

Abstract
Background
A challenge of understanding the mechanisms underlying cognition including neurodevelopmental and neuropsychiatric disorders is mainly given by the potential severity of cognitive disorders for the quality of life and their prevalence. However, the field has been focused predominantly on protein coding variation until recently. Given the importance of tightly controlled gene expression for normal brain function, the goal of the study was to assess the functional variation including non-coding variation in human genome that is likely to play an important role in cognitive functions. To this end, we organized and utilized available genome-wide datasets from genomic, transcriptomic and association studies into a comprehensive data corpus. We focused on genomic regions that are enriched in regulatory activity—overlapping transcriptional factor binding regions and repurpose our data collection especially for identification of the regulatory SNPs (rSNPs) that showed associations both with allele-specific binding and allele-specific expression. We matched these rSNPs to the nearby and distant targeted genes and then selected the variants that could implicate the etiology of cognitive disorders according to Genome-Wide Association Studies (GWAS). Next, we use DeSeq 2.0 package to test the differences in the expression of the certain targeted genes between the controls and the patients that were diagnosed bipolar affective disorder and schizophrenia. Finally, we assess the potential biological role for identified drivers of cognition using DAVID and GeneMANIA.
Results
As a result, we selected fourteen regulatory SNPs locating within the loci, implicated from GWAS for cognitive disorders with six of the variants unreported previously. Grouping of the targeted genes according to biological functions revealed the involvement of processes such as ‘posttranscriptional regulation of gene expression’, ‘neuron differentiation’, ‘neuron projection development’, ‘regulation of cell cycle process’ and ‘protein catabolic processes’. We identified four rSNP-targeted genes that showed differential expression between patient and control groups depending on brain region: NRAS—in schizophrenia cohort, CDC25B, DDX21 and NUCKS1—in bipolar disorder cohort.
Conclusions
Overall, our findings are likely to provide the keys for unraveling the mechanisms that underlie cognitive functions including major depressive disorder, bipolar disorder and schizophrenia etiopathogenesis.
Electronic supplementary material
The online version of this article (10.1186/s12868-018-0414-3) contains supplementary material, which is available to authorized users.
Keywords: Genetic of cognition, Major depressive disorder, Bipolar affective disorder, Schizophrenia, Autism spectrum disorders, SNPs, Functional variants, Gene regulation
Background
Looking back over the past decade of human genomics, one can therefore assert that the successful completion of Human Genome Project [1] and 1000 genomes [2] pilot project produced a remarkable increase in our knowledge on genetic variants. Among these, the most common type of variation are single nucleotide polymorphisms abbreviated to SNPs. One estimate is that there are 150 million SNPs in the human genome. However, most SNPs lack any functional significance and only a small fraction of base substitutions can have phenotypic manifestations appearing as changes in the amino acid sequence of the resulting protein product, or changes to the level of gene expression [3, 4]. These functional SNPs play a vital role with respect to inter individual’s disease susceptibility and drug response [5, 6]. The framework of the Genome-Wide Association Studies, GWAS, [7] has dominated the investigation of the correlation between the phenotype and certain genetic variants. Presently, more than 16,000 SNPs and small insertions/deletions have been associated with specific human outcomes, diseases and traits according to the GWAS Catalog by the US National Human Genome Research Institute (NHGRI) [7, 8]. It should be noted that about 85% of potentially functional variants are expected to be located in non-coding regions, and a smaller number thereof is believed to act through the regulation of gene expression [9, 10].
The problem is it seems impossible to distinguish the association signals detected from a causative variant and from a number of tag SNPs that are likely part of a larger region of linkage disequilibrium [11]. Moreover, several causal variants may converge to create the significant GWAS signals, which are related to one common tag SNP [12]. Thus, the association of any genetic variants with the disease does not necessarily mean the functionality of these variants. The GWAS-implicated associations accordingly, can be difficult to transfer into the understanding of molecular mechanisms that underlie the phenotypic outcome. In general, GWAS signals have rarely been tracked to causal polymorphisms thus far. This adds to the complexity of the development of effective methods for disease treatment and prevention [13].
A second problem is the significant heterogeneity of natural human populations that can be taken care of through proper quality control and study setup including extended cohorts of patients and controls. Using the data from The Encyclopedia of DNA Elements, ENCODE, [14] can play an important role in contributing to the latter issue. Since the ENCODE Project was initiated with the aim to find all functional elements in the genome, it has accumulated numerous data on chromatin and transcribed genes obtained from various cell lines and tissues, and based on these, candidate regulatory SNPs may be found. In particular, available ChIP-seq data on allele-specific binding of different transcription factors (TFs) could be considered as a clear sign that the SNPs with regulatory potential are located within the genome regions occupied by these factors [15–17]. ChIP-seq data on allele-specific binding of active chromatin marks can also provide important insights towards the localization of regulatory variants particularly in combination with allele-specific expression profiles from RNA-seq [15, 16]. Thus, the study of allele-specific events of any kind seems very valuable for identifying the functional regulatory consequences of non-coding SNPs. Notably; these allow analyzing the functionality of a significant amount of SNPs utilizing a relatively small amount of experimental datasets [17].
There is overwhelming evidence for the existence of substantial genetic influences on general and specific cognitive abilities, and brain-behaviour relationships in healthy and pathological conditions [18, 19]. Nevertheless, until recently the genetics of cognition was constrained by the lack of information. The neurological mutations with rather severe cognitive effects have been practically prevalent throughout the known variants [20]. The current advances in the identification of genetic variation when integrated with the results of genome-wide expression analyses now allow to investigate the molecular-genetic mechanisms that drive cognition and disorders thereof [21]. Therefore, the goal of this work was to reveal novel drivers of certain human neurodevelopmental and neuropsychiatric traits, and/or disorders including major depression, schizophrenia, bipolar affective disorder and autism spectrum disorders. The methodology applied focuses on adopting genome-wide datasets (ChIP-Seq, ChIA-PET and RNA-Seq data) to find the functional SNP variants in the human genome and unravel the underlying mechanisms that are likely to promote cognitive deficits.
