2) and NADH as substrates, delivering steady-state kinetic parameters similar to previously published results.37 Quinazolinones 1–3, 7, and 11 were found to have submicromolar IC50 values against the MtNdh (Figure 6). Compounds 1, 7, and 11 showed the highest potency, with IC50 values from 7 to 26 nM, values 3-orders of magnitude lower than that for the previously characterized NDH-2 inhibitor chlorpromazine (CPZ; 10–25 μM; Figure 6)24 but superior to the quinolinyl pyrimidines discovered in a target-based screening effort against the MTb NDH-2.38 No time-dependent inhibition was observed for these compounds. The mode of inhibition against MtNdh was further investigated using compound 1: double reciprocal plots of enzyme activity under varying concentrations of 1 and either Q2 or NADH suggested patterns indicative of a noncompetitive mode of inhibition with both substrates (Ki values of 30–40 nM), although the effect on slope was less evident when varying Q2 (Figure 7). This is similar to previous studies with a thiophenazine-based NDH-2 inhibitor39 and suggests that inhibition occurs through binding of the compound at an allosteric site, independent of the main substrate binding pockets.
Figure 6.
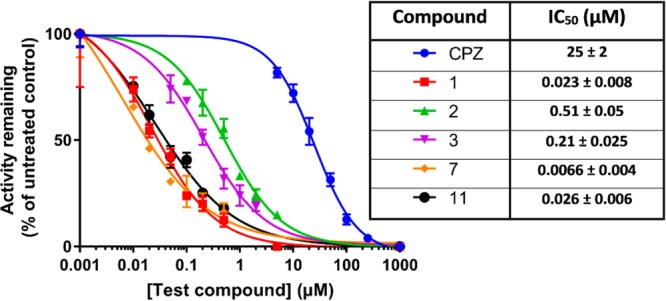
Inhibition of recombinant MBP-MtNdh by select quinazolinones. Steady-state enzyme activity was measured by monitoring absorbance changes at 340 nm due to NADH oxidation, as described in the Experimental Section. NADH and Q2 were fixed at 250 and 40 μM, respectively. Each data point is the mean ± SD of at least 3 independent measurements. Inset table shows calculated IC50 values.
Figure 7.

Double-reciprocal plot of inhibition kinetics of MBP-MtNdh by 1. NADH (left) or Q2 (right) concentrations were varied, in the presence of fixed concentrations of 40 μM Q2 (A) or 250 μM NADH (B). Data points are the average of at least 3 independent measurements. Solid lines represent best-fit values of the data to the general Michaelis–Menten reversible inhibition equation, using nonlinear regression analysis.
In conclusion, the nonproton pumping type II NADH dehydrogenase (NDH-2) has been shown to play a critical role in the respiratory metabolism of bacteria. The 2-mercapto-quinazolinone scaffold is a potent inhibitor of the mycobacterial NDH-2, and although in this study MTb whole-cell SAR only obtained limited improvement in potency, this information, together with the recently disclosed structural information on the enzyme from the bacterium Caldalkalibacillus thermarum,40 will aid in the future rational structure-based development of new and improved NDH-2 inhibitors. Critical for any structure-guided inhibitor design is an understanding of the ligand-binding pocket of the NDH-2 protein. The low amino acid identity between the C. thermarum and MTb NDH-2 proteins (30%) combined with the finding that this inhibitor likely binds to an allosteric site with no allosteric ligand-binding sites identified on the existing bacterial NDH-2 structure40 suggests that cocrystallization of the compound with the MTb NDH-2 protein will be essential to facilitate rational drug design. During revision of this paper, the SAR of a similar mercapto-quinazolinone scaffold41 was reported with a complementary paper describing the importance of the mycobacterial type II NADH dehydrogenase despite the nonessentiality of the individual ndh and ndhA genes.42
Experimental Section
Chemistry Summary
All commercially available reagents, solvents, and starting materials were purchased from Aldrich Chemical Co. (UK). Where necessary, a Biotage FLASH 25+ column chromatography system was used to purify mixtures; reagent-grade solvents used for chromatography were purchased from Fisher Scientific (UK), and flash column chromatography silica cartridges were obtained from Biotage (UK). Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator UV254, from Merck). Developed plates were air-dried and analyzed under a UV lamp (UV 254/365 nm). Microwave irradiation was conducted using a BIOTAGE INITIATOR unit. The machine consists of a continuous focused microwave power delivery system with operator-selectable power output (0–400 W at 2.45 GHz). All 1H and 13C NMR spectra were recorded on a Bruker ARX-500 spectrometer (500 and 300 MHz for 1H and 13C NMR, respectively). Chemical shifts (δ) are reported in ppm relative to the residual solvent peak or internal standard (tetramethylsilane), and coupling constants (J) are reported in hertz (Hz). Data are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, m = multiplet), and integration. LC–MS analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicrOTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer, where both instruments were connected to an Agilent diode array detector. LC–MS chromatographic separations were conducted with a Waters X bridge C18 column, 50 mm × 2.1 mm, 3.5 μm particle size; mobile phase of water/acetonitrile + 0.1% HCOOH or water/acetonitrile + 0.1% NH3; linear gradient from 80:20 to 5:95 over 3.5 min and then held for 1.5 min; flow rate of 0.5 mL·min–1. All assay compounds had a measured purity of ≥95% (by TIC and UV) as determined using this analytical LC–MS system. High resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer.
General Method for the Preparation of Amide Coupling (1, 4–11, 14–16, 19–27)
To a solution of 2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid (1.0 equiv) in a 1:1 ratio of acetonitrile (1.5 mL) and DMF (1.5 mL) were added EDC·HCl (1.0 equiv), HOAT (1.2 equiv), or HATU (1.0 equiv) and primary/secondary amine (1.0 equiv), and the mixture was stirred for 30 min; DIPEA (2.0 equiv) was added to the reaction mixture and stirred at room temperature overnight. If a precipitate had formed this was filtered off, washed with acetonitrile, and air-dried. If the reaction mixture had remained homogeneous, the solution was purified directly by preparative HPLC under acidic conditions, using 0.1% HCOOH in water/acetonitrile (5–95%) gradient elution to afford the product.
N-Cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (1)
To a solution of acid of 2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid (30) (500 mg, 2.1 mmol, 1.0 equiv) in 1:1 ratio of acetonitrile (1.5 mL) and DMF (1.5 mL) were added EDC·HCl (1.2 equiv), HOAT (1.2 equiv), or HATU (804 mg, 2.1 mmol, 1.0 equiv) in DCM (3 mL) and cyclohexylamine (209 mg, 2.1 mmol, 1.0 equiv), and the mixture was stirred for 30 min; DIPEA (0.73 mL, 2.1 mmol, 2.0 equiv) was added to the reaction mixture and stirred at room temperature overnight. The reaction mixture was purified by preparative HPLC under acidic conditions, using 0.1% HCOOH in water/acetonitrile (5–95%) gradient elution to afford a white solid of N-cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide as a white powder (450 mg, 66% yield), C16H19N3O2S, MW 317.41; 1H NMR (500 MHz, DMSO) δ 12.66 (brs, 1H), 8.14 (d, J = 7.9 Hz, 1H), 8.04 (dd, J = 1.3, 7.9 Hz, 1H), 7.80–7.76 (m, 1H), 7.50 (d, J = 7.9 Hz, 1H), 7.45–7.41 (m, 1H), 3.92 (s, 2H), 3.59–3.51 (m, 1H), 1.76–1.64 (m, 4H), 1.55 (dd, J = 3.5, 9.1 Hz, 1H), 1.30–1.12 (m, 5H). LCMS m/z 318.12 [M + 1]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H20N3O2S, 318.1270; found, 318.1284.
N-Cycloheptyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (4)
White powder (21 mg, 23% yield), C17H21N3O2S, MW 331.43; 1H NMR (500 MHz, DMSO) δ 12.65 (brs, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.02 (dd, J = 1.3, 7.9 Hz, 1H), 7.77–7.72 (m, 1H), 7.48–7.37 (m, 2H), 3.88 (s, 2H), 3.73 (ddd, J = 4.2, 8.6, 16.9 Hz, 1H), 1.60–1.34 (m, 12H). LCMS m/z 332.14 [M + 1]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H22N3O2S, 332.1427; found, 332.1439.
N-Cyclopentyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (5)
White powder (39 mg, 24% yield), C15H17N3O2S, MW 303.38; 1H NMR (500 MHz, DMSO) δ 8.23 (d, J = 7.1 Hz, 1H), 8.04 (dd, J = 1.3, 8.0 Hz, 1H), 7.79–7.75 (m, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.45–7.41 (m, 1H), 4.03–3.98 (m, 1H), 3.91 (s, 2H), 1.82–1.76 (m, 2H), 1.67–1.62 (m, 2H), 1.53–1.36 (m, 4H). LCMS m/z 304.12 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C15H18N3O2S, 304.1114; found, 304.1107.
