

Abstract
Permanent cerebral blood flow reduction results in brain injury (stroke), whereas transient ischemic stress results in preconditioning, which can ameliorate the extent of irreversible brain injury from subsequent ischemia—the phenomena of ischemic tolerance. Neurogenesis in the brain occurs after both ischemic injury and the brief ischemia resulting in preconditioning. As neurogenesis is regarded as having an intrinsic neuroprotective role in the brain, we investigated the possible role of these endogenous progenitor cells in the induction of ischemic tolerance. Methylazoxymethanol acetate (MAM) was injected in wild-type mice to attenuate precursor cell proliferation and ganciclovir was used to diminish newly generated cells in GFAP/HSV-TK mice. Both MAM and ganciclovir significantly attenuated ischemia-induced progenitor cell proliferation in the subventricular zone, dentate gyrus, penumbra, and corpus callosum as quantified by 5-bromo-2′-deoxyuridine- or Ki-67-positive cells. Attenuation of ischemia-induced progenitor cell proliferation in the brain blocked the induction of ischemic tolerance. Further the number of TUNEL (TdT-mediated dUTP nick end labeling)-positive cells was considerably increased in MAM-treated animals, whereas MAM did not cause cell death in sham-operated controls. The results of this study suggest a role for endogenous progenitors in the protective effect of ischemic tolerance.
Keywords: ischemic tolerance, MAM, progenitor cells, proliferation
Introduction
Ischemic, traumatic, and inflammatory brain injury can upregulate proliferation of endogenous progenitor cells and induce differentiation and migration of these cells (Ben-Hur et al, 2003; Jin et al, 2005; Naylor et al, 2005; Zhang et al, 2001). In the normal adult brain, these proliferating precursors migrate from the rostral subventricular zone toward the olfactory bulb via the rostral migratory stream (Jin et al, 2005) or from the subgranular zone (Jin et al, 2005) to the adjacent granule cell layer of dentate gyrus (DG) (Bernabeu and Sharp, 2000; Jin et al, 2001). After acute focal ischemic brain injury, proliferation of progenitor cells is enhanced and an ameliorative role for these cells in ischemic brain injury has been suggested (reviewed by Martino and Pluchino, 2006). Ischemic brain injury may result from a transient or permanent reduction in cerebral blood flow with resultant infarction; however, the extent of irreversible brain injury after ischemia can be attenuated by prior ischemic stress applied below the threshold of damage thereby producing preconditioning. Such preconditioning results in tolerance to subsequent severe ischemia. Although the neuroprotective mechanism of preconditioning is not known (Stenzel-Poore et al, 2003), the ischemic preconditioning stimulus can induce proliferation of progenitor cells in the brain. The presence of newly generated neuronal and glial precursors (Naylor et al, 2005) after transient or prolonged ischemia has been interpreted as a restorative response of the brain to injury (Martino and Pluchino, 2006). Applying preconditioning 3 days before the stroke can significantly reduce the infarct volume and stimulate the transcription of trophic factors in the ipsilateral cerebral cortex and hippocampus (Naylor et al, 2005). The phenomenon of tolerance can be a powerful neuroprotectant with 100% protection and no CA1 neuronal loss in at least 50% of the animals subjected to global ischemia, and tolerance in focal ischemia shows a 50% reduction in neuronal cell loss in the cortex and striatum (Liu et al, 1998). Increases in the number of proliferating progenitor cells in the brain can occur through direct administration of progenitors by transplantation of neuronal or mesenchymal stem cells (Chen et al, 2001; Ukai et al, 2007) or indirectly by administration of growth factors or granulocyte colony-stimulating factor (Kuhn et al, 1997; Schabitz et al, 2003). Both direct and indirect approaches to increase progenitor cell presence in acute brain ischemia attenuate the size of infarct and ameliorate the neurologic deficits after the stroke. However, no study has directly evaluated the effect of progenitor cell induction in tolerance as neuroprotective. Accordingly, in the present study of ischemic tolerance, using a filament occlusion rodent focal ischemia model, the attenuation of progenitor cell proliferation was induced using two different methods: a DNA methylating agent methylazoxymethanol acetate (MAM) (Shors et al, 2001) was administered to diminish the number of newly generated cells and ganciclovir (GCV) was used to specifically diminish multipotent glial fibrillary acidic protein (GFAP)-positive proliferating cells in GFAP/HSV-TK mice (Delaney et al, 1996). Reducing the number of precursor cells revealed the functional importance of these multipotent proliferating cells in the phenomenon of tolerance.
Materials and methods
Animal Groups
All animal procedures were conducted in a facility certified by the Association for Assessment and Accreditation of Laboratory Animal Care in accordance with protocols approved by the Institutional Animal Care and Use Committee and the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals. Wild-type, male mice strain C57/BL6 (aged 7 to 9 weeks) were used to investigate the effect of progenitor cell ablation in tolerance. An additional group of GFAP/HSV-TK heterozygous transgenic mice (n = 3, HSV-TK; Jackson Laboratories, West Grove, PA, USA) was added (Imura et al, 2003; Shors et al, 2001) to support the result achieved in the wild-type animals. All mice were acclimatized for at least 1 to 2 weeks before study. They were kept under single ventilation condition, 12 h light/dark cycles, and with free access to food and water during study.