Results
Algorithm overview
An overview of the bioinformatic algorithm is shown in Fig. 1 as a flow chart. Further details regarding each step are described in the “Methods” section
Integrating ChIP-seq data from multiple human cell lines. The motivation was to select functional variants through the comprehensive bioinformatics analysis. At the first stage, we collected and incorporated all ChIP-Seq data for human cell lines of different origin that were available at the time of download (July 2015).
Genomic alignment and data filtering. The goal of this technical step was to align raw input reads against the human genome. We kept only the hits that passed our primary quality filtering (in particular, alignment coverage) to ensure further accurate identification of the allele-specific events. To avoid the alignment biases that favored the reads containing the reference allele during further bias binding analysis [22] we realigned the ChIP-Seq reads to specific alternative genome sequences at an interim stage.
SNP calling, identifying SNPs in the regulatory regions (OTFRs) from the ChIP-Seq data. Obviously, the search and analysis of functional variants, especially non-coding ones is the major challenging task. In an effort to succeed, the first selection criteria was the location of the heterozygous SNPs within previously defined regulatory genome regions—Overlapping Transcriptional Factor binding Regions, here and further abbreviated to OTFRs [23]. After SNP calling, only polymorphic sites that survived further filtering were analyzed within OTFRs.
Identifying associated allele-specific binding events. At this step, we assessed the representation of different alleles of the selected heterozygous SNPs in the ChIP DNA. The motivation was that the SNPs with a statistically significant allele-specific signal (asymmetric SNPs) could influence the functional activity of the OTFRs in the human genome.
Identifying targeted genes. We assumed here that the asymmetric SNPs that fit the promotor, intronic and untranslated regions (UTRs) of human genome could directly contribute to the changes in the expression of their nearby genes. The available data on chromatin interactions (ChIA-PET with an RNA pol II antibody) performed for HCT-116, K562 and MCF-7 human cell lines were used in order to determine other possible gene targets that were located distantly from the asymmetric SNP position in the genome.
Identifying potentially regulatory variants (regulatory SNPs, abbreviated here and further to rSNPs). In this step, we selected the asymmetric SNPs that were associated with significant expression differences of their targeted genes through the analysis of several RNA-Seq datasets: the RNA-Seq data for HCT-116, K562 and MCF-7 cells from ENCODE and human RNA-Seq data from the International Cancer Genome Consortium, ICGC [24]. In the event the identified asymmetric SNPs are associated with significant expression differences of their targeted genes and are found in the population, these effects on the expression can continue in terms of phenotypic differences, including neuropsychiatric traits. To avoid a reference allele mapping bias [16, 25, 26], the RNA-Seq reads were realigned to specific alternative genome sequences. Then the heterozygous markers, namely the heterozygous SNPs mapped in the coding regions of the targeted genes were collected through RNA-Seq data analysis. Next, the significant (p < 0.05) allele-specific expression bias was assessed for the corresponding target genes using the selected markers. The resulting variants were further considered as rSNPs. If there was no SNP ID available for the resulting variant, we provided the designation like chr10:70716212, where chrN is human chromosome and the latter number—the rSNP position on the chromosome, bp. The targeted genes for the selected rSNP panel (point 5 from the Algorithm list) were recognized as candidate genes that could contribute to phenotypic outcome.
Link the rSNPs to the risk of a spectrum of cognitive disorders. At this step, we collected GWAS-implicated associations for a spectrum of traits related to cognition and cognitive disorders. Next, we cross-referenced the list of associations from GWAS with the rSNP list. Particularly, we assessed the overlap between the list of rSNPs and the list of the loci from − 10,000 to + 10,000 bp around each GWAS- implicated SNP index. Then we specified MAF (minor allele frequencies) values for GWAS indexes and for selected regulatory variants independently through the open-source (dbSNP). We continued with only those previously annotated rSNP variants that had MAF values close to those given by dbSNP for GWAS-implicated indexes. The latter argued the case that the selected rSNPs were closely linked to the GWAS-implicated loci and thus may have a role in cognitive functions and suggest a higher risk of cognitive disabilities or disorders.
Identifying asymmetry in the expression of the targeted genes. To ensure the regulatory potential of the selected rSNPs we utilized the RNA-Seq datasets for two brain regions: the part of frontal cortex and the part of anterior cingulate from the patients that were diagnosed schizophrenia and bipolar affective disorder available by Xiao et al. [27]. Here we assessed the targeted genes that were differentially expressed between certain patient groups and controls depending on brain region.
Define the potential biological role of the selected regulatory variants. Further, we assessed the composite functional gene–gene interactions between the targeted genes and the genes most related to the original targeted list by GeneMANIA [28]. We also conducted the gene-annotation enrichment analysis and functional annotation clustering using The Database for Annotation, Visualization and Integrated Discovery, DAVID tools [29, 30].
Fig. 1.

The flowchart representing the graphical overview of bioinformatic pipeline. Light-grey rounded-rectangular shapes present the utilization of raw data. asSNP—SNP that is associated with significant allele-specific binding bias, rSNP—regulatory SNP, DE—differential expression, MAF—minor allele frequency, ENCODE—encyclopedia of DNA elements, ICGC—international cancer genome consortium
Identify SNPs that are associated with allele-specific binding and their targeted genes
Chromatin immunoprecipitation sequencing (ChIP-seq) data of TFs, histone marks and other chromatin-associated factors often need to be interpreted in the context of gene regulation. In the present study, the task required first predicting the allele-specific binding from raw data as a straightforward way to home in on regulatory variation. According to this purpose, SRA ChIP-Seq datasets for 27 human cell lines and samples (Additional file 1) and ENCODE ChIP-Seq datasets for the HCT-116, K562 and MCF-7 cells were similarly analyzed resulting in the identification of 298367 unique heterozygous SNPs within the OTFR regions. Then we selected 14,436 SNPs that were defined as asymmetric—associated with a statistically significant allele-specific signal, and therefore could affect the functional activity of the regulatory regions in the human genome. Next step we analyzed the locations of the asymmetric variants in the human genomic regions and identified their nearby and, in possible cases, distant targeted genes—the genes with the expression that might be modified by the asymmetric SNP variant (see “Methods” section for details). As a result, 12,109 from the totaled analyzed asymmetric SNPs were suggested to affect the expression of 9876 targeted genes and entered further analyses.