N-Cyclobutyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (6)
White powder (76 mg, 66% yield), C14H15N3O2S, MW 289.35; 1H NMR (400 MHz, DMSO) δ 12.67 (s, 1H), 8.54 (d, J = 7.7 Hz, 1H), 8.05–8.02 (m, 1H), 7.81–7.75 (m, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 4.19 (dd, J = 8.1, 16.1 Hz, 1H), 3.91 (s, 2H), 2.20–2.10 (m, 2H), 1.97–1.85 (m, 2H), 1.67–1.57 (m, 2H). LCMS m/z 290.04 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C14H16N3O2S, 290.0957; found, 290.0962.
N-(4,4-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (7)
To a solution of 2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid 30 (108 mg, 0.4 mmol, 1.0 equiv) in DCM (3 mL) were added HATU (160 mg, 0.4 mmol, 1.0 equiv) and 4,4-difluorocyclohexan-1-amine (57 mg, 0.4 mmol, 1.0 equiv) and the mixture was stirred for 30 min; DIPEA (0.074 mL, 0.4 mmol, 2.0 equiv) was added to the reaction mixture and stirred at room temperature overnight. The reaction mixture was purified by preparative HPLC under acidic conditions, using 0.1% HCOOH in water/acetonitrile (5–95%) gradient elution to afford a white solid of N-(4,4-difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide as a white powder 7 (115 mg, 75% yield), C16H17F2N3O2S, MW 353.39; 1H NMR (500 MHz, DMSO) δ 12.64 (s, 1H), 8.39 (d, J = 6.9 Hz, 1H), 8.02 (dd, J = 1.2, 7.9 Hz, 1H), 7.77–7.72 (m, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 3.90 (s, 2H), 3.77 (t, J = 4.0 Hz, 1H), 2.04–1.75 (m, 6H), 1.56–1.47 (m, 2H). LCMS m/z 354.14 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H18F2N3O2S, 354.1082; found, 354.1091.
N-(4,4-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide HCl Salt Preparation (7)
N-(4,4-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (60 mg, 0.17 mmol, 1.0 equiv) in ethyl acetate (10 mL) and ethanol (3 mL) was heated up to 80 °C, until dissolved (90%). The solution was cooled immediately, followed by the addition of 2 M HCl in diethyl ether (1 mL, 747 mg, 20.49 mmol, 120 equiv) and stirred for a further 15 min, where white precipitates were formed. The precipitate was filtered and dried to afford the N-(4,4-difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide HCl salt (50 mg, 75% yield), C16H17F2N3O2S·HCl, MW 389.85; 1H NMR (500 MHz, DMSO) δ 8.32 (d, J = 7.6 Hz, 1H), 8.04 (dd, J = 1.2, 7.9 Hz, 1H), 7.80–7.76 (m, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.45–7.41 (m, 1H), 3.95 (s, 2H), 2.52–2.50 (m, 1H), 2.05–1.99 (m, 2H), 1.97–1.76 (m, 4H), 1.57–1.48 (m, 2H). LCMS m/z 354.14 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H18F2N3O2S, 354.1082; found, 354.1091.
N-(2,2-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (8)
White powder (93 mg, 62% yield), C16H17F2N3O3S2, MW 353.39; 1H NMR (500 MHz, DMSO) δ 12.64 (s, 1H), 8.46 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.79–7.75 (m, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.43 (dd, J = 7.5 Hz, 1H), 4.03–4.01 (m, 3H), 2.08–2.03 (m, 1H), 1.86–1.63 (m, 4H), 1.49–1.38 (m, 3H). LCMS m/z 354.10 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H18F2N3O3S2, 354.1082; found, 354.1094.
N-(3,3-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (9)
White powder (83 mg, 55% yield), C16H17F2N3O3S2, MW 353.39; 1H NMR (500 MHz, DMSO) δ 12.61 (brs, 1H), 8.31 (d, J = 7.9 Hz, 1H), 8.04 (dd, J = 1.3, 7.9 Hz, 1H), 7.80–7.76 (m, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 3.94 (s, 2H), 3.91–3.75 (m, 1H), 2.21–2.15 (m, 1H), 1.98–1.95 (m, 1H), 1.81–1.68 (m, 4H), 1.45–1.24 (m, 2H). LCMS m/z 354.10 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H18F2N3O3S2, 354.1082; found, 354.1095.
N-(Cyclohexylmethyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (10)
White powder (34 mg, 25% yield), C17H21N3O2S, MW 331.43; 1H NMR (500 MHz, DMSO) δ 12.64 (s, 1H), 8.16 (s, 1H), 8.05–8.02 (m, 1H), 7.79–7.75 (m, 1H), 7.50 (dd, J = 3.1, 8.1 Hz, 1H), 7.44–7.40 (m, 1H), 3.93 (d, J = 2.5 Hz, 2H), 2.95–2.92 (m, 2H), 1.67–1.58 (m, 5H), 1.38–1.34 (m, 1H), 1.12–1.07 (m, 3H), 0.87–0.79 (m, 2H). LCMS m/z 332.21 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H22N3O2S, 332.1427; found, 332.1431.
N-((4,4-Difluorocyclohexyl)methyl)-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (11)
White powder (120 mg, 77% yield), C17H19N3F2O2S, MW 367.41; 1H NMR (500 MHz, DMSO) δ 12.64 (s, 1H), 8.27 (t, J = 5.8 Hz, 1H), 8.04 (dd, J = 1.3, 7.9 Hz, 1H), 7.80–7.76 (m, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.45–7.42 (m, 1H), 3.95 (s, 2H), 3.02 (t, J = 6.4 Hz, 2H), 1.96–1.87 (m, 2H), 1.74–1.52 (m, 5H), 1.17–1.08 (m, 2H). LCMS m/z 368.01 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H20N3F2O2S, 368.1238; found, 368.1255.
N-Cyclohexyl-N-methyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (14)
White powder (35 mg, 25% yield), C17H21N3O2S, MW 331.43; 1H NMR (500 MHz, DMSO) δ 12.60 (brs, 1H), 8.04 (dd, J = 1.6, 7.9 Hz, 1H), 7.80–7.74 (m, 1H), 7.49–7.40 (m, 2H), 4.34 (s, 1H), 4.23 (s, 1H), 3.80–3.76 (m, 1H), 3.01 (s, 2H), 2.74 (s, 1H), 1.76 (t, J = 12.0 Hz, 3H), 1.62–1.52 (m, 2H), 1.52–1.43 (m, 2H), 1.38–1.23 (m, 2H), 1.15–1.05 (m, 1H). LCMS m/z 332.15 [M + H]+. HRMS (m/z): [M + H]+ calcd for C17H22N3O2S, 332.1427; found, 332.1440.
2-((4-Oxo-3,4-dihydroquinazolin-2-yl)thio)propanoic Acid (56)
To a solution of 2-thioxo-2,3-dihydroquinazolin-4(1H)-one 29 (500 mg, 2.8 mmol, 1.0 equiv) were added 2-bromopropanoic acid (557 mg, 3.6 mmol, 1.3 equiv) and triethylamine (2.3 mL, 16.8 mmol, 6.0 equiv) in DMF (5 mL), and the mixture was stirred for 12 h at 75 °C. Once the reaction has completed, it was poured onto crushed ice and acidified with 1 N HCl, where the product precipitated out of the solution. The precipitate was filtered and dried to yield 2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)propanoic acid 56 (548 mg, 78% yield), C11H10N2O3S, MW 250.27; 1H NMR (500 MHz, DMSO) δ 12.66 (s, 1H), 8.04 (q, J = 3.10 Hz, 1H), 7.70 (m, 1H), 7.50 (d, J = 8.00 Hz, 1H), 7.44 (m, 1H), 4.57 (q, J = 7.3 Hz, 2H), 1.55 (d, J = 7.30 Hz, 3H). LCMS m/z 251.00 [M + H]+ 100%.
N-Cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)propanamide (15)
White powder (45 mg, 35% yield), C17H21N3O2S, MW 331.43; 1H NMR (500 MHz, DMSO) δ 12.59 (brs, 1H), 8.17 (d, J = 7.7 Hz, 1H), 8.04 (dd, J = 1.4, 7.9 Hz, 1H), 7.81–7.77 (m, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.45–7.42 (m, 1H), 4.54 (q, J = 7.0 Hz, 1H), 3.56–3.48 (m, 1H), 1.76–1.61 (m, 4H), 1.53–1.50 (m, 4H), 1.29–1.10 (m, 5H). LCMS m/z 332.10 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H22N3O2S, 332.1427; found, 332.1432.