Focal Ischemia Model
In brief, animals were anesthetized using 1.5% isoflurane in 30% O2 and 70% NO2. In each mouse, the cerebral blood flow was monitored before, during, and after surgery with a laser Doppler monitor (Perimed AB, Stockholm, Sweden), using a flexible probe affixed to the right (ipsilateral) parietal region on the skull (~1 mm caudal/~3 mm lateral to the bregma). To ensure a consistent histopathologic result, the body temperature of mice was monitored with a rectal probe and kept at 37°C±0.5°C during and after surgery for as long as the animal was under the influence of anesthetics (drowsy and not active), using a homeothermic blanket and a heating lamp (Harvard Apparatus, Holliston, MA, USA). Focal ischemia, transient middle cerebral artery occlusion (MCAO), was induced by inserting a filament (6-0, Doccol, Redlands, CA, USA) into the internal carotid to occlude the orifice of the middle cerebral artery via an external carotid artery approach as described previously. To assure uniform ischemia and reperfusion, the study animals were required to have a cerebral blood flow reduced to 20% of normal or less and a return to baseline after reperfusion of the middle cerebral artery. Control animals underwent sham operation. In brief, mice were anesthetized, carotid artery exposed, a silk thread was placed under the common carotid artery, and external carotid artery was ligated, but no filament was inserted into the internal carotid artery. All animals received a single dose of 50 mg/kg 5-bromo-2′-deoxyuridine (BrdU, an analog of thymidine; Sigma, St Louis, MO, USA) intraperitoneally, 24 h before euthanization (Miller and Nowakowski, 1988). The duration of MCAO was 15 mins for the preconditioning stimulus and 60 mins for prolonged ischemia to produce stroke or as a challenge to establish the presence of ischemic tolerance after preconditioning.
Methylazoxymethanol Acetate and Ganciclovir Treatment
Animals were divided into four study groups (n = 4 each): (i) control (sham operation, OP), (ii) preconditioning (15 mins MCAO), (iii) stroke (60 mins MCAO), and (iv) ischemic tolerance (preconditioning followed by stroke after 72 h). The MAM C57/BL6 mice were treated with daily subcutaneous injection of MAM (5 mg/kg; Midwest Research Institute, Kansas, MO, USA), with a single injection a day (Figure 2F) for 7 days to diminish the number of newly generated cells (Shors et al, 2001). This interval was selected, as longer application of the drug can affect learning and memory and a higher dosage (7 or 15 mg/kg) can cause severe weight loss (Shors et al, 2001). In the GFAP/HSV-TK mice (Bush et al, 1999; Imura et al, 2003), the number of newly generated neurospheres was attenuated by administration of 100 mg/kg GCV (Roche, Berkeley, CA, USA) applied subcutaneously for 7 days, using an Alzet osmotic pump (model 1007D, Alzet, Cupertino, CA, USA) (Delaney et al, 1996; Imura et al, 2003). Control animals were treated with an equal amount of phosphate-buffered saline (Sigma) (Figure 2F).
Figure 2.
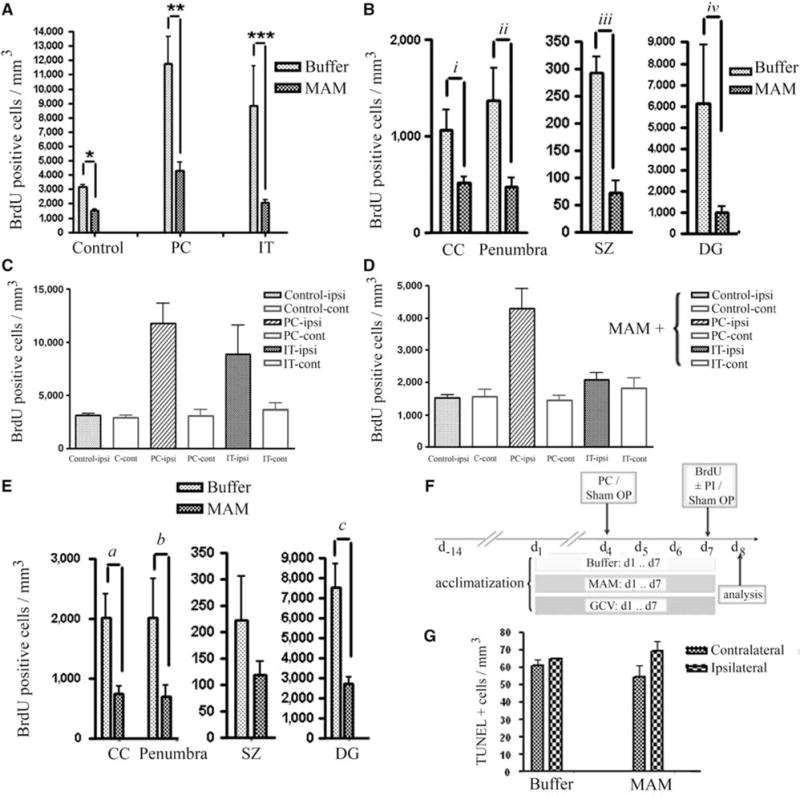
Quantification analysis of BrdU-positive progenitor cells. (A) Reduction of BrdU-positive cells (cells/mm3) after MAM treatment in control animals (sham OP), preconditioning (PC), and ischemic tolerance (IT). (B) Attenuation of BrdU-positive cells with MAM treatment occurred in CC, DG, subependymal zone (SZ) as well as in the penumbra (number of BrdU-positive cells present at the hypoperfused area in the vicinity of ischemic border, excluding subependymal zone (SZ), CC, and DG), after tolerance induction (a preconditioning stimulus (15 mins MACO) followed by prolonged ischemia (60 mins MACO after 72 h)). (C, D) The number of BrdU-positive cells is greater in ipsilateral than in contralateral sides in buffer-treated (C) and MAM-treated (D) animals in response to preconditioning (PC, 15 mins MACO), ischemic tolerance (IT, 15 mins MACO followed by 60 mins MACO after 72 h) and in buffer-treated controls (note scale difference between C and D). Significance showed in Panel “A”. (E) Comparison of the BrdU-positive cells in CC, penumbra, SZ, and DG after preconditioning stimulus (15 mins MACO) in buffer- and MAM-treated animals. (F) Time line: all the animals were acclimatized for 2 weeks. Buffer, MAM, or GCV (GCV) was applied subcutaneously for 7 days and BrdU (50 mg/kg) was injected intraperitoneally at day 7. (G) Bars show the number of TUNEL-positive cells in both contralateral and ipsilateral sides of the brain of control animals (treated with buffer) or animals treated with MAM for 7 days. No significant changes were observed in the number of TUNEL-positive cells in MAM-treated animals compared with the control. *P = 0.0029, **0.009, ***P = 0.0048, iP = 0.0023, iiP = 0.0217, iiiP = 0.0088, ivP = 0.0059, aP = 0.0297, bP = 0.0139, and cP = 0.0059.