Integrate with gene expression profiling (RNA-Seq) data
We assumed that the allele-specific expression of the targeted genes observed through the RNA-Seq data analysis could be largely attributable to the regulatory impact of the associated polymorphic variants. At this stage, we employed three ENCODE RNA-Seq datasets for HCT-116, K562 and MCF-7 cells. The asymmetric SNPs that were mapped within transcribed genomic regions were also analyzed using the ICGC human RNA-Seq dataset (Methods). Out of 12,109 asymmetric SNPs with allele-specific expression analyzed using heterozygous SNP markers (Methods), 1633 variants (nearly 13%) had evidence of allele-specific expression differences (p < 0.05 by binomial test) of the corresponding targeted genes. These variants were considered rSNPs that were further analyzed as candidate susceptibility factors in cognitive disorders both with their targeted genes (Additional file 2: Table S1).
Integrate with genome-wide association (GWAS) data
To assess the biological role of identified rSNPs, we examined their overlap with the loci that have been associated with human cognitive disorders or disabilities through GWAS (Methods). For each of resulting 1174 GWAS-implicated index SNPs we examined the presence of identified rSNPs within the − 10,000 to + 10,000 bp window. We assumed here that the certain rSNP is in one linkage group with the index SNP since 1 centimorgan is approximately equal to DNA region of 1 million base pairs [31]. We also examined if these rSNPs have MAF values close to those of GWAS-implicated index SNPs to ensure the linkage (see “Methods” section for details).
This identified fourteen unique rSNPs within GWAS-implicated loci that were associated with risk of cognitive disorders and totalled twelve targeted genes (Additional file 2: Table S2). These regulatory variants were regarded the candidate disease drivers, including a potential impact on schizophrenia, depression and autism spectrum disorders developing by leading to changes in the expression of corresponding target genes. Figure 2 represents the locations of the selected rSNPs in the human genome.
Fig. 2.
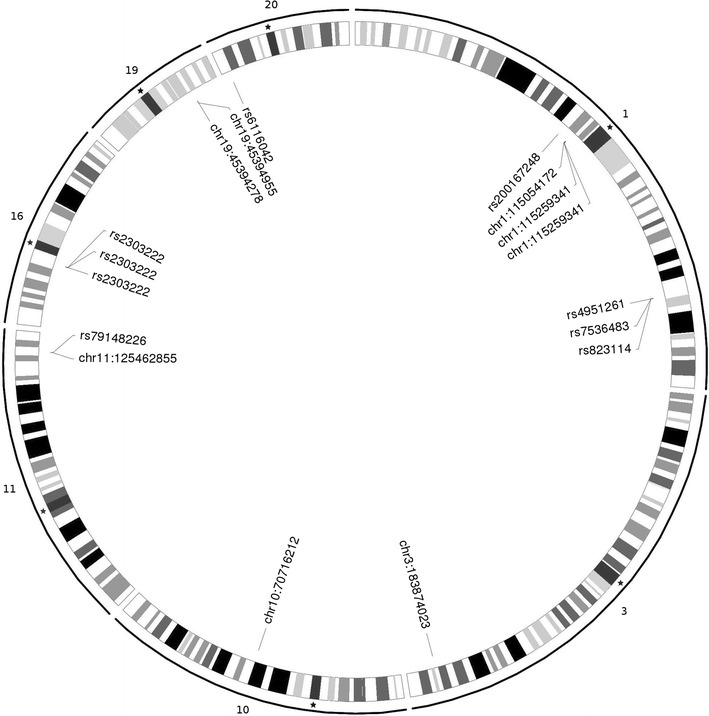
Circular visualization of the distribution of the identified rSNPs in the human genome from R. Grey lines with the numbers along the circumference: individual human chromosomes; the black asterisks: the locations of the centromeres. The IDs for the certain rSNPs are listed in the callouts. The chr(chromosome number):the number(rSNP position on the chromosome, bp) callouts are given for not-annotated variants (according to dbSNP)
Target gene annotation based on gene ontology and biological pathways
Figure 3 shows that physical interactions, co-expression and certain co-localization are apparent among potentially affected genes, such as TOMM40, DDX21, NRAS and NUCKS1. Among the targeted genes, the protein products of NRAS and RAB25 have common structural domain and were identified as interacting partners by GeneMANIA [32]. The totaled list of query genes and the interaction gene–gene network details are given in Additional file 2: Tables S3 and S4, respectively.
Fig. 3.