N-Cyclohexyl-N-methyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)propanamide (16)
White powder (40 mg, 29% yield), C18H23N3O2S, MW 345.46; 1H NMR (500 MHz, DMSO) δ 12.60 (brs, 1H), 8.07–8.02 (m, 1H), 7.82–7.75 (m, 1H), 7.50–7.41 (m, 2H), 4.24–4.18 (m, 1H), 3.05 (s, 2H), 2.75 (s, 1H), 1.80–1.70 (m, 3H), 1.64–1.54 (m, 1H), 1.54–1.47 (m, 6H), 1.36–1.20 (m, 2H), 1.14–1.02 (m, 2H). LCMS m/z 346.10 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C18H24N3O2S, 346.1584; found, 346.1608.
N-Cyclohexylglycine (53)
Cyclohexylamine 51 (100 mg, 1.0 mmol, 1.0 equiv) and ethyl 2-hydroxyacetate 52 (104.9 mg, 1.0 mmol, 61.0 equiv) were stirred neat in a round-bottom flask for 12 h. The solvent of the crude mixture was evaporated completely, and the residue was purified via flash column chromatography eluting with 10% DCM in methanol to afford N-Cyclohexylglycine 53 (100 mg, 63% yield), C8H15NO2, MW 157.21; 1H NMR (500 MHz, DMSO) δ 6.34 (brs, 1H), 4.05–4.04 (m, 2H), 3.82–3.77 (m, 1H), 3.77–3.69 (m, 1H), 1.86–1.83 (q, J = 5.28 Hz, 2H), 1.67–1.63 (m, 2H), 1.57–1.53 (m, 1H), 1.35–1.26 (m, 2H), 1.15–1.07 (m, 3H). LCMS m/z 158.21 [M + H]+ 100%.
2-(Methylthio)quinazolin-4(3H)-one (54)
A solution of iodomethane (398 mg, 2.8 mmol, 1.0 equiv) in methanol (8 mL) was added slowly to a closed vessel containing a solution of 2-thioxo-2,3-dihydroquinazolin-4(1H)-one 29 (500 mg, 2.8 mmol, 1.0 equiv) in 1% aqueous NaOH (8 mL) under N2 atmosphere and stirred for 1 h at room temperature. The reaction mixture was then cooled to 0 °C, and the pH was adjusted to 7 with an aqueous solution of 1 N HCl. The solvent was removed under reduced pressure, and the solid was filtered, washed with water/methanol, and dried to afford 2-(methylthio)quinazolin-4(3H)-one 54 (532 mg, 98% yield), C9H8N2OS, MW 192.24; 1H NMR (500 MHz, DMSO) δ 12.62 (s, 1H), 8.09 (q, J = 3.13 Hz, 1H), 7.81 (m, 1H), 7.59 (d, J = 8.10 Hz, 1H), 7.47 (m, 1H), 2.63 (s, 3H). LCMS m/z 193.18 [M + H]+ 100%.
N-Cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)amino)acetamide (17)
2-Amino-N-cyclohexyl-acetamide 55 (40.63 mg, 0.26 mmol, 1.0 equiv), 2-(methylthio)quinazolin-4(3H)-one 54 (50 mg, 0.26 mmol, 1.0 equiv) were suspended in ethyl acetate (2 mL) in a sealed microwave vial and irradiated (0–400 W at 2.65 GHz) at 140 °C for 2 h. Upon completion, the reaction was filtered and the filtrate was extracted with diethyl ether to afford N-cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)amino)acetamide as a white powder 17 (4.4 mg, 4.4% yield), C16H20N4O2, MW 300.36; 1H NMR (500 MHz, DMSO) δ 8.10 (d, J = 7.7 Hz, 1H), 7.58–7.56 (m, 1H), 7.33 (d, J = 8.7 Hz, 1H), 7.22–7.18 (m, 1H), 3.95 (s, 2H), 1.76–1.66 (m, 4H), 1.56–1.53 (m, 1H), 1.31–1.07 (m, 6H). LCMS m/z 301.0 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H21N4O2, 301.1659; found, 301.1665.
N-Cyclohexyl-2-((4-oxo-3,4-dihydroquinazolin-2-yl)oxy)acetamide (18)
2-(Methylthio)quinazolin-4(3H)-one 54 (50 mg, 0.26 mmol, 1.0 equiv) and N-cyclohexylglycine 53 (21.7 mg, 0.12 mmol, 1.0 equiv) were dissolved in THF (2 mL) and heated to 80 °C. Upon reaction completion, the crude product was directly purified via prep-HPLC Gilson purification using 0.1% ammonia in water/acetonitrile 5–95% gradient elution to afford the product as a white powder 18 (10 mg, 24% yield), C16H19N3O3, MW 301.34; 1H NMR (500 MHz, DMSO) δ 12.36 (brs, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.9 (d, 1H), 7.73 (d, J = 8.7 Hz, 1H), 7.40–7.33 (m, 1H), 3.77 (s, 2H), 3.60–3.56 (m, 1H), 1.76–1.66 (m, 4H), 1.57–1.53 (m, 1H), 1.31–1.10 (m, 6H). LCMS m/z 302.1 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H20N3O32, 302.1499; found, 302.1487.
N-Cyclohexyl-3-(4-oxo-3,4-dihydroquinazolin-2-yl)propanamide (19)
White powder (110 mg, 80% yield), C17H21N3O2, MW 299.37; 1H NMR (500 MHz, DMSO) δ 12.18 (s, 1H), 8.08 (dd, J = 1.3, 7.9 Hz, 1H), 7.85–7.76 (m, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.48–7.45 (m, 1H), 3.55–3.48 (m, 1H), 2.83 (t, J = 7.4 Hz, 2H), 2.58 (t, J = 7.4 Hz, 2H), 1.73–1.64 (m, 4H), 1.55–1.52 (m, 1H), 1.28–1.09 (m, 5H). LCMS m/z 300.10 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H22N3O2, 300.1707; found, 300.1721.
2-((3-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic Acid (50)
2-Mercapto-3-methylquinazolin-4(3H)-one 49 (220 mg, 1.14 mmol, 1.0 equiv), 2-bromoacetic acid (206 mg, 1.48 mmol, 1.3 equiv), and triethylamine (0.95 mL, 6.8 mmol, 6.0 equiv) were stirred in DMF (5 mL) for 12 h at 75 °C. Upon reaction completion, the reaction mixture was poured onto crushed ice and acidified with 1 N HCl. The precipitate formed was collected and dried to afford the product 2-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid 50 (250 mg, 87% yield), as a white powder, C11H10N2O3S, MW 250.27; 1H NMR (500 MHz, DMSO) δ 12.89 (s, 1H), 8.08 (q, J = 3.05 Hz, 1H), 7.79 (m, 1H), 7.46 (m, 2H), 4.11 (s, 2H), 3.55 (s, 3H). LCMS m/z 251.04 [M + H]+ 100%.
N-Cyclohexyl-2-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (20)
White powder (20 mg, 15% yield), C17H21N3O2S, MW 331.42; 1H NMR (500 MHz, DMSO) δ 8.15 (d, J = 7.9 Hz, 1H), 8.08 (d, J = 7.9 Hz, 1H), 7.82–7.78 (m, 1H), 7.52 (d, J = 8.2 Hz, 1H), 7.47–7.43 (m, 1H), 3.98 (s, 2H), 3.54 (s, 4H), 1.76–1.65 (m, 4H), 1.55 (d, J = 12.8 Hz, 1H), 1.28–1.11 (m, 5H). LCMS m/z 332.15 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C17H22N3O2S, 332.1427; found, 332.1436.
2-(Quinazolin-2-ylthio)acetic Acid (44)
2-Chloroquinazoline 43 (0.2 g, 1.22 mmol) and sodium thioglycolic acid 39b (1 equiv) were dissolved in DMF, and triethylamine (6.0 equiv) was added. The reaction mixture was heated to 100 °C overnight. The resulting precipitate was filtered and used without any further purification to afford the product 2-(quinazolin-2-ylthio)acetic acid 44 (0.153 g, 57% yield) as a white powder, C10H8N2O2S, MW 220.25; 1H NMR (500 MHz, DMSO) δ 9.41 (d, J = 1.5 Hz, 1H), 8.09 (m, 1H), 7.95–7.93 (m, 1H), 7.79–7.77 (m, 1H), 7.66–7.61 (m, 1H), 4.05 (s, 2H). LCMS m/z 221.1 [M + H]+ 100%.
N-Cyclohexyl-2-(quinazolin-2-ylthio)acetamide (21)
White powder (21 mg, 10% yield), C16H19N3OS, MW 301.41; 1H NMR (300 MHz, DMSO) δ 9.36 (m, 1H), 8.05–8.0 (m, 2H), 7.79–7.76 (m, 1H), 3.95 (s, 2H), 3.52–3.48 (m, 1H), 2.49–2.48 (m, 2H), 1.72–1.32 (m, 5H), 1.25–1.21 (m, 3H). LCMS m/z 302.1 [M + H]+ 100%.