Ischemic Lesion Size Measurement
Animals were fully anesthetized before decapitation. Brains were removed from the skull and immersed in prechilled isopentane (Sigma) and kept at −80°C before use. Cryosections were prepared at 14 μm thickness, using a cryostat (Leica Microsystems, Bannockburn, IL, USA). To measure the infarct size and edema in different study groups, brain sections were stained with cresyl violet and analyzed at least at six levels from + 0.50±0.20 to −2.40±0.20 mm from bregma. Digital images were then analyzed in Adobe Photoshop. The volume of infarct was calculated using the following formula: infarct volume = (volume of left hemisphere (volume of right hemisphere–measured infarct volume))/volume of left hemisphere. The brain swelling was determined using the following formula: swelling (edema) = (volume of right hemisphere volume of left hemisphere)/volume of left hemisphere (Shuaib et al, 2002).
Histologic Examination and Analysis
The 14-μm-thick coronal frozen sections were fully rehydrated in phosphate-buffered saline, fixed, and permeabilized in prechilled methanol. 5-Bromo-2′-deoxyuridine and Ki-67 staining was applied on separate brain slices to quantify proliferating cells in different study groups. At least six brain slices were considered for each brain and, for stereological analysis, sections were selected from three different regions (which were in the infarct region) as follows: region I = + 0.50±0.20 mm, region II = −0.50±0.20 mm, and region III = −2.40±0.20 mm from bregma. In brief, for BrdU staining, DNA molecules were denatured using HCl (0.1 N; Sigma) at 37°C and the acid was neutralized by two washes (10 mins) of borate buffer (0.1 mol/L; Sigma). Primary anti-BrdU antibody (1:100; Sigma) was applied for 1 h at room temperature and staining was evaluated after exposing sections to a fluorescein isothiocyanate-coupled anti-mouse IgG antibody (1:100; Jackson Laboratories) for 45 mins at room temperature. For Ki-67 immunostaining, the anti-Ki-67 (polyclonal rabbit; Abcam, Cambridge, MA, USA) primary antibody was applied for 1 h at room temperature and donkey anti-rabbit IgG (1:200; Jackson Laboratories) secondary antibody was applied for 45 mins at room temperature (Haapasalo et al, 2005). Subsequently, the immunolabeling was visualized using a DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA, USA). All of the sections were washed before and after each incubation with a blocking solution containing phosphate-buffered saline, 0.1% bovine serum albumin, and 5% serum. Negative control staining was performed as described above but omitting the respective primary antibody. To evaluate cell death, TUNEL (TdT-mediated dUTP nick end labeling) staining was performed using fluorescein-dUTP to label DNA strand breaks according to the manufacturer’s instructions (Roche). Each section was stained with DAPI for nuclear staining and sections were examined by fluorescence microscopy (Leica Micro-systems) using either bright-field or excitation/emission wavelengths of 495/515 nm for green fluorophores.
In many conditions, estimation of the total number (N) of target cells would be obtained by multiplying the reference volume of the region (Vref) and the calculated numerical density (Nv) (N = VrefNv) and fractionator methods can be used by sampling only a portion of the sections, in which the estimates will be unbiased. In our study, the distribution of the proliferating precursors (BrdU +/Ki-67 +) as well as TUNEL-positive cells was not homogeneous (because of migration of some cells and different extents of damage). Therefore, sampling procedures would not ensure that all objects of interest in the sections would have equal probabilities of being included in the sample. For this reason, all cells were counted in the obtained sectional plane using the Bioquant software or manually (n~10 fields with ×40 objective), to obtain an unbiased two-dimensional counting frame (West, 1999). All the transections intercepted by the upper and left border were counted, whereas those intercepted by the lower and right border were excluded. The ipsilateral and contralateral hemisphere fields were chosen from at least six different regions of the brain (sets of parallel sections with a known separation distance). Two sections from each region, I = + 0.50±0.20, II = −0.50±0.20, and III = −2.40±0.20 mm from bregma, were counted. Subsequently, the number of cells multiplied by the number of transections and finally the total number of proliferating (or TUNEL-positive) cells was calculated per 1 mm3. Using sets of six parallel sections (with a pair of related sections) with a known separation distance allowed us an unbiased estimation of the distribution of proliferating precursor (as well as TUNEL-positive) cells present in the ipsilateral and contralateral ischemic hemisphere in each region.
Statistical Analysis
The statistical significance between different study groups was determined using analysis of variance followed by Bonferroni post hoc test and the Student’s t-test was used for comparison between two study groups. P < 0.05 was considered statistically significant.
Results
Identification of 5-Bromo-2′-deoxyuridine-Positive Progenitor Cells
To identify proliferating precursor cells in the brain tissue, mice were injected intraperitoneally with a single dose of 50 mg/kg BrdU in all study groups. Frozen brain sections (14 μm) were then stained to visualize BrdU incorporation. The total number of BrdU-positive cells in each area of interest in the brain was counted, quantified in mm3, and compared with the appropriate control. The number of BrdU-positive proliferating progenitor cells is increased three-fold after tolerance and four-fold after preconditioning compared with the control animals (compare Figures 1B versus 1C and Figure 2A). However, in animals pretreated with MAM (5 mg per kg per day × 7), a significantly reduced number of BrdU-positive cells is found in all groups compared with their respective controls (Figures 1 and 2). In sham-operated animals, the number of BrdU-positive cells was significantly (P = 0.0029) decreased after MAM treatment to 48% of buffer-treated controls; after preconditioning, the number of BrdU-positive cells in the MAM-treated group was decreased to 37% (P = 0.009); and after tolerance, to less than 24% of the respective control (P = 0.0048) (Figure 2A). Region of interest data show that there is considerable upregulation in the number of BrdU-positive cells at the corpus callosum (CC), in the penumbral region, at the subependymal zone, and DG after tolerance, which was significantly attenuated after MAM treatment (Figure 2B; CC (P = 0.0023), penumbra (P = 0.0217), subependymal zone (P = 0.0088), and DG (P = 0.0059)). The same result was achieved after MAM treatment in the presence of preconditioning but in the absence of stroke (Figure 2E; CC (P = 0.0297), penumbra (P = 0.0139), and DG (P = 0.0059)). Methylazoxymethanol acetate reduced the number of BrdU-positive cells in the control animals by 52% (Figure 2A). The effect of preconditioning and tolerance on upregulation of BrdU-positive cells occurred bilaterally but was greater in the ipsilateral hemisphere either with (Figure 2D) or without MAM treatment (Figure 2C). To confirm that MAM merely prevents progenitor cells from proliferation and does not induce cell death, we performed TUNEL assay and found no statistically significant alteration between buffer-treated and MAM-treated control animals (Figure 2G).