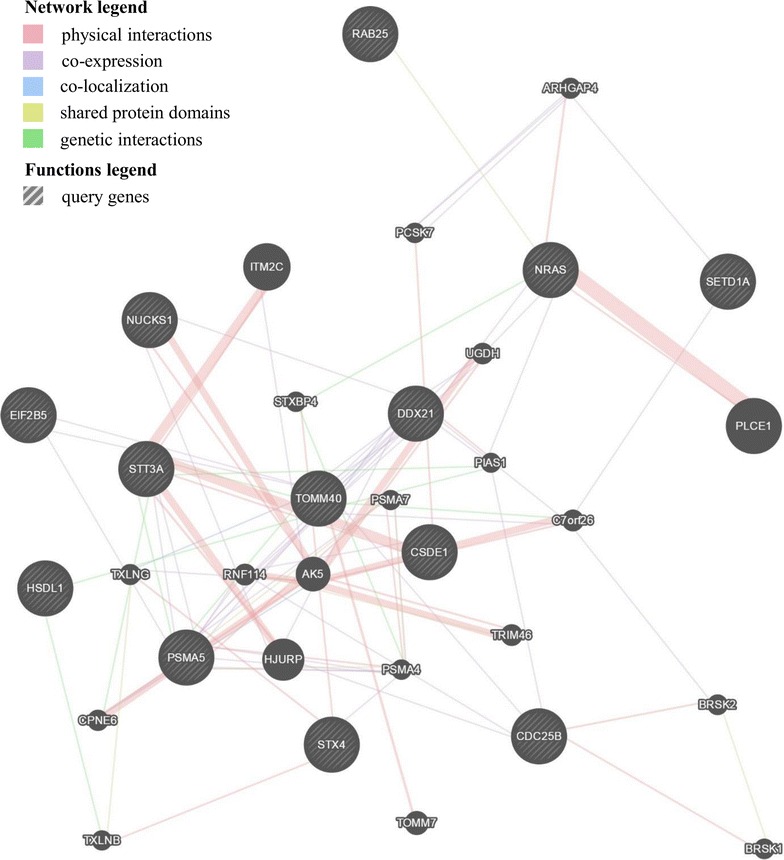
Function prediction of the targeted gene panel by GeneMANIA. The initial list of targeted genes and the type of connections between genes/proteins are illustrated in the functions and networks legends, respectively
The functions of the potentially affected proteins together with the other associated proteins (considering physical and genetic interactions by GeneMANIA) were analyzed using a DAVID software. Pathway analyses revealed 15 nominally enriched gene-sets, which showed partial overlap in terms of the underlying genes. The enriched gene-sets included cell cycle, regulation of protein catabolic processes, innate immune response activating cell surface receptor signaling pathway and stimulatory C-type lectin receptor signaling pathway (Additional file 2: Table S5). The results show that these genes are also involved in the positive regulation of protein modification by small protein conjugation or removal; posttranscriptional regulation of gene expression; neuron projection development; cell morphogenesis involved in neuron differentiation and multiply signaling pathways. Thus, in determining the functions of the identified rSNPs in terms of biological behaviors of the potentially targeted genes, we can speculate that they are possibly important in protein metabolism (including catabolism, phosphorylation, transmembrane transport and import into mitochondrial matrix), regulation of cell cycle, regulation of gene expression, neuron differentiation and development. Accordingly, the PSMA5 protein, PSMA4 and PSMA7 proteins that interact with the protein products of the targeted gene panel are associated with the hsa03050: Proteasome biological pathway by KEGG (Additional file 2: Table S5). The stimulatory C-type lectin receptor signaling pathway enriched in targeted genes is involved in regulating of immunopathogenesis [33] and guiding the dendritic cells in immunity [34].
Analysis of effects on human transcriptomes
We further screened a panel of human cases of schizophrenia and bipolar disorder available by Chun Xu and colleagues [27] for expression of rSNP targeted genes when depending on the tissue and the patient cohort.
As a result of analysis by DeSeq 2.0, NRAS and CDC25B targeted genes happened to be DE in the frontal cortex of schizophrenia-vs-controls and bipolar-disorder–vs-controls groups, respectively when considering a significance threshold of adjusted P value ≤ 0.1 after correcting for multiple testing. Both NRAS and CDC25B genes were higher expressed in the frontal cortex of the certain patient cohorts when compared to controls (logFC of 0,2 and 0,3, respectively). Two other targeted genes happened to be DE in the anterior cingulate of bipolar disorder patients vs controls: DDX21 (adjusted P-value < 0.00012; logFC = −0.7) and NUCKS1 (adjusted P-value < 0.1; logFC = −0.28). No targeted gene was successful to survive the correction for multiple testing in the anterior cingulate of schizophrenia patients vs controls (when considering a significance threshold of adjusted P-value ≤ 0.1).
Discussion
The terms ‘neurodevelopmental and neuropsychiatric disorders’ can cover, to varying degrees, diverse disease classifications including autism, schizophrenia, bipolar disease, etc., that are leading causes of disability worldwide with environmental, genetic and epigenetic risk factors to produce a range of phenotypes in each complex case. Despite seemingly distinct primary diagnoses, the considerable phenotypic heterogeneity, as well as a significant clinical overlap between the subtypes of the certain disease, has been reported [35] as well as an overlap between some genome-wide significant SNPs for different diseases has been observed [36]. The recent data, primarily genome-wide association studies (GWAS), provides the evidence for genetic risk factors. These include genome-wide association with (1) schizophrenia (such as the major histocompatibility complex, MHC, region at 6p22-p21 [37]; (2) depression [38]; (3) bipolar disorder [39], including the variants within ANK3 [40], NCAN [41], CACNA1C and ODZ4 [42] and (4) specific chromosomal regions: 2q, 5, 7q, 15q, 16p [43, 44] and risk genes [45, 46] for autism. Yet, the exact etiology of these disorders remains unknown. It is therefore plausible to believe that different “biological” subgroups of the disease exist, with different underlying genetic etiologies mapping on specific pathways, which underlie specific dysfunctions. Moreover, GWAS often fail to identify replicable common variants [43, 47] or known candidate genes within the GWAS-implicated loci [48, 49]. Assuming that functional alterations affect quantifiable processes including regulatory processes, the comprehensive analysis of genome-wide data is poised to deliver crucial insights into the nature of cognition and the neurodevelopmental and neuropsychiatric disorders thereof.
In this study focusing on the identification of functional risk variants for major neurodevelopmental and neuropsychiatric disorders, we chose to investigate regulatory variation including non-coding, that might strongly influence gene regulation and thus provide relevant information about underlying pathways and molecular mechanisms. Overall, our findings suggested that among the total of identified rSNPs, fourteen fall within the GWAS-implicated loci that are associated with the risk of cognitive disorders (Table 1). Moreover, we identified the corresponding targeted genes that are involved in several processes that seem to be critical for the neurodegeneration and brain dysfunction (Additional file 2: Table S5).
Table 1.