2-((4-Aminoquinazolin-2-yl)thio)acetic Acid (46)
2-Chloroquinazolin-4-amine 45 (0.2 g, 1.11 mmol, 1.0 equiv), sodium thioglycolic acid 39b (0.103 g, 1.11 mmol, 1.0 equiv), and triethylamine were dissolved in DMF (2 mL) (0.93 mL, 6.68 mmol, 6.0 equiv). The reaction mixture was heated to 100 °C overnight. The resulting precipitate was filtered to afford a white powder (0.2 g, 76% yield) and used without further purification. LCMS (ESI) m/z 236.1 [M + H]+
2-((4-Aminoquinazolin-2-yl)thio)-N-cyclohexylacetamide (22)
White powder (21 mg, 10% yield), C16H20N4OS, MW 316.42; 1H NMR (300 MHz, DMSO) δ 8.13 (dd, J = 3.0, 6.0 Hz, 1H), 7.97 (d, J = 6 Hz, 1H), 7.87 (brs, 2H), 7.77–7.69 (m, 1H), 7.51–7.49 (m, 1H), 7.38–7.34 (m, 1H), 3.75 (s, 2H), 3.55–3.45 (m, 1H), 1.76–1.47 (m, 5H), 1.23–1.09 (m, 5H). LCMS m/z 317.1 [M + H]+ 100%.
2-((4-Oxo-3,4,5,6,7,8-hexahydroquinazolin-2-yl)thio)acetic Acid (40)
2-Sulfanyl-5,6,7,8-tetrahydro-4-quinazolinol 38 (0.1 g, 5.49 mmol, 1.0 equiv), chloroacetic acid 39a (1.3 equiv), and triethylamine (6 equiv) were stirred in DMF at room temperature overnight. Once the reaction was complete, the reaction mixture was poured over crushed ice and acidified with 1 N HCl. The white precipitate was collected by filtration and washed with water to afford the product as a white powder (750 mg, 58% yield) which was used without further purification, C10H12N2O3S, MW 240.28; 1H NMR (300 MHz, DMSO) δ 12.59 (brs, 1H), 3.93 (s, 2H), 2.45 (t, J = 3.0 Hz, 2H), 2.29 (t, J = 3.0 Hz, 2H), 1.70–1.63 (m, 4H). LCMS (ESI) m/z 239.1 [M – H]+ 100%.
N-Cyclohexyl-2-((4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-yl)thio)acetamide (23)
White powder (43 mg, 17% yield), C16H23N3O2S, MW 321.44; 1H NMR (300 MHz, DMSO) δ 12.47 (brs, 1H), 8.00 (brs, 1H), 3.80 (s, 2H), 3.58–3.49 (m, 1H), 2.47 (t, J = 3.0 Hz, 2H), 2.31 (t, J = 3.0 Hz, 2H), 1.75–1.54 (m, 9H), 1.29–1.13 (m, 5H). LCMS m/z 320.1 [M – H]+ 100%.
2-((4-Oxo-4,5,6,7-tetrahydro-3H-cyclopenta[d]pyrimidin-2-yl)thio)acetic acid (42)
2-Thioxo-1,2,3,5,6,7-hexahydro-4H-cyclopenta[d]pyrimidin-4-one 41 (1.0 g, 5.94 mmol, 1.0 equiv), chloroacetic acid 39a (1.3 equiv), and triethylamine (6 equiv) were stirred in DMF (2 mL) at room temperature overnight. Once the reaction was complete, it was poured over crushed ice and acidified with 1 N HCl. The white precipitate was collected by filtration and washed with water to afford the product as a white powder (800 mg, 59% yield), which was used without further purification, C9H10N2O3S, MW 226.05; 1H NMR (300 MHz, DMSO) δ 12.65 (brs, 1H), 3.96 (s, 2H), 2.71 (t, J = 3.0 Hz, 2H), 2.58 (t, J = 3.0 Hz, 2H), 1.97–1.93 (m, 2H). LCMS m/z 227.1 [M + H]+ 100%.
N-Cyclohexyl-2-((4-oxo-4,5,6,7-tetrahydro-3H-cyclopenta[d]pyrimidin-2-yl)thio)acetamide(24)
White powder (59 mg, 22% yield), C15H23N3O2S, MW 307.41; 1H NMR (300 MHz, DMSO) δ 12.59 (brs, 1H), 8.03 (s, 1H), 3.84 (s, 2H), 3.58–3.49 (m, 1H), 2.71 (t, J = 3.0 Hz, 2H), 2.60 (t, J = 3.0 Hz, 2H), 2.0–1.92 (m, 2H), 1.75–1.52 (m, 5H), 1.29–1.16 (m, 5H). LCMS (ESI) m/z 308.1 [M + H]+ 100%.
2-((4,5-Dimethyl-6-oxo-1,6-dihydropyrimidin-2-yl)thio)acetic Acid (48)
5,6-Dimethyl-2-thiouracil 47 (0.5 g, 3.20 mmol), chloroacetic acid 39a (0.39 g, 4.16 mmol), and triethylamine (2.62 mL, 1.94 mmol) were stirred in DMF (2 mL) at room temperature overnight. Once the reaction was complete, it was poured over crushed ice and acidified with 1 N HCl. The white precipitate was collected by filtration and washed with water to afford the product as a white powder (0.36 g, 54% yield) which was used without further purification, C8H10N2O3S, MW 214.24; 1H NMR (300 MHz, DMSO) δ 12.56 (brs, 1H), 3.89 (s, 2H), 2.14 (s, 3H), 1.85 (s, 3H). LCMS (ESI) m/z 213.1 [M – H]; HPLC purity 98%.
N-Cyclohexyl-2-((4,5-dimethyl-6-oxo-1,6-dihydropyrimidin-2-yl)thio)acetamide (25)
White powder (0.047 g, 17% yield), C14H21N3O2S, MW 295.40; 1H NMR (400 MHz, DMSO-d6) δ 7.99 (dd, J = 20.3, 8.1 Hz, 1H), 3.80 (s, 2H), 3.11–2.97 (m, 1H), 2.20 (s, 3H), 1.89 (s, 3H), 1.78–1.64 (m, 5H), 1.33–1.12 (m, 5H). LCMS (ESI) m/z 296.1 [M + H]+ 100%.
6-Fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (33)
2-Amino-5-fluorobenzoic acid 32 (1 g, 6.5 mmol, 1.0 equiv) and thiourea (2.1 g, 25.8 mmol, 6.0 equiv) were heated neat in a round-bottom flask to 180 °C for 3 h with stirring. The reaction was cooled, and 25 mL of water was added. After stirring for 10 min, the formed precipitate was filtered off, washed with cold water, and dried to afford 6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one 33 (0.45 g, 35.9% yield) as a yellow powder, C8H5FN2OS, MW 196.2; 1H NMR (500 MHz, DMSO) δ 9.72 (s, 1H), 7.51–7.39 (m, J = 8.5 Hz, 1H), 7.37–7.30 (m, J = 8.6 Hz, 1H), 7.19–7.16 (m, J = 8.85 Hz, 1H). LCMS m/z 197.78 [M + H]+ 100%.
2-((6-Fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic Acid (34)
6-Fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one 33 (414 mg, 2.1 mmol, 1.0 equiv), 2-bromoacetic acid (586.39 mg, 4.2 mmol, 2.0 equiv), and triethylamine (1.76 mL, 12.6 mmol, 6.0 equiv) were stirred in DMF (5 mL) at 75 °C for 12 h. Upon reaction completion, the reaction mixture was poured over crushed ice and acidified with 1 N HCl. The precipitate was filtered and dried to afford 2-((6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid 34 (150 mg, 27.9% yield), C10H7FN2O3S, MW 254.24; 1H NMR (500 MHz, DMSO) δ 12.88 (s, 1H), 7.72 (q, J = 3.87 Hz, 1H), 7.65 (m, 1H), 7.54 (q, J = 4.62 Hz, 1H), 4.03 (s, 2H). LCMS m/z 254.10 [M + H]+ 100%.
N-(4,4-Difluorocyclohexyl)-2-((6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (26)
To a solution of 2-((6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetic acid 34 (100 mg, 0.39 mmol, 1.0 equiv) in a 1:1 ratio of acetonitrile (1.5 mL) and DMF (1.5 mL) were added EDC·HCl (90 mg, 0.74 mmol, 1.2 equiv), HOAT (64 mg, 0.47 mmol, 1.2 equiv), and 4,4-difluorocyclohexan-1-amine (67 mg, 0.39 mmol, 1.0 equiv), and the mixture was stirred for 30 min. DIPEA (0.13 mL, 0.78 mmol, 2.0 equiv) was added, and the resulting mixture was stirred at room temperature overnight. The crude mixture was purified by preparative HPLC under acidic conditions, using 0.1% HCOOH in water/acetonitrile (5–95%) gradient elution to afford N-(4,4-difluorocyclohexyl)-2-((6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide as a white powder 26 (12 mg, 8.2% yield), C16H16N3F3O2S, MW 371.86; 1H NMR (500 MHz, DMSO) δ 12.76 (brs, 1H), 8.39–8.36 (m, 1H), 7.71–7.60 (m, 2H), 7.52 (dd, J = 5.0, 8.9 Hz, 1H), 3.88 (s, 2H), 3.77 (d, J = 8.5 Hz, 1H), 2.08–1.77 (m, 6H), 1.55–1.47 (m, 2H). LCMS m/z 372.3 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C16H17N3F3O2S, 372.0988; found, 372.0987.