Figure 1.
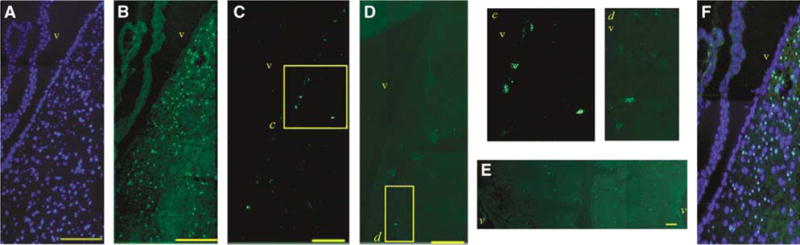
Identification of BrdU-positive cells. (A, B) Immunohistochemistry after tolerance. (A) DAPI staining. (B) BrdU staining of cells at the ipsilateral subependymal zone (25 μm from the margin of the ventricle in adult brain considered as subependymal zone). (C) BrdU-positive cells at the subependymal zone of control group. (D) Subependymal zone BrdU immunoreactivity was significantly decreased after MAM treatment. (E) BrdU immunostaining more prominent on the ipsilateral side of the brain (right) (CC, −0.50±0.20 mm from bregma) after tolerance induction. (F) Panels A and B merged. Brain sections in panels A–D were obtained from 0.50±0.20 mm from bregma. The insets in panels C and D (c and d) are magnified and presented on the right. v: ventricle. Bars: 100 μm.
Identification of Ki-67-Positive Progenitor Cells
Ki-67 expression in individual cells was analyzed in fresh frozen sections of brains after tolerance in MAM-treated animals and the result was compared with the buffer-treated control group (Figures 3 and 4). The expression pattern and accumulation of Ki-67-positive proliferating cells (Figures 4 and 6) were similar to the result achieved after BrdU immunolabeling. Region of interest data showed a considerable number of Ki-67-positive cells at the area of CC, subependymal zone, and DG after tolerance, which was significantly reduced by 50% or more in MAM-treated animals after 7 days (P < 0.05; Figure 4A). Similar to the BrdU staining observations, the number of Ki-67-positive cells at the ipsilateral side of the brain was greater than that at the contralateral side of the brain (Figure 4B). The appearance of Ki-67-positive cells at the ipsilateral CC was twice as high as that at the contralateral side (P = 0.025) and similar data were observed at the ipsilateral DG after tolerance (Figure 4B). Changes in the number of positively stained cells were significant at the area of penumbra (P = 0.0002) (Figure 4B).
Figure 3.
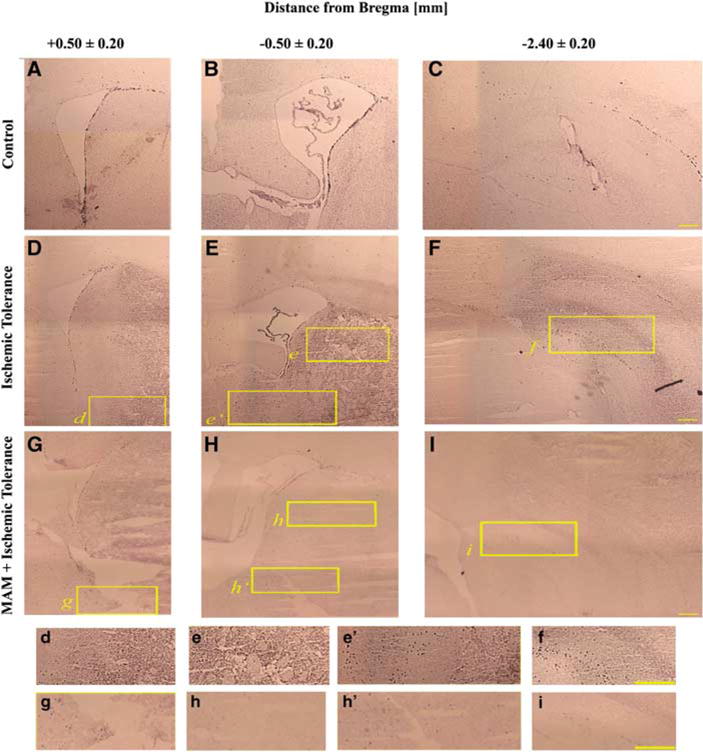
Identification of Ki-67-positive cells. Immunohistochemistry showing that the number of Ki-67-positive cells is increased after tolerance (D–F) compared with the controls (A–C). However, the number of proliferating progenitors was decreased in MAM-treated animals (MAM + tolerance (G–I)). Panels A, D, and G are brain slices from + 0.50±0.20 mm, B, E, and H from −0.50±0.20 mm, and C, F, and I from −2.40±0.20 mm from the bregma. The insets (marked with lowercase letters) are magnified and are presented in the lower panels. Bar: 1 mm.
Figure 4.