The rSNPs selected for the GWAS traits related to human diseases and cognitive disorders with their targeted genes
| rSNP ID | Targeted gene | GWAS index ID | GWAS-implicated trait |
|---|---|---|---|
| chr10:70716212 | DDX21 | rs2017305 | Depression (quantitative trait) |
| rs200167248 | PSMA5 | rs12049330 | Major depressive disorder |
| chr11:125462855 | STT3A | rs548181 | Combined |
| chr11:125462855 | STT3A | rs548181 | Schizophrenia |
| rs79148226 | STT3A | rs548181 | Schizophrenia |
| rs79148226 | STT3A | rs548181 | Combined |
| chr1:115054172 | CSDE1 | rs3827735 | Autism |
| chr1:115054172 | CSDE1 | rs11102807 | Autism |
| chr1:115259341 | CSDE1 | rs10489525 | Autism |
| chr1:115259341 | NRAS | rs10489525 | Autism |
| chr1:115259341 | CSDE1 | rs8453 | Autism |
| chr1:115259341 | NRAS | rs8453 | Autism |
| rs4951261 | NUCKS1 | rs823114 | Parkinson’s disease |
| rs823114 | NUCKS1 | rs823114 | Parkinson’s disease |
| rs7536483 | NUCKS1 | rs823128 | Parkinson’s disease |
| rs2303222 | SETD1A | rs11865038 | Parkinson’s disease |
| rs2303222 | AC135050.2 | rs11865038 | Parkinson’s disease |
| rs2303222 | STX4 | rs11865038 | Parkinson’s disease |
| chr19:45394278 | TOMM40 | rs115881343 | Cognitive decline (age-related) |
| chr19:45394278 | TOMM40 | rs2075650 | Cognitive decline |
| chr19:45394955 | TOMM40 | rs115881343 | Cognitive decline (age-related) |
| chr19:45394955 | TOMM40 | rs2075650 | Cognitive decline |
| rs6116042 | CDC25B | rs3761218 | Bipolar disorder |
| chr3:183874023 | EIF2B5 | rs1969253 | Major depressive disorder |
The chromosome position in chr:number format is given in place of rSNP ID for six of the variants that are not reported in the Database of Single Nucleotide Polymorphisms (dbSNP, Build ID: {138}). Here chr is human chromosome and the number represents the rSNP position on the chromosome, bp; GWAS index—the ID for the GWAS-implicated SNP that is associated with the specific cognitive trait; combined: the GWAS-implicated associations for all from the list: autism spectrum disorder, attention deficit-hyperactivity disorder, bipolar disorder, major depressive disorder, and schizophrenia
Overall, recent studies have shown the histone modifications such as phosphorylation, methylation, acetylation, and ubiquitination, can recruit chromatin remodeling protein complexes or alter the structure of chromatin to impact gene expression [50, 51]. In particular, lysine methylation regulates the activation (H3K4, H3K36, and H3K79) as well as repression (H3K9, H3K27, and H4K20) of transcription (reviewed in [52]). The last decade has been marked by an increased interest in relating epigenetic mechanisms to gene transcription, protein synthesis, and synaptic plasticity and distally on learning, memory, complex human behaviors and other cognitive functions [53–57]. In the case of schizophrenia it was shown that a number of genes mapping to risk loci may regulate the gene expression through epigenetic mechanisms [58]. Thus, in terms of potential regulatory consequences, the most interesting result seems to be the identification of SETD1A and DDX21 genes that are targeted by rs2303222 and chr10:70716212 variants in our study. These two rSNPs, rs2303222 and chr10:70716212, were found mapped within the loci from − 10,000 to + 10,000 bp around rs11865038 (GWAS-implicated association for Parkinson’s disease) and rs2017305 (GWAS-implicated association for depression), respectively (Additional file 2: Table S2).
The SET1 family of histone methyltransferases is responsible for depositing the H3K4 methylation mark on promoters of active genes [59, 60]. Particularly, the mutations that modify SETD1A function were documented to contribute to neurodevelopmental disorders, including autism and schizophrenia [61, 62] and also to gene silencing [63]. The DDX21 gene encodes a nucleolar protein that is a putative RNA helicase characterized by the conserved DEAD box motif (Asp-Glu-Ala-Asp). This DDX21 helicase is believed to play important roles in coordinating ribosomal RNA transcription and processing, in RNA editing and RNA transport [64, 65]. Data indicate that DDX21 was confirmed to associate with SET8 methyltransferase [66] and is implicated in a number of human diseases [67]. The SET8 interactor protein specifically catalyzes mono-methylation of K20 on histone H4 (H4K20me1) and thus has been implicated in important processes including gene transcriptional regulation, cell cycle control and maintenance of the genome integrity [68, 69]. Thus, our results make a compelling contribution to the case for the interfaces between regulatory variation and the epigenetic mechanisms to be involved in the pathogenesis of neurodevelopmental and neurodegenerative disorders.
The DDX21, rs2303222 targeted gene for RNA helicase, was also shown to be involved in nuclear and mitochondrial splicing. It is worth to note that two other rSNPs—chr19:45394278 and chr19:45394955 novel variants without available SNP ID showed an evidence to contribute to the mitochondria function. These were both associated with cognitive decline according to GWAS and affect the expression of shared TOMM40 targeted gene, encoding the channel-forming subunit of the TOM translocase complex that is essential for import of protein precursors into mitochondria. Overall, this is in line with the hypothesis that there is an association of autism [70, 71], bipolar disorder [72, 73], schizophrenia and other neuropsychiatric diseases [74–76] with impairments in multiply aspects of mitochondrial function including mitochondrial trafficking that affect neuronal synaptic transmission, neuronal growth and consequently neuronal plasticity and connectivity.
The rs2303222 variant targets one more gene, STX4, and our results suggest that this candidate contributes exclusively to Parkinson’s disease. The corresponding protein, syntaxin 4, is involved in synaptic plasticity in hippocampal neurons [77] but has not been previously documented to be associated with Parkinson’s disease, although the synaptic plasticity in the motor cortex was linked to skill learning in mice [78].