2-Thioxo-2,3-dihydropyrido[3,4-d]pyrimidin-4(1H)-one (36)
2-Aminopyridine-4-carboxylic acid 35 (1 g, 7.2 mmol, 1.0 equiv) and thiourea (2.8 g, 36.2 mmol, 5.0 equiv) were heated neat in a round-bottom flask to 180 °C for 3 h with stirring. The reaction was cooled, and 25 mL of water was added. After stirring for 10 min, the formed precipitate was filtered off, washed with cold water, and dried to afford 2-thioxo-2,3-dihydropyrido[3,4-d]pyrimidin-4(1H)-one 36 (1.11 g, 85% yield) as a yellow powder, C7H5FN2OS, MW 179.2; 1H NMR (500 MHz, DMSO) δ 12.89 (brs, 1H), 12.70 (brs, 1H), 8.73 (s, 1H), 8.49 (d, J = 4.85 Hz, 1H), 7.79 (d, J = 0.80 Hz, 1H). LCMS m/z 180.11 [M + H]+ 100%.
2-((4-Oxo-3,4-dihydropyrido[3,4-d]pyrimidin-2-yl)thio)acetic Acid (37)
2-Thioxo-2,3-dihydropyrido[3,4-d]pyrimidin-4(1H)-one 36 (500 mg, 2.79 mmol, 1.0 equiv), 2-bromoacetic acid (775.39 mg, 5.5 mmol, 2.0 equiv), and triethylamine (2.33 mL, 16.7 mmol, 6.0 equiv) were stirred in DMF (5 mL) at 75 °C for 12 h. Upon reaction completion, the reaction mixture was poured over crushed ice and acidified with 1 N HCl, and the precipitate was collected and dried to afford 2-((4-oxo-3,4-dihydropyrido[3,4-d]pyrimidin-2-yl)thio)acetic acid 37 (600 mg, 90.6% yield), C9H7N3O3S, MW 237.24; 1H NMR (500 MHz, DMSO) δ 12.89 (s, 1H), 8.85 (d, J = 0.65 Hz, 1H), 8.57 (d, J = 5.00 Hz, 1H), 7.88 (d, J = 0.80 Hz, 1H), 4.08 (s, 2H). LCMS m/z 238.15 [M + H]+ 100%.
N-(4,4-Difluorocyclohexyl)-2-((4-oxo-3,4-dihydropyrido[3,4-d]pyrimidin-2-yl)thio)acetamide (27)
White powder (64 mg, 42% yield), C15H16N4O2F2S, MW 354.38; 1H NMR (500 MHz, DMSO) δ 13.02 (s, 1H), 8.86 (s, 1H), 8.57 (d, J = 5.2 Hz, 1H), 8.29 (d, J = 7.4 Hz, 1H), 7.87 (d, J = 5.0 Hz, 1H), 3.98 (s, 2H), 3.31 (s, 1H), 2.09–1.99 (m, 2H), 1.98–1.86 (m, 2H), 1.86–1.77 (m, 2H), 1.58–1.48 (m, 2H). LCMS m/z 355.22 [M + H]+ 100%. HRMS (m/z): [M + H]+ calcd for C15H17N4O2F2S, 355.1034; found, 355.1033.
Intrinsic Clearance (Cli) Experiments
Test compound (0.5 μM) was incubated with female CD1 mouse liver microsomes (Xenotech LLC; 0.5 mg/mL, 50 mM potassium phosphate buffer, pH 7.4), and the reaction started with addition of excess NADPH (8 mg/mL, 50 mM potassium phosphate buffer, pH 7.4). Immediately, at time zero, and then at 3, 6, 9, 15, and 30 min, an aliquot (50 μL) of the incubation mixture was removed and mixed with acetonitrile (100 μL) to stop the reaction. Internal standard was added to all samples; the samples were centrifuged to sediment precipitated protein, and the plates were then sealed prior to UPLC-MS/MS analysis using a Quattro Premier XE (Waters Corporation, USA). XLfit (IDBS, UK) was used to calculate the exponential decay and consequently the rate constant (k) from the ratio of peak area of test compound to internal standard at each time point. The rate of intrinsic clearance (Cli) of each test compound was then calculated using the following calculation.
where V (mL/mg protein) is the incubation volume/mg protein added and microsomal protein yield is taken as 52.5 mg protein/g liver. Verapamil (0.5 μM) was used as a positive control to confirm acceptable assay performance.
Aqueous Solubility
The aqueous solubility of the test compounds was measured using laser nephelometry. Compounds were subject to serial dilution from 10 to 0.5 mM in DMSO. An aliquot was then mixed with Milli-Q water to obtain an aqueous dilution plate with a final concentration range of 250–12 μM, with a final DMSO concentration of 2.5%. Triplicate aliquots were transferred to a flat bottomed polystyrene plate which was immediately read on the NEPHELOstar (BMG Lab Technologies). The amount of laser scatter caused by insoluble particulates (relative nephelometry units, RNU) was plotted against compound concentration using a segmental regression fit, with the point of inflection being quoted as the compounds aqueous solubility (μM).
CHI LogD Determination
The chromatographic hydrophobicity index (CHI) was determined according the method previously described.43,44 Briefly, test compounds were prepared as 0.5 mM solutions in 50:50 acetonitrile/water and analyzed by reversed-phase HPLC-UV (wavelength 254 nm) using a Phenomenex Luna C18 100 Å, 150 × 4.6 mm, 5 μm column with a gradient of aqueous phase (50 mM ammonium acetate (pH 7.4)) and mobile phase (acetonitrile). By plotting the retention time of a set of reference compounds against known CHI values, the CHI value of test compounds was calculated according to their retention time.
Plasma Protein Binding Experiments
In brief, a 96-well equilibrium dialysis apparatus was used to determine the free fraction in plasma for each compound (HT Dialysis LLC, Gales Ferry, CT). Membranes (12–14 kDaA cutoff) were conditioned in deionized water for 60 min, followed by conditioning in 80:20 deionized water/ethanol overnight and then rinsed in water and isotonic buffer before use. Mouse plasma from appropriate species was removed from the freezer and allowed to thaw on the day of experiment. Thawed plasma was then centrifuged (Allegra X12-R, Beckman Coulter, USA) and spiked with test compound (final concentration 10 μg/mL), and 150 μL aliquots (n = 3 replicate determinations) were loaded into the 96-well equilibrium dialysis plate. Dialysis against isotonic buffer (150 μL) was carried out for 5 h in a temperature controlled incubator at ca. 37 °C (Barworld scientific Ltd., UK) using an orbital microplate shaker at 100 rpm (Barworld scientific Ltd., UK). At the end of the incubation period, 50 μL aliquots of plasma or buffer were transferred into a 96-well deep plate and the composition in each well was balanced with control fluid (50 μL), such that the volume of buffer to plasma is the same. Sample extraction was performed by the addition of 200 μL of acetonitrile containing an appropriate internal standard. Samples were allowed to mix for 1 min and then centrifuged at 3000 rpm in 96-well blocks for 15 min (Allegra X12-R, Beckman Coulter, USA) after which 150 μL of supernatant was removed to 50 μL of water. All samples were analyzed by UPLC-MS/MS on a Quattro Premier XE Mass Spectrometer (Waters Corporation, USA). The unbound fraction was determined as the ratio of the peak area in buffer to that in plasma.
PAMPA Assay
The permeability was performed using a 96-well precoated BD Gentest PAMPA plate (BD Biosciences, U.K.). Each well was divided into two chambers: donor and acceptor, separated by a lipid-oil-lipid trilayer constructed in a porous filter. The effective permeability, Pe, of the compound was measured at pH 7.4. Stock solutions (5 mM) of the compound were prepared in DMSO. The compound was then further diluted to 10 μM in phosphate buffered saline at pH 7.4. The final DMSO concentration did not exceed 5% v/v. The compound dissolved in phosphate buffered saline was then added to the donor side of the membrane, and phosphate buffered saline without compound was added to the acceptor side. The PAMPA plate was left at room temperature for 5 h; after which time, an aliquot (100 μL) was then removed from both acceptor and donor compartments and mixed with acetonitrile (80 μL) containing an internal standard: donepezil at 50 ng/mL). The samples were centrifuged (10 min, 5 °C, 3270g) to sediment precipitated protein and sealed prior to UPLC-MS/MS analysis using a Quattro Premier XE (Waters Corp, USA). Pe was calculated as shown in the below equation.
where CA(t) = peak area of compound present in acceptor well at time (t) = 18 000 s; Cequi = [CD(t) × VD + CA(t) × VA]/(VD + VA); A = filter area; VD = donor well volume; VA = acceptor well volume; t = incubation time; CD(t) = peak area of compound present in donor well at time (t) = 18 000 s. Recovery of compound from donor and acceptor wells was calculated, and data was only accepted when recovery exceeded 70%.