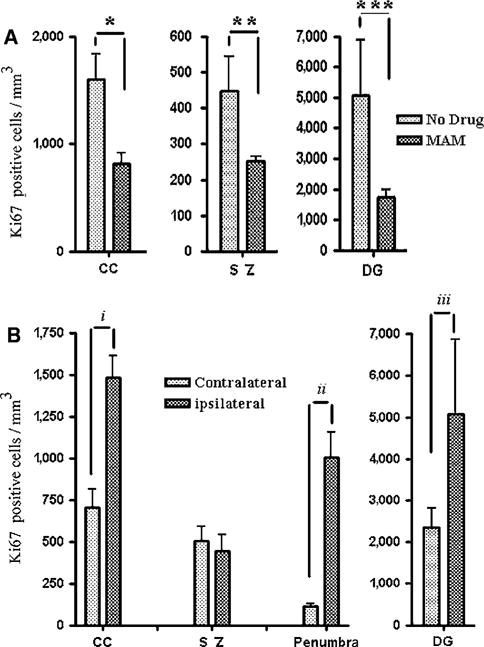
Quantification of Ki-67-positive cells. (A) The number of Ki-67-positive cells by region of interest after tolerance in MAM-treated animals as compared with the controls. The number of Ki-67-positive cells was significantly reduced at CC, subependymal zone (SZ), as well as dentate granule cells (DG). (B) Similar to BrdU immunostaining, the number of Ki-67-positive cells was significantly higher in the ipsilateral CC, penumbra, and DG compared with the contralateral side after tolerance. No statistically significant differences were observed at the subependymal zone (SZ), presumably because of migration of these cells toward the margin of ischemic core and penumbra. *P = 0.027, **P = 0.0066, ***P = 0.010, iP = 0.025, iiP = 0.0002, and iiiP = 0.016.
Figure 6.
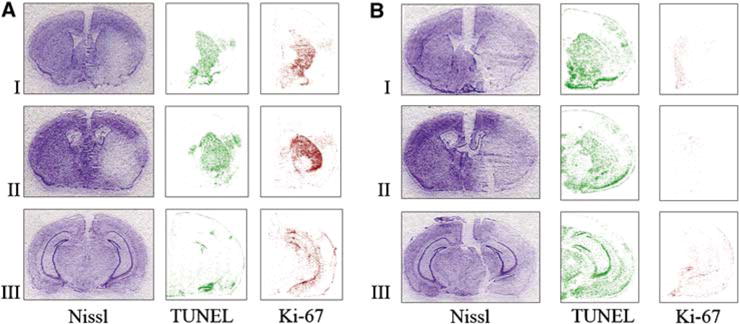
Comparison of the extent of injury (Nissl) and number of proliferating (Ki-67) and apoptotic cells (TUNEL) in MAM-treated animals (B) and control group (A) after attempting to induce tolerance: a preconditioning stimulus (15 mins MACO) followed by prolonged ischemia (60 mins MACO after 72 h). Nissl staining shows that the infarct size is significantly larger in MAM animals subjected to the ischemic tolerance paradigm (B) compared with buffer-treated controls, which show ischemic tolerance (A). The number of TUNEL-positive cells was increased after MAM treatment, whereas the number of Ki-67-positive proliferating progenitors was significantly reduced in MAM-treated animals (B). Brain sections were obtained from the following regions: I = + 0.50±0.20, II = −0.50±0.20, and III = −2.40±0.20 mm from the bregma.
Histologic Determination of Infarct Volume and Topographic Analysis
The extent of ischemic injury in each study group was evaluated in Nissl-stained sections, and the mean value of infarct volume, infarct percentage, and edema were compared between controls and MAM (5 mg per kg per day, subcutaneously)-treated animals after 15 mins preconditioning, stroke (60 mins MCAO), and tolerance (15 mins preconditioning followed by stroke). The mean quantification of infarct volume after 15 mins preconditioning was 1.56±1.04 mm3 (mean±SEM) in the control group, which was not significantly different from 4.61±1.63 mm3 in MAM-treated animals (Figure 5A). After inducing tolerance, the infarct volume significantly (P = 0.001) increased from 26.09±4.41 mm3 in buffer-treated animals to 51.45±1.63 mm3 in MAM-treated animals (Figure 5A).
Figure 5.
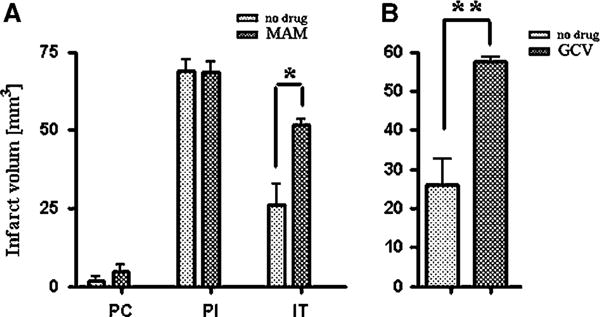
Quantification of infarct size. (A) Infarct volume after 15 mins preconditioning (PC), after prolonged ischemia (PI; 60 mins MCAO), and after ischemic tolerance (15 mins MACO followed by 60 mins MACO after 72 h in MAM-treated C57/BL6 mice and buffer-treated controls. Preconditioning results in ischemic tolerance (speckled bars, PI versus IT). Animals treated with MAM for 7 days before PI did not show the protective effect of preconditioning (hatched bars, PI versus IT = NS). (B) Ischemic tolerance is blocked in GCV-treated HSV-TK animals (7 days treatment before PI) compared with buffer-treated controls. Both groups were treated identically with 15 mins preconditioning MCAO followed with 60 mins PI after 72 h (time line: see Figure 2F). *P = 0.001 and **P = 0.0336.
After the attempt at tolerance induction in the MAM-treated group, the volume of edema was significantly (P < 0.05) increased compared with control (21.16±3.14 versus 6.13±0.74 mm3, respectively) (not shown).