Another interesting result regarding all three rs4951261, rs823114 and rs7536483 as rSNPs (regulatory variants) is the shared NUCKS1 targeted gene encoding a protein that links energy homeostasis, glucose metabolism and transcription. There is an evidence that NUCKS1 can regulate the recruitment of the RNA polymerase II enzyme and the chromatin accessibility in the specific promoter regions [79].
We also recognized shared targeted STT3A gene for chr11:125462855 and rs79148226 regulatory variants associated with GWAS-implicated loci for schizophrenia and schizophrenia combined with autism. The significant associations of the variants in STT3A locus with the schizophrenia as well as the potential role in pathogenic mechanisms were previously documented [80]. Further, the mutations in STT3A are being considered in the differential diagnosis for congenital disorders of N-linked glycosylation (CDG-N-linked) pathway. So it was shown by Freeze and colleagues [81] that the STT3A mutation significantly impairs glycosylation of the biomarker transferrin in a previously unreported case of inherited glycosylation disorder characterized with broad clinical features including microcephaly, cerebellar atrophy, intellectual disability and seizures. The available literature can be used to argue that STT3A functions is important for efficient protein folding and anterograde trafficking [82]. It’s important that the alterations in the proteostasis network including protein folding contribute to abnormal protein aggregation in the pathology of various neurodegenerative diseases [83–85], however the STT3A mutations may directly affect or may not directly affect neurodevelopment and cognitive function.
Summing the evidence, human studies revealed that the brain is particularly sensitive to changes in dosage of various proteins including from regulators to synaptic proteins to the coordinators of the transport and metabolism of brain mRNAs [86, 87]. Dynamic changes that are required for synaptic plasticity, a cellular correlate for learning and memory, rely on protein synthesis and protein degradation. Thus, either of these cellular processes must be finely balanced as significant impairments could result in pathologies.
In our study, novel chr3:183874023 variant within the locus for major depressive disorder was matched to EIF2B5 gene. This gene encodes a subunit of eukaryotic translation initiation factor 2B (EIF2B), which has a role in protein synthesis as an essential regulator [88]. To give another example, among the nominally enriched targeted gene-sets, the regulation of protein catabolic processes was identified in the present study, in particular through the effects of rs200167248 on PSMA5 target. This gene was associated with major depressive disorder in the study (Table 1) and encodes a member of the peptidase T1A family, that is a 20S core alpha proteasome subunit [89].
Interestingly, a partial overlap in Gene Ontology terms for the underlying PSMA5 gene was found here between the ‘protein catabolic processes’ and the ‘innate immune response’ gene-sets. Another targeted gene that was associated with innate immune response is NRAS target for chr1:115259341 novel regulatory variant for autism. The protein product for NRAS shuttles between the Golgi apparatus and the plasma membrane [90]. This finding may be in correspondence to the evidence that alterations in immune response were recognized among individuals diagnosed with autism spectrum disorders [91, 92]. The chr1:115259341 variant in NRAS falls also to the promotor region of CDSE1 (cold shock domain containing E1) gene. Interestingly, CDSE1 is a distant target for the chr1:115054172 variant in 5′UTR of TRIM33. Both chr1:115259341 and chr1:115054172 rSNPs are located within GWAS-implicated locus for autism (Additional file 2: Table S2). Here our findings are in accordance with the published findings that suggest NRAS-CSDE1 as candidate genes mapping the previously reported linkage region (1p13.2) for autism [93]. CSDE1, also known as UNR (upstream of N-ras), is an RNA-binding protein that may contribute to post-transcriptional control of gene expression in several ways: acting as an activator or inhibitor of translation initiation, stabilizing mRNA or promoting mRNA turnover [94–97]. Thus, the novel regulatory variants of chr1:115259341 and chr1:115054172 are likely to play an important role in the proper control of brain gene expression and, consequently in cognitive functions.
Finally, our findings demonstrate that changes in the expression of a number of candidate genes identified in the study may contribute to the etiopathogenesis of schizophrenia and bipolar disorder. This could be due to the effects of the related rSNPs, regardless of whether the GWAS associations with the certain trait was significant. (In this regard, testing differential expression of the targeted genes between the patients with distinct genotypes appears to provide interesting data. The available datasets have unfortunately not allowed us to conduct the analysis.)
Our data proposed, as mentioned in the “Results” section, that the physical interactions and co-expression do exist among rSNP-targeted genes, including DDX21 and NUCKS1. Interestingly, both these genes were found lower expressed in the anterior cingulate [98] of patients with bipolar disorder when compared to controls. This finding may represent a role of DDX21 in gene transcriptional regulation [64] and of NUCKS1 in the DNA damage response [99] as candidate pathways in disease etiopathogenesis. It is worth to note, that the associated rSNPs (Table 1) do not fall in GWAS-implicated loci directly for bipolar disorder. We also could find no evidence of DDX21 and NUCKS1 expression changes to bipolar disorder or depression in papers. This is possible because the heterogeneities of disease phenotypes [100, 101].
Further schizophrenia and bipolar disorder patients have shown higher expression of two different targeted genes: NRAS and CDC25B, respectively, in the frontal cortex than controls. NRAS- encoded protein, an intrinsic GTPase of Ras superfamily, is generally associated with cancer advance and progression [102, 103] but is also important in neurodevelopmental disorders for the role to transduce signal from activated receptors further to MAPK cascade [104]. As a proof of concept of candidate pathways, deregulation of CDC25 phosphatase proteins also has an essential role in cell-cycle-driven neuronal death [104]. Moreover, the members of CDC25 family were documented as possible targets that have therapeutic potential in disease [105, 106]. Thus, our findings relating DE targeted genes might lead to new insights to explore the possible links for regulatory variants in different brain regions of schizophrenia and bipolar disorder. However, a role for identified regulatory variants and their gene targets in cognition and disorders thereof is yet to be seen in details.