Animal Care Assurance
All mouse studies performed at the University of Dundee were performed under the authority of a project license (PPL70/8346) issued by the Home Office under the Animals (Scientific Procedures) Act 1986, as amended in 2012 (and in compliance with EU Directive EU/2010/63). License applications are approved by the University’s Ethical Review Committee (ERC) before submission to the Home Office. The ERC develops and oversees policy on all aspects of the use of animals on University premises and is a subcommittee of the University Court, its highest governing body.
Mouse Pharmacokinetics
Test compound was dosed as a bolus solution intravenously at 3 mg free base/kg (dose volume: 5 mL/kg; dose vehicle: 10% DMSO; 40% PEG400; 50% saline) to female C57Bl/6 mice (n = 3) or dosed orally by gavage as a suspension at 10 or at 5, 30, or 100 mg free base/kg (dose volume: 10 mL/kg; dose vehicle: 1.0% carboxy methyl cellulose) to female C57Bl/6 mice (n = 3/dose level). Blood samples were taken from each mouse tail vein at predetermined time points postdose, mixed with nine volumes of distilled water, and stored frozen until UPLC-MS/MS analysis. Pharmacokinetic parameters were derived from the blood concentration time curve using PK Solutions software v 2.0 (Summit Research Services, USA).
Strains and Media
Mycobacterium tuberculosis H37Rv (ATCC 27294) was used for all work with this organism unless otherwise indicated. The activity of the compound on cell wall synthesis and the bc1 complex was measured using the piniBAC reporter strain and the cydC::aph strains, respectively, using previously described methods.35,45 Broth-based medium (7H9/ADC/Tw) for MTb consisted of Middlebrook 7H9 (Becton Dickinson) supplemented with 10% ADC [albumin (50 g/L)/dextrose (20 g/L)/NaCl (8.1 g/L)], 0.2% glycerol] and 0.05% Tween 80. Solid growth medium for MTb was Middlebrook 7H11 (Becton Dickinson) supplemented with OADC [ADC with 0.06% oleic acid]. Potencies of compounds by MIC determination were performed as previously described.45 The ΔndhA strain was generated by insertion of the aph gene into the native BamHI restriction site 873 nucleotides downstream of the start codon using the previously described suicide vector method46 to effect legitimate recombination in MTb.
Measurement of Intracellular ATP Levels
MTb H37Rv was grown to early log phase (OD650 nm of 0.2) and diluted 20-fold in the same medium (7H9/ADC/Tw). 100 μL aliquots of cells were added to each well of sterile white polystyrene 96-well plates (Corning Inc., Corning, NY) containing 2 μL drug dilutions in DMSO in triplicate. ATP was measured at different time points as indicated by a bioluminescent assay using the BacTiter-Glo reagent (Promega, Madison, WI) as recommended by the manufacturer.
Bioenergetics
The extracellular flux analysis assays were performed as described previously.21,36 In short, M. tuberculosis H37Rv were grown in 7H9 (10% OADC, 0.01% tyloxapol) to an OD of 0.8. Bacilli were pelleted, suspended in unbuffered carbon-source-free XF 7H9 media, and seeded into the XF culture plate, at a density of 2 × 106 bacilli/well. During the XF screen, the oxygen consumption rate (OCR) was measured, noninvasively and in real time. OCR data points are representative of the average OCR during 3 min of continuous measurement in the transient microchamber, with the error being calculated from the OCR measurements taken from eight replicate wells by the Wave Desktop 2.3 software (Agilent). During the assay carbon sources (glucose and palmitate at a final concentration of 0.2%), the mercapto-quinazolinones (Compounds 1 and 7 at final concentrations of 39 and 45 μM, respectively) and CCCP (final concentration of 3 μM) were added to the assay media at the indicated time.
Inverted Membrane Vesicle (IMV)
IMVs were isolated, and the ATP generation assay was performed as described previously.36 In brief, MTb mc26230 (cultured in 7H9, 0.01% tyloxapol, 10% OADC, 0.2% casamino acids, and 24 μg/mL pantothenic acid) were collected via centrifugation, incubated with lysozyme, and lysed via bead beating in a buffer containing 10 mM HEPES, 50 mM KCl, 5 mM MgCl2, and 10% glycerol. IMVs were isolated by differential centrifugation. The ATP synthesis assay was performed using a Roche Bioluminescence Assay Kit CL II. Membrane vesicles were provided either 250 μM NADH or 1 mM succinate as an electron donor, 50 μM ADP, and 5 mM phosphate in the above buffer containing the luciferase/luciferin reagents. Luminescence was measured in a 384-well plate using a BioTek Synergy H4 plate reader.
Recombinant Rv1854c Purification
The ORF corresponding to Rv1854c was codon optimized for E. coli, synthesized (ThermoFisher), and cloned between the NcoI and HindIII sites of pMALc5x, affording a vector for the IPTG-inducible overexpression of MtNdh with an N-terminal maltose-binding protein (MBP) fusion tag. This vector was transformed into E. coli BL21 (DE3) pLysS, and protein expression was achieved in 1 L of media (ZYM + 0.1% glucose, 0.1% glycerol, 2 mM MgSO4, 34 μg/mL chloramphenicol, 50 μg/mL carbenicillin) following induction with 0.3 mM IPTG at 37 °C for 4 h. Cells were harvested, resuspended in 40 mL of buffer A (50 mM HEPES, 100 mM KPO4, pH 7.1, 5% (w/v) glycerol) with added 5 U/mL DNase, complete protease inhibitor, 5 μM FAD, 10 mM MgCl2, and 0.25% CHAPS, and sonicated on ice to lyse. Following centrifugation at 20 000g for 30 min, the soluble fraction was incubated with 2 mL of settled volume of pre-equilibrated (buffer A + 0.25% CHAPS) amylose resin for 30 min at 4 °C and then applied to a gravity flow column. The resin was washed with 15 CV buffer A + 0.25% CHAPS and then 15 CV buffer A without CHAPS. Bound protein was eluted with 10 mL of buffer A + 20 mM maltose. The same purification procedure was repeated twice on the initial resin flow-throughs; all eluates were combined and concentrated using spin columns. The concentrated prep was then passed through a Superdex 75 10/300 column (running buffer was buffer A), and relevant fractions were collected and concentrated. Aliquots of the resulting weakly yellow solution were flash frozen and stored at −80 °C until needed. Protein concentration was measured by spectrophotometric flavin (FAD) analysis, as described in ref (47).
Ndh Activity Assays
MtNdh catalytic activity was measured by monitoring NADH oxidation at 340 nm (ε = 6220 M–1 cm–1) in a UV–vis spectrophotometer at RT. Reactions (1 mL) consisted of 0.1 M HEPES, pH 7.0, 10% DMSO, 0.1–0.5 nM recombinant MBP-MtNdh, and varying concentrations of Q2 and NADH. Reactions were started by addition of NADH, following ∼1 min preincubation of all other reaction components. Kinetic data was taken at steady state within the first 1 min of the reaction. Background NADH oxidation in the absence of Q2 was measured (typically <5% of rate with Q2 present) and subtracted from all subsequent measurements, as required. All kinetic data was plotted and analyzed using GraphPad Prism software.
Acknowledgments
This work was funded in part by the Intramural Research Program of NIAID (AI000693-25), by grants from the Foundation for the National Institutes of Health (BARRY11HTB0) with support from the Bill & Melinda Gates Foundation (OPP1024021), and by the South African Medical Research Council (SAMRC) with funds received from Strategic Health Innovation Partnerships (SHIP) unit of the SAMRC, by the National Research Foundation of South Africa, and OPP1066891 “A Centre of Excellence for Lead Optimization for Diseases of the Developing World”, a joint award from the Wellcome Trust and the Bill & Melinda Gates Foundation. A.J.C.S. is a Burroughs Welcome Investigator in the Pathogenesis of Infectious Diseases. P.M.F. was a Howard Hughes Medical Institute Medical Research Fellow. We thank David Gray, James Roberts, and Alastair Pate for support with data analysis, compound handling, and data management.