For topographic analysis, representative images of three sections through the region of ischemia were stained with Nissl, TUNEL, and Ki-67 after ischemic tolerance in control (Figure 6A) and MAM-treated animals (Figure 6B). The degree of neuroprotection from tolerance can be seen in the Nissl-stained sections. Ki-67 immunoreactivity is attenuated after MAM treatment. The results of Ki-67 staining were similar to those of BrdU staining (data not shown). TUNEL staining is increased in MAM-treated animals.
Effect of Ganciclovir in GFAP/HSV-TK Mice
Ganciclovir treatment, which reduces the number of newly generated neurospheres (Delaney et al, 1996) in HSV-TK mice (100 mg per kg per day, Alzet osmotic pump), attenuated the protective effect of tolerance. In Nissl-stained 14 μm frozen brain sections, the infarct volume in GCV-treated HSV-TK animals (58.50±0.36 mm3) was significantly greater than that in the control group (26.09±4.41 mm3; P = 0.001) (Figure 5B). The mean volume of edema after the attempt at inducing tolerance was also significantly increased in GCV-treated HSV-TK animals from 6.13±0.74 to 24.61±1.13 mm3 compared with the non treated animals (P = 0.0012) (not shown).
Discussion
Neurogenesis occurs normally in developing central nervous system and is subjected to regulation via growth factors (Kuhn et al, 1997), physiotherapy (Kronenberg et al, 2006), neurotransmitters (Bernabeu and Sharp, 2000), and learning and memory (Gould et al, 1999). In mature brains, neurogenesis can be triggered by pathologic events such as stroke (Naylor et al, 2005), seizures (Nakagawa et al, 2000; Parent et al, 1997), and neuroinflammatory states (Picard-Riera et al, 2002). The proliferating precursors present at the subependymal zone and DG after stroke have the potential to differentiate and express mature neuronal markers over time (Dempsey et al, 2003). Glial progenitors are mainly present at the striatum, cortex, and the margin of the infarct (Dempsey et al, 2003; Nakagawa et al, 2000; Parent et al, 1997).
Some putative therapeutic factors can ameliorate the size of an infarct and improve neurologic outcome after stroke. These include administration of growth and stimulating factors, neurotrophic factors, and granulocyte colony-stimulating factor (Dempsey et al, 2003; Kuhn et al, 1997; Schabitz et al, 2003; Sehara et al, 2007). The neuroprotective effect varies with the method and time point of administration (Tenjin et al, 1995). The transplantation of stem cells into the brain can also be neuroprotective (Ukai et al, 2007). The exact role of growth factors in protecting the brain after a stroke is not clear, but it is recognized that these molecules do stimulate precursor cells to proliferate. Further, these molecules can reduce apoptosis by upregulating antiapoptotic molecules such as Bcl2 and by reducing proapoptotic molecules such as Bax (Schabitz et al, 2000). An increase in the number of progenitor cells can also be achieved by direct transplantation of either neuronal or mesenchymal stem cells, which can consequently ameliorate infarct size and improve functional performance (Chen et al, 2001; Horita et al, 2006; Ukai et al, 2007). Multipotent mesenchymal stem cells can successfully integrate into the penumbra of the ischemic brain (Ukai et al, 2007) and have the potential to differentiate into neuronal (NeuN +) and glial (GFAP +, MBP +, CNPase +) progenitors in vivo as well as in vitro (Sanchez-Ramos et al, 2000; Snyder et al, 1997). However, the site of implantation (Modo et al, 2002) and a restricted time window are recognized as effecting such transplants (Schmidt et al, 2007). Multipotent progenitor cells induced in the brain by any of these mechanisms have been assumed to have intrinsic neuroprotective properties by either cell replacement or the release of neuroprotective molecules (Martino and Pluchino, 2006).
A subthreshold ischemic stimulus (preconditioning) can also reduce infarct size and ameliorate neurologic deficits and, being subthreshold, does not induce cell death (Stenzel-Poore et al, 2003). Such preconditioning also increases the number of proliferating precursors (Jin et al, 2001). Our previous observations showed that preconditioning alone (10 mins MCAO) induced no infarction (Jin et al, 2001). Applying a single dose of BrdU, an analog of thymidine, 24 h before killing results in BrdU incorporation into the replicated DNA molecules and labeling of proliferating progenitor cells (Miller and Nowakowski, 1988). To address the question of the potential role of progenitor cell proliferation in the phenomenon of ischemic tolerance, we used a DNA methylating agent, MAM, to reduce the number of proliferating cells in vivo (Shors et al, 2001). In the present study, we show that MAM can significantly reduce the number of proliferating precursors (BrdU-positive cells) in the brain in agreement with previously published data (Shors et al, 2001). We therefore asked if reducing the number of BrdU-positive cells to less than 40% compared with the controls might hamper the protective effect observed in the brain during ischemic tolerance. The MAM-treated animals had a marked reduction in progenitor proliferation in response to the preconditioning stimulus (Figure 2A) and a markedly attenuated tolerance response after preconditioning (Figure 5A). As shown previously, the progenitor cell proliferation in the setting of focal ischemia occurs bilaterally (Jin et al, 2001). In the infarcted brains, we observed that proliferating progenitor cells are more numerous in the ipsilateral rather than the contralateral hemisphere after focal ischemia—a finding compatible with precursor migration from the healthy hemisphere to participate in defense mechanisms activated in the ischemic hemisphere, as has been previously described (Jin et al, 2001, 2005). This might explain the marked increase in the number of BrdU- and Ki-67-positive cells observed at the penumbra.