Conclusions
Much attention has focused on unravelling the mechanisms by which genetic variation can determine divergence in gene expression levels and, consequently, the phenotypic outcome, yet we are still far from an integrated, evidence-based understanding of the etiopathogenesis of cognitive disorders. Summing up, in the current study, we present novel findings that expand the repertoire of functional variation in human genome, recognize the targeted genes and provide an evidence relevant to disease-associated effects of the identified rSNPs on cognition including on bipolar affective disorder, major depressive disorder and schizophrenia.
Methods
Data collection
To investigate the potentially functional SNPs in human genome and further interpret the underlying mechanisms we collected the chromatin immunoprecipitation sequencing (ChIP-Seq, ChIA-PET) and transcriptional profiling (RNA-Seq) data available within the framework of international validated projects such as the Encyclopedia of DNA Elements Project, ENCODE [14] and the International Cancer Genome Consortium, ICGC [24]. In total, we reprocessed 617 ChIP-seq for 29 unique samples and human RNA-seq data sets from two different studies. The samples were from Illumina HiSeq platform. SRA files were converted to fastq format by the fastq-dump tool.
ChIP-seq datasets were performed using antibodies towards histone epigenetic markers (anti-H3K27ac, anti-H3K4 me1, anti-H3K4 me2, anti-H3K4 me3, anti-H3K27 me3), transcriptional factors and a few other chromatin-associated proteins (Additional file 1) and obtained from the following: ENCODE and SRA [107]. ChIA-PET and RNA-Seq datasets for HCT-116, MCF-7 and K562 cells were obtained from ENCODE.
The human RNA-Seq datasets were obtained from the International Cancer Genome Consortium (ICGC) Controlled Data (EGAD00001000215) by Seshagiri et al. [108] and from the NIH Short Read Archive (SRP035524) by Xiao et al. [27].
Trimming low quality positions
The Trimmomatic-3.2.2 program [109] was applied for the data pre-processing and removing the adapter sequences. To reduce false positives, only the genomic regions that were covered by at least 10 high-quality reads were further analyzed. We also excluded bases with Phred ≤ 20.
Human reference sequences
We used the human genome build 37 (GRCh37) assembly based on the Genome Reference Consortium Human genome build 37. The genome sequence was downloaded from the UCSC Genome Center [110].
It was shown, that mapping the reads to a single reference genome can significantly affect the outcome of analyses of allele-specificity, both for RNA-seq and ChIP-seq experiments [22, 25]. This will cause the reads representing the reference allele to be preferentially mapped. Thus, in order to avoid mapping bias towards reference alleles we constructed the specific alternative genome sequences by replacing the reference bases at the polymorphic sites with the bases representing the alternate alleles from the collection of all heterozygous SNPs identified directly from ChIP-Seq data. The alternate reference genomes were built independently for the HCT-116, K562 and MCF-7 cell lines. To realign the reads before the search for allele-specific events each specific alternative reference was used independently to utilize the raw data from the corresponding human cell line.
Data alignment to reference genome sequences
Bowtie2 [111] or TopHAT2 [112] software was applied to map raw paired-end reads to reference genome. Then SAMtools 1.2 [113] and Picard tools were used to discard the duplicated reads and PCR/optical artifacts. We continued with uniquely mapped reads at QMAP > 25 threshold (SAMtools) to reduce the share of sequencing and alignment errors.
Determination of genomic regions
Our previous analysis of the ChIP-Seq data for multiply original human cell lines resulted in defining a certain set of potentially regulatory regions in the human genome specified as OTFRs [23]. Each of OTFRs (Overlapping Transcriptional Factor binding Regions) showed the associations with specific phenotypic outcome and contained binding sites for two or more transcriptional factors. Thus, to identify functional SNP variants we analyzed only the reads that mapped to OTFRs after realignment of the available raw ChIP-Seq data.
Categories of gene elements, such as intronic regions and 3′\5′ UTRs as well as the transcription start sites for annotated genes (TSSs) were obtained from the GRCh37 annotation data [114]. Promoter regions were set as from 1,8 kbp upstream to 1,8 kbp downstream of all annotated TSSs.
SNPs calling
After preprocessing and alignment, all the ChIP-Seq reads that mapped within the OTFRs were filtered with the depth of 10 and a mapping quality of 25 set as threshold and then processed using SAMtools pileup, PerlScript and R [115]. As a result, we discovered 298,367 heterozygous SNPs directly from ChIP-Seq data.
Quality-control metrics for SNPs
To ensure accurate identification of the allele-specific events, all discovered SNPs were subjected to primary filtering. The SNPs in the following categories were eliminated: SNPs within sex chromosomes, mitochondrial DNA and repeat regions [116], SNPs within 5 bp of the regions that map to insertions/deletions, clustered SNPs (that is, those within 10 bp of two other SNPs) and SNPs with significantly different coverage of the reference and alternative alleles (p < 0.05 by binomial test). After this initial quality control, only the reads mapped to the heterozygous sites with at least three alleles that were identified from at least at two samples and two reference genome sequences were further analysed to avoid somatic mutations.
Analysis of the allele-specific binding events
After an alignment to the alternative genomes described above by Bowtie2 [111], ChIP-Seq reads that were specifically mapped to the specific allele of each heterozygous SNP were counted using SAMtools Perl library. The significant (p < 0.01) differences for read counts between the reference and the alternative alleles were assessed using a two-sided binomial test (implemented in R). The resulting p-values were adjusted for multiple testing by Benjamini–Hochberg adjustment.
Determination of the targeted genes
To predict the potential targeted (affected) genes nearby the rSNP position we considered that the rSNPs that fall into the intronic, 3′\5′ UTRs and promoter regions may affect the expression specifically of these genes.