Glossary
Abbrevations
- NDH-2
type II NADH dehydrogenase
- MTb
Mycobacterium tuberculosis
- MtNdh
MTb NDH-2
- SAR
structure–activity relationship
- Kan
kanamycin
- INH
isoniazid
- RIF
rifampicin
- MDR-TB
multidrug resistant TB
- XDR-TB
extensively drug-resistant tuberculosis
- MoA
mechanism of action
- MIC
minimum inhibitory concentration
- AUC
area under the curve
- IMVs
inverted membrane vesicles
- OCR
oxygen consumption rate
- LLE
ligand-lipophilicity efficiency
- ADME
absorption, distribution, metabolism, and excretion
- met-ID
metabolite identification
- cLogP
calculated logP
- Cli
intrinsic clearance
- CFU
colony-forming units
- OXPHOS
oxidative phosphorylation
- CCCP
carbonyl cyanide m-chlorophenyl hydrazine
- ETC
electron transport chain
- CPZ
chlorpromazine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00275.
Compound syntheses and characterization, Met-ID studies of compounds 1 and 7 with microsomes with and without GSH, graphs showing metabolic transformations in Met-ID studies, pharmacokinetic studies, anaerobic cidal and intramacrophage activity of compound 1, and whole genome sequencing methods and data table (PDF)
Author Present Address
¤ P.M.F.: Department of Internal Medicine, University of Pittsburgh, 1218 Scaife Hall, 3550 Terrace Street, Pittsburgh, Pennsylvania 15261, USA.
Author Contributions
α D.M. and P.C.R. contributed equally.
Author Contributions
H.I.M.B., D.F.W., K.A., and K.Y.R. performed biological experiments on MTb. T.R.I. and J.S. performed sequencing analysis of resistant mutants. G.A.P. performed enzyme assays. D.A.L., P.M.F., and A.J.C.S. performed and analyzed bioenergetics. D.M., P.C.R., T.B., J.R.H., K.G., C.S.M., T.-S.F., L.J.S., and K.C. synthesized compounds. P.S., L.E., J.R., Y.S., L.F., M.O.-C., O.E., and K.D.R. performed ADME/PK analysis. H.I.M.B., S.R.G., C.E.B., P.C.R., P.G.W., and V.M. helped with data interpretation. P.C.R., H.I.M.B., and S.R.G. wrote the manuscript. D.F.W., T.-S.F., C.S.M., C.E.B., and D.M. edited the manuscript which was reviewed by all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. (2015) Global tuberculosis report 2015, World Health Organization, Geneva, Switzerland; ISBN 978-92-4-156505-9. [Google Scholar]
- WHO. (2010) Treatment of tuberculosis: guidelines, World Health Organization, Geneva, Switzerland; ISBN 978-92-4-154783-3. [Google Scholar]
- Horsburgh C. R. J.; Barry C. E. I.; Lange C. (2015) Treatment of Tuberculosis. N. Engl. J. Med. 373 (22), 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Iseman M. D. (1993) Treatment of Multidrug-Resistant Tuberculosis. N. Engl. J. Med. 329 (11), 784–791. 10.1056/NEJM199309093291108. [DOI] [PubMed] [Google Scholar]
- Migliori G. B.; Loddenkemper R.; Blasi F.; Raviglione M. C. (2007) 125 years after Robert Koch’s discovery of the tubercle bacillus: the new XDR-TB threat. Is “science” enough to tackle the epidemic?. Eur. Respir. J. 29 (3), 423–427. 10.1183/09031936.00001307. [DOI] [PubMed] [Google Scholar]
- Velayati A. A.; Masjedi M. R.; Farnia P.; Tabarsi P.; Ghanavi J.; ZiaZarifi A. H.; Hoffner S. E. (2009) Emergence of New Forms of Totally Drug-Resistant Tuberculosis Bacilli: Super Extensively Drug-Resistant Tuberculosis or Totally Drug-Resistant Strains in Iran. Chest 136 (2), 420–425. 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- Schluger N. W. (2009) Drug-Resistant Tuberculosis. Chest 136 (2), 333–335. 10.1378/chest.09-0785. [DOI] [PubMed] [Google Scholar]
- Palomino J. C.; Martin A. (2014) Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 3 (3), 317–340. 10.3390/antibiotics3030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A. I.; Gillespie S. H.; Hoelscher M.; Philips P. P. J.; Cole S. T.; Abubakar I.; McHugh T. D.; Schito M.; Maeurer M.; Nunn A. J. (2014) New antituberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. Lancet Infect. Dis. 14 (4), 327–340. 10.1016/S1473-3099(13)70328-1. [DOI] [PubMed] [Google Scholar]
- Selassie A. W.; Pozsik C.; Wilson D.; Ferguson P. L. (2005) Why Pulmonary Tuberculosis Recurs: A Population-based Epidemiological Study. Annals of Epidemiology 15 (7), 519–525. 10.1016/j.annepidem.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Owens J. P.; Fofana M. O.; Dowdy D. W. (2013) Cost-effectiveness of novel first-line treatment regimens for tuberculosis. International Journal of Tuberculosis and Lung Disease 17 (5), 590–596. 10.5588/ijtld.12.0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhardt C.; Vernon A.; Raviglione M. C. (2010) New drugs and new regimens for the treatment of tuberculosis: review of the drug development pipeline and implications for national programmes. Curr. Opin. Pulm. Med. 16 (3), 186–193. 10.1097/MCP.0b013e328337580c. [DOI] [PubMed] [Google Scholar]
- Cole S. T. (2016) Inhibiting Mycobacterium tuberculosis within and without. Philos. Trans. R. Soc., B 371 (1707), 20150506. 10.1098/rstb.2015.0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R.; O’Malley T.; Liao R.; Guinn K. M.; Hickey M. J.; Mohaideen N.; Murphy K. C.; Boshoff H. I. M.; Mizrahi V.; Rubin E. J.; Sassetti C. M.; Barry C. E.; Sherman D. R.; Parish T.; Sacchettini J. C. (2013) Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium tuberculosis. PLoS One 8 (9), e75245. 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. J.; Reddy M. C.; Ioerger T. R.; Rothchild A. C.; Dartois V.; Schuster B. M.; Trauner A.; Wallis D.; Galaviz S.; Huttenhower C.; Sacchettini J. C.; Behar S. M.; Rubin E. J. (2013) Tryptophan Biosynthesis Protects Mycobacteria from CD4 T-Cell-Mediated Killing. Cell 155 (6), 1296–1308. 10.1016/j.cell.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. P. S.; Alonso S.; Rand L.; Dick T.; Pethe K. (2008) The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105 (33), 11945–11950. 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus M. A.; Gerrick E. R.; Xu W.; Park S. W.; Long J. E.; Boutte C. C.; Rubin E. J.; Schnappinger D.; Ehrt S.; Fortune S. M.; Sassetti C. M.; Ioerger T. R. (2017) Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio 8 (1), e02133-16. 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikal A.; Hards K.; Cheung C.-Y.; Menorca A.; Timmer M. S. M.; Stocker B. L.; Cook G. M. (2016) Activation of type II NADH dehydrogenase by quinolinequinones mediates antitubercular cell death. J. Antimicrob. Chemother. 71 (10), 2840–2847. 10.1093/jac/dkw244. [DOI] [PubMed] [Google Scholar]
- Hong W. D.; Gibbons P. D.; Leung S. C.; Amewu R.; Stocks P. A.; Stachulski A.; Horta P.; Cristiano M. L. S.; Shone A. E.; Moss D.; Ardrey A.; Sharma R.; Warman A. J.; Bedingfield P. T. P.; Fisher N. E.; Aljayyoussi G.; Mead S.; Caws M.; Berry N. G.; Ward S. A.; Biagini G. A.; O’Neill P. M.; Nixon G. L. (2017) Rational Design, Synthesis, and Biological Evaluation of Heterocyclic Quinolones Targeting the Respiratory Chain of Mycobacterium tuberculosis. J. Med. Chem. 60 (9), 3703–3726. 10.1021/acs.jmedchem.6b01718. [DOI] [PubMed] [Google Scholar]