To confirm the results of BrdU immunolabeling of replicated DNA molecules in proliferating cells, we used immunoreactivity of brain slices for Ki-67 antigen. The Ki-67 antigen expression is cell cycle dependent, its antigen reactivity is not detected during DNA repair processes (Hall et al, 1993), and it is expressed exclusively either in the nuclear matrix or at the perinuclear region of proliferating cells (Verheijen et al, 1989). This large nuclear protein is preferentially expressed at late interphase, which encompasses G1, S, and G2, as well as mitosis (prophase, prometaphase, anaphase, and telophase). The Ki-67 antigen is absent in resting (G0) cells and has a detectable biologic half-life of less than an hour, as its immunoreactivity is rapidly decreased during anaphase and telophase (Schluter et al, 1993). Our data show that the number of Ki-67-positive cells is increased after preconditioning or tolerance, particularly in the CC, DG, and penumbra, in a pattern similar to that observed with BrdU staining. Further, MAM significantly reduced the number of Ki-67-positive cells as well as BrdU-stained cells (Figure 4). However, MAM does not alter cell fate. Even high dosage (14 mg per kg per day) and prolonged administration (2 weeks) of this methylating agent can only slightly reduce the number of DCX-positive cells compared with the controls (Dupret et al, 2005) The results observed with Ki-67 and BrdU staining show that progenitor cell proliferation is indeed upregulated by the preconditioning stimulus and subsequently by ischemic tolerance. Further, progenitor cell proliferation is regionally induced in the brain, including the subependymal zone, CC, and DG.
We also applied an alternate method to simulate the effect of MAM on minimizing the number of proliferating precursors. Other investigators have used low-dose X-irradiation (Mizumatsu et al, 2003); however, this method can alter glial ratio profile and cause dose-dependent apoptosis as well as cognitive impairments, which might interfere with the exclusive effect of proliferating progenitors after ischemia (Mizumatsu et al, 2003; Monje et al, 2002). Accordingly, we chose to reduce the number of proliferating progenitor cells in a transgenic animal model of GFAP/HSV-TK mice (Bush et al, 1999). In these animals, the herpes simplex virus thymidine kinase gene (HSV-Tk) is inserted into the first exon of the GFAP promoter cassette (Bush et al, 1999). Proliferating cells that express the transgene-derived HSV-TK can metabolize the antiviral drug GCV to toxic nucleotide analogs, which disturb nucleic acid synthesis and induce cell death in vitro as well as in vivo (Bush et al, 1999). By using this model, progenitor cell ablation can be regulated by GCV administration (Bush et al, 1999), which reduces the number of GFAP-expressing and dividing cells at the onset of sphere formation (Imura et al, 2003). In these animals, GCV can abolish the formation of multipotent neurospheres rather than eliminating the GFAP-expressing progeny derived from the non-GFAP-expressing cells. It is known that neuronal stem cells are heterogeneous with respect to GFAP expression during embryogenesis (Suslov et al, 2002) or in the postnatal and mature brain (Imura et al, 2003). However, it is not clear if multiple lineages of neuronal stem cells are involved in the process of neurogenesis or if a single lineage can gradually adopt (or lose) GFAP expression. Nevertheless, our data show that 7-day administration of GCV completely blocks the protective effect of ischemic tolerance, which supports the data achieved after MAM treatment.
To conclude, the data shown here and those reported previously indicate that endogenous precursors proliferate in the setting of focal ischemia and migrate to the vicinity of the ischemic region, including migration from the contralateral hemisphere to the site of injury. Although the mechanism is unknown, this cell population appears to actively participate in mechanisms that prevent expansion of infarct size (Martino and Pluchino, 2006). Using two different mechanisms to attenuate ischemia-induced upregulation of endogenous progenitor cells and two different markers of the progenitor cell population, we show here that progenitor cell proliferation is an effector of the neuroprotection produced by preconditioning and tolerance. Our observations showed the effect of early proliferating precursors regardless of their potential ability to differentiate into neuronal or glial cells. These data support our hypothesis that depletion of precursor number is the main reason that the protective effect of tolerance is reduced in this study. The cell lineage effecting neuroprotection is not determined, but as it would take some time for newly generated cells to differentiate into neuronal or glial cells and the time course of tolerance is relatively short, we assume that multipotent progenitor cells mediate tolerance. We further assume that the neuroprotective effect is produced not by the structural replacement of cellular elements but rather through a neurotrophic mechanism such as the release of trophic factors that act in the preconditioned state (Naylor et al, 2005). Pharmacological knockdown of the precursor cell population and attenuation of its proliferative response to ischemia are associated with the loss of ischemic tolerance effect (Figure 6). This study supports the concept that proliferating progenitors have a major effect on the induction of ischemic tolerance.
Acknowledgments
We thank Dr D Boison for generous donation of GFAP/HSV-TK mice, A Wilz and R Gala for their technical assistance, and Dr R Meller and Dr T Lusardi for their advice on histologic and topographic analysis.
This study was supported by NIH Grant NS24728-14.
References
- Ben-Hur T, Ben-Menachem O, Furer V, Einstein O, Mizrachi-Kol R, Grigoriadis N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol Cell Neurosci. 2003;24:623–31. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- Bernabeu R, Sharp FR. NMDA and AMPA/kainate glutamate receptors modulate dentate neurogenesis and CA3 synapsin-I in normal and ischemic hippocampus. J Cereb Blood Flow Metab. 2000;20:1669–80. doi: 10.1097/00004647-200012000-00006. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–8. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- Delaney CL, Brenner M, Messing A. Conditional ablation of cerebellar astrocytes in postnatal transgenic mice. J Neurosci. 1996;16:6908–18. doi: 10.1523/JNEUROSCI.16-21-06908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey RJ, Sailor KA, Bowen KK, Tureyen K, Vemuganti R. Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J Neurochem. 2003;87:586–97. doi: 10.1046/j.1471-4159.2003.02022.x. [DOI] [PubMed] [Google Scholar]
- Dupret D, Montaron MF, Drapeau E, Aurousseau C, Le Moal M, Piazza PV, Abrous DN. Methylazoxymethanol acetate does not fully block cell genesis in the young and aged dentate gyrus. Eur J Neurosci. 2005;22:778–83. doi: 10.1111/j.1460-9568.2005.04262.x. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–5. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Haapasalo J, Mennander A, Helen P, Haapasalo H, Isola J. Ultrarapid Ki-67 immunostaining in frozen section interpretation of gliomas. J Clin Pathol. 2005;58:263–8. doi: 10.1136/jcp.2004.018606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall PA, McKee PH, Menage HD, Dover R, Lane DP. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 1993;8:203–7. [PubMed] [Google Scholar]
- Horita Y, Honmou O, Harada K, Houkin K, Hamada H, Kocsis JD. Intravenous administration of glial cell line-derived neurotrophic factor gene-modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in the adult rat. J Neurosci Res. 2006;84:1495–504. doi: 10.1002/jnr.21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci. 2003;23:2824–32. doi: 10.1523/JNEUROSCI.23-07-02824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA. 2001;98:4710–5. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Mao XO, Childs J, Peel A, Logvinova A, Banwait S, Greenberg DA. Comparison of ischemia-directed migration of neural precursor cells after intrastriatal, intraventricular, or intravenous transplantation in the rat. Neurobiol Dis. 2005;18:366–74. doi: 10.1016/j.nbd.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Bick-Sander A, Bunk E, Wolf C, Ehninger D, Kempermann G. Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol Aging. 2006;27:1505–13. doi: 10.1016/j.neurobiolaging.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci. 1997;17:5820–9. doi: 10.1523/JNEUROSCI.17-15-05820.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–78. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino G, Pluchino S. The therapeutic potential of neural stem cells. Nat Rev Neurosci. 2006;7:395–406. doi: 10.1038/nrn1908. [DOI] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 1988;457:44–52. doi: 10.1016/0006-8993(88)90055-8. [DOI] [PubMed] [Google Scholar]
- Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res. 2003;63:4021–7. [PubMed] [Google Scholar]
- Modo M, Stroemer RP, Tang E, Patel S, Hodges H. Effects of implantation site of stem cell grafts on behavioral recovery from stroke damage. Stroke. 2002;33:2270–8. doi: 10.1161/01.str.0000027693.50675.c5. [DOI] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–62. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- Nakagawa E, Aimi Y, Yasuhara O, Tooyama I, Shimada M, McGeer PL, Kimura H. Enhancement of progenitor cell division in the dentate gyrus triggered by initial limbic seizures in rat models of epilepsy. Epilepsia. 2000;41:10–8. doi: 10.1111/j.1528-1157.2000.tb01498.x. [DOI] [PubMed] [Google Scholar]
- Naylor M, Bowen KK, Sailor KA, Dempsey RJ, Vemuganti R. Preconditioning-induced ischemic tolerance stimulates growth factor expression and neurogenesis in adult rat hippocampus. Neurochem Int. 2005;47:565–72. doi: 10.1016/j.neuint.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Evercooren AB. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA. 2002;99:13211–6. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Ramos J, Song S, Cardozo-Pelaez F, Hazzi C, Stedeford T, Willing A, Freeman TB, Saporta S, Janssen W, Patel N, Cooper DR, Sanberg PR. Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp Neurol. 2000;164:247–56. doi: 10.1006/exnr.2000.7389. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Kollmar R, Schwaninger M, Juettler E, Bardutzky J, Scholzke MN, Sommer C, Schwab S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke. 2003;34:745–51. doi: 10.1161/01.STR.0000057814.70180.17. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–7. doi: 10.1161/01.str.31.9.2212. [DOI] [PubMed] [Google Scholar]
- Schluter C, Duchrow M, Wohlenberg C, Becker MH, Key G, Flad HD, Gerdes J. The cell proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell Biol. 1993;123:513–22. doi: 10.1083/jcb.123.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt UR, Wagner DC, Foerschler A, Kranz A, Bulawina L, Kamprad M, Egger D, Emmrich F, Boltze J. Human umbilical cord blood cell treatment of stroke: MRI-controlled evaluation of the therapeutic time window. Society for Neuroscience; San Diego: 2007. abstract 661.10. [Google Scholar]
- Sehara Y, Hayashi T, Deguchi K, Zhang H, Tsuchiya A, Yamashita T, Lukic V, Nagai M, Kamiya T, Abe K. G-CSF enhances stem cell proliferation in rat hippocampus after transient middle cerebral artery occlusion. Neurosci Lett. 2007;418:248–52. doi: 10.1016/j.neulet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–6. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Xu Wang C, Yang T, Noor R. Effects of nonpeptide V(1) vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke. 2002;33:3033–7. doi: 10.1161/01.str.0000039405.31526.06. [DOI] [PubMed] [Google Scholar]
- Snyder EY, Yoon C, Flax JD, Macklis JD. Multipotent neural precursors can differentiate toward replacement of neurons undergoing targeted apoptotic degeneration in adult mouse neocortex. Proc Natl Acad Sci USA. 1997;94:11663–8. doi: 10.1073/pnas.94.21.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–37. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci USA. 2002;99:14506–11. doi: 10.1073/pnas.212525299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenjin H, Anderson RE, Meyer FB. Treatment with basic fibroblastic growth factor following focal cerebral ischemia does not prevent neuronal injury. J Neurol Sci. 1995;128:66–70. doi: 10.1016/0022-510x(94)00208-6. [DOI] [PubMed] [Google Scholar]
- Ukai R, Honmou O, Harada K, Houkin K, Hamada H, Kocsis JD. Mesenchymal stem cells derived from peripheral blood protects against ischemia. J Neurotrauma. 2007;24:508–20. doi: 10.1089/neu.2006.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen R, Kuijpers HJ, van Driel R, Beck JL, van Dierendonck JH, Brakenhoff GJ, Ramaekers FC. Ki-67 detects a nuclear matrix-associated proliferation-related antigen. II. Localization in mitotic cells and association with chromosomes. J Cell Sci. 1989;92(Part 4):531–40. doi: 10.1242/jcs.92.4.531. [DOI] [PubMed] [Google Scholar]
- West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Zhang L, Chopp M. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 2001;105:33–41. doi: 10.1016/s0306-4522(01)00117-8. [DOI] [PubMed] [Google Scholar]