We also considered the possibility that each rSNP may affect a promotor of a distant gene, may be outside the associated risk region. To identify such distantly affected genes, we took advantage of an analysis of the recently available ChIA-PET data (“Data collection” section). A minimum of 20 paired ChIA-PET mapped reads that mapped to the certain genomic region was required. The filtering by at least 10 ChIA-PET reads mapped to the genomic regions in both directions was applied to minimize the mapping errors. Next, the average combined area of ChIP-Seq RNA Pol II peaks was calculated in order to determine the effective size of the human genome. Then we built the contact matrix for the regions of ± 1000 bp from the positions of the SNPs associated with allele-specific binding bias and promoter regions of known human genes. The contacts that fit the intersecting and genomic regions and the interchromosomal contacts were excluded from the analysis. Pearson’s agreement criterion (p < 0.001) was applied to assess the reliable contacts.
Definition of heterozygous SNP markers through RNA-Seq data analysis
RNA sequencing enables defining the allele-specific expression by measuring the sequence reads that are unambiguously mapped to each of the two gene alleles and, accordingly, assessing the preferential expression of the certain allele in a diploid genome. In this case, at least one exonic heterozygous SNP must fit the usable RNA-Seq reads. The asymmetric SNPs that fell in the gene promotors and UTRs and were discovered from RNA-Seq data were directly used for allele-specific expression analysis. Alternatively, we discovered the heterozygous SNP markers within the coding regions of the analyzed targeted genes through the analysis of the ICGC human RNA-Seq dataset (EGAD00001000215).
Analysis of the allele-specific expression events
RNA reads were realigned to each of the used human reference genomes using TopHAT2 [112]. From the mRNA sequence reads and the location of each assymetric SNPs, the appropriate base read at the location of each polymorphic site was extracted. Then both SAMtools pileup and custom-made R scripts were used to extract allele counts for each SNP. A minimum of 10 RNA reads crossing the heterozygous SNP position was required. An exon was considered to have allele-specific expression if the proportion of the expression between two alleles was significantly greater than 1.5 or less than 1/1.5 (P-value ≤ 0.05). The Fisher exact test was used to examine the significance. The resulting p-values were adjusted for multiple testing by Benjamini-Hochberg adjustment.
Collecting the GWAS-implicated associations
We used the ‘Alzheimer’s disease’, ‘autism’, ‘autism spectrum disorder’, ‘antipsychotic’, ‘anxiety’, ‘bipolar disorder’, ‘cognitive’, ‘depression’, ‘depressive disorder’, ‘Parkinson’s disease’, ‘posttraumatic’ and ‘schizophrenia’ signatures for the GWAS Catalog query to define GWAS-implicated loci that could be related to cognitive disorders.
Determination of closely linked SNPs
The threshold for difference in the minor allele frequencies (MAF) was set ≤ 15% for considering that two analyzed SNPs fall within one linkage group. These served to choose the linked GWAS-implicated variants for the identified rSNPs when integrating with genome-wide association (GWAS) data. Here the variants, that were reported by the Database of Single Nucleotide Polymorphisms, dbSNP [117] were subjected to a direct allele frequency analysis.
Functional annotation of targeted genes
The tools of the DAVID Bioinformatics Resources (the Database for Annotation, Visualization and Integrated Discovery, DAVID) [29] were used to provide functional interpretation of targeted gene list derived from the study. All remaining parameters were kept at their default values. The results of functional enrichment analysis are given in the Additional file 2: Table S5.
GeneMANIA [32] web interface was also used to identify direct (physical binding) and indirect (functional) interacting partners of targeted genes based on genomic and co-expression data as well as published experimental data. The input was a list of twelve targeted genes (Table 1) which was then extended by GeneMANIA. Data sets were collected from publicly available databases according to the pipeline described in detail in [118]. A resulting functional association network illustrating the relationships among the genes is presented on the Fig. 3 (see Additional file 2: Tables S3, S4 for details).
Differential gene expression (DEG) analysis
Once the target genes are predicted, investigating gene expression levels in the case of disease is useful as well to assess the effects of multiple genetic variants on gene function. The RNA-Seq data for the patients from the analyzed cohorts (see “Data collection” section) were realigned to the human reference genomes at an intermediate stage. Then the DeSeq2 Bioconductor package [119] was applied to the data on certain tissues from the patient groups and controls. The resulting p-values were adjusted for multiple testing by Benjamini–Hochberg adjustment. Genes with Benjamini–Hochberg adjusted p-value < 0.01 were considered as significant.
R code
All statistics and circos imaging was done in R, version 3.1.0 [115]. The custom-made scripts that were applied to perform the analyses and generate the plots are available upon request.
Additional files
Additional file 1. Contains the accession list 1 with the accession numbers for utilized ChIP-Seq datasets (by NCBI archive).
Additional file 2. Contains the Tables S1–S5, as listed below: Table S1. (The identified rSNPs) contains the IDs and information on the totalled regulatory variants identified in the study. Table S2. (The rSNPs that were found associated with cognitive disorders) contains the data on the identified regulatory variants that fell within the − 10,000 to + 10,000 bp window around GWAS-implicated SNPs for analyzed traits related to cognition and cognitive disorders. Table S3. (The list of genes in GeneMANIA query) contains the totalled annotation of the targeted genes and their interactors by GeneMANIA. Table S4. (The interactions within the targeted gene list by GeneMANIA) contains the information on the composite functional gene–gene interactions between the targeted genes and the genes most related to the original targeted list by GeneMANIA. Table S5. (Functional annotation of targeted genes) contains DAVID results for targeted genes and their potential interaction partners by GeneMANIA.
About this supplement
This article has been published as part of BMC Neuroscience Volume 19 Supplement 1, 2018: Selected articles from Belyaev Conference 2017: neuroscience. The full contents of the supplement are available online at https://bmcneurosci.biomedcentral.com/articles/supplements/volume-19-supplement-1.
Authors’ contributions
This study was designed by LOB, NPB and TIM. LOB performed study conceptualization, data collecting, and was a major contributor to the study methodology and data analysis. EEK analyzed and interpreted the data, prepared figures, tables, and additional files, and was a major contributor in writing the manuscript. IIB performed formal data analysis and data visualization from R, prepared figures and tables including supplementary material and drafted the manuscript. EYL analyzed the data and drafted