- Yano T.; Kassovska-Bratinova S.; Teh J. S.; Winkler J.; Sullivan K.; Isaacs A.; Schechter N. M.; Rubin H. (2011) Reduction of Clofazimine by Mycobacterial Type 2 NADH:Quinone Oxidoreductase: A PATHWAY FOR THE GENERATION OF BACTERICIDAL LEVELS OF REACTIVE OXYGEN SPECIES. J. Biol. Chem. 286 (12), 10276–10287. 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosa A.; Lamprecht D. A.; Arora K.; Barry C. E.; Boshoff H. I. M.; Ioerger T. R.; Steyn A. J. C.; Mizrahi V.; Warner D. F. (2017) Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrob. Agents Chemother. 61 (10), e01338-17. 10.1128/AAC.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alland D.; Steyn A. J.; Weisbrod T.; Aldrich K.; Jacobs W. R. (2000) Characterization of the Mycobacterium tuberculosis iniBAC Promoter, a Promoter That Responds to Cell Wall Biosynthesis Inhibition. J. Bacteriol. 182 (7), 1802–1811. 10.1128/JB.182.7.1802-1811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naran K.; Moosa A.; Barry C. E.; Boshoff H. I. M.; Mizrahi V.; Warner D. F. (2016) Bioluminescent Reporters for Rapid Mechanism of Action Assessment in Tuberculosis Drug Discovery. Antimicrob. Agents Chemother. 60 (11), 6748–6757. 10.1128/AAC.01178-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein E. A.; Yano T.; Li L.-S.; Avarbock D.; Avarbock A.; Helm D.; McColm A. A.; Duncan K.; Lonsdale J. T.; Rubin H. (2005) Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. U. S. A. 102 (12), 4548–4553. 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48 (1), 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- McAdam R. A.; Quan S.; Smith D. A.; Bardarov S.; Betts J. C.; Cook F. C.; Hooker E. U.; Lewis A. P.; Woollard P.; Everett M. J.; Lukey P. T.; Bancroft G. J.; Jacobs W. R.; Duncan K. (2002) Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulenceb. Microbiology 148 (10), 2975–2986. 10.1099/00221287-148-10-2975. [DOI] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; DeJesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. (2011) High-Resolution Phenotypic Profiling Defines Genes Essential for Mycobacterial Growth and Cholesterol Catabolism. PLoS Pathog. 7 (9), e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Young R. J. (2015) Molecular Property Design: Does Everyone Get It?. ACS Med. Chem. Lett. 6 (7), 722–725. 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseru G. M.; Makara G. M. (2009) The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 8 (3), 203–212. 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- Brown M. F.; Avery M.; Brissette W. H.; Chang J. H.; Colizza K.; Conklyn M.; DiRico A. P.; Gladue R. P.; Kath J. C.; Krueger S. S.; Lira P. D.; Lillie B. M.; Lundquist G. D.; Mairs E. N.; McElroy E. B.; McGlynn M. A.; Paradis T. J.; Poss C. S.; Rossulek M. I.; Shepard R. M.; Sims J.; Strelevitz T. J.; Truesdell S.; Tylaska L. A.; Yoon K.; Zheng D. (2004) Novel CCR1 antagonists with improved metabolic stability. Bioorg. Med. Chem. Lett. 14 (9), 2175–2179. 10.1016/j.bmcl.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Swallow S. (2015) Fluorine in Medicinal Chemistry. Prog. Med. Chem. 54, 65–133. 10.1016/bs.pmch.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Dalvie D. (2015) Predicting Toxicities of Reactive Metabolite–Positive Drug Candidates. Annu. Rev. Pharmacol. Toxicol. 55 (1), 35–54. 10.1146/annurev-pharmtox-010814-124720. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Walker D. P.; Bauman J.; Price D. A.; Baillie T. A.; Kalgutkar A. S.; Aleo M. D. (2011) Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States. Chem. Res. Toxicol. 24 (9), 1345–1410. 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- Hartkoorn R. C.; Uplekar S.; Cole S. T. (2014) Cross-Resistance between Clofazimine and Bedaquiline through Upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58 (5), 2979–2981. 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K.; Ochoa-Montaño B.; Tsang P. S.; Blundell T. L.; Dawes S. S.; Mizrahi V.; Bayliss T.; Mackenzie C. J.; Cleghorn L. A. T.; Ray P. C.; Wyatt P. G.; Uh E.; Lee J.; Barry C. E.; Boshoff H. I. (2014) Respiratory Flexibility in Response to Inhibition of Cytochrome c Oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58 (11), 6962–6965. 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht D. A.; Finin P. M.; Rahman M. A.; Cumming B. M.; Russell S. L.; Jonnala S. R.; Adamson J. H.; Steyn A. J. C. (2016) Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 7, 12393. 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T.; Rahimian M.; Aneja K. K.; Schechter N. M.; Rubin H.; Scott C. P. (2014) Mycobacterium tuberculosis Type II NADH-Menaquinone Oxidoreductase Catalyzes Electron Transfer through a Two-Site Ping-Pong Mechanism and Has Two Quinone-Binding Sites. Biochemistry 53 (7), 1179–1190. 10.1021/bi4013897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirude P. S.; Paul B.; Roy Choudhury N.; Kedari C.; Bandodkar B.; Ugarkar B. G. (2012) Quinolinyl Pyrimidines: Potent Inhibitors of NDH-2 as a Novel Class of Anti-TB Agents. ACS Med. Chem. Lett. 3 (9), 736–740. 10.1021/ml300134b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T.; Li L.-S.; Weinstein E.; Teh J.-S.; Rubin H. (2006) Steady-state Kinetics and Inhibitory Action of Antitubercular Phenothiazines on Mycobacterium tuberculosis Type-II NADH-Menaquinone Oxidoreductase (NDH-2). J. Biol. Chem. 281 (17), 11456–11463. 10.1074/jbc.M508844200. [DOI] [PubMed] [Google Scholar]
- Heikal A.; Nakatani Y.; Dunn E.; Weimar M. R.; Day C. L.; Baker E. N.; Lott J. S.; Sazanov L. A.; Cook G. M. (2014) Structure of the bacterial type II NADH dehydrogenase: a monotopic membrane protein with an essential role in energy generation. Mol. Microbiol. 91 (5), 950–964. 10.1111/mmi.12507. [DOI] [PubMed] [Google Scholar]
- Harbut M. B.; Liu R.; Yang B.; Yano T.; Vilcheze C.; Lockner J.; Guo H.; Franzblau S. G.; Petrassi M.; Jacobs W. R.; Rubin H.; Chatterjee A. K.; Wang F. (2018) Small molecules targeting Mycobacterium tuberculosis type II NADH dehydrogenase with antimycobacterial activity. Angew. Chem., Int. Ed. 57, 3478. 10.1002/anie.201800260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C.; Weinrick B.; Leung L. W.; Jacobs W. R. (2018) Plasticity of Mycobacterium tuberculosis NADH dehydrogenases and their role in virulence. Proc. Natl. Acad. Sci. U. S. A. 115, 1599. 10.1073/pnas.1721545115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camurri G.; Zaramella A. (2001) High-throughput liquid chromatography/mass spectrometry method for the determination of the chromatographic hydrophobicity index. Anal. Chem. 73 (15), 3716–3722. 10.1021/ac001388j. [DOI] [PubMed] [Google Scholar]
- Valko K.; Nunhuck S.; Bevan C.; Abraham M. H.; Reynolds D. P. (2003) Fast Gradient HPLC Method to Determine Compounds Binding to Human Serum Albumin. Relationships with Octanol/Water and Immobilized Artificial Membrane Lipophilicity. J. Pharm. Sci. 92 (11), 2236–2248. 10.1002/jps.10494. [DOI] [PubMed] [Google Scholar]
- Park Y.; Pacitto A.; Bayliss T.; Cleghorn L. A. T.; Wang Z.; Hartman T.; Arora K.; Ioerger T. R.; Sacchettini J.; Rizzi M.; Donini S.; Blundell T. L.; Ascher D. B.; Rhee K.; Breda A.; Zhou N.; Dartois V.; Jonnala S. R.; Via L. E.; Mizrahi V.; Epemolu O.; Stojanovski L.; Simeons F.; Osuna-Cabello M.; Ellis L.; MacKenzie C. J.; Smith A. R. C.; Davis S. H.; Murugesan D.; Buchanan K. I.; Turner P. A.; Huggett M.; Zuccotto F.; Rebollo-Lopez M. J.; Lafuente-Monasterio M. J.; Sanz O.; Diaz G. S.; Lelièvre J.; Ballell L.; Selenski C.; Axtman M.; Ghidelli-Disse S.; Pflaumer H.; Bösche M.; Drewes G.; Freiberg G. M.; Kurnick M. D.; Srikumaran M.; Kempf D. J.; Green S. R.; Ray P. C.; Read K.; Wyatt P.; Barry C. E.; Boshoff H. I. (2017) Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis. 3 (1), 18–33. 10.1021/acsinfecdis.6b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshoff H. I. M.; Reed M. B.; Barry C. E.; Mizrahi V. (2003) DnaE2 Polymerase Contributes to In Vivo Survival and the Emergence of Drug Resistance in Mycobacterium tuberculosis. Cell 113 (2), 183–193. 10.1016/S0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- Aliverti A., Curti B., and Vanoni M. A. (1999) Identifying and Quantitating FAD and FMN in Simple and in Iron-Sulfur-Containing Flavoproteins. In Flavoprotein Protocols (Chapman S. K., and Reid G. A., Eds.), pp 9–23, Humana Press, Totowa, NJ; DOI: 10.1385/1-59259-266-x:9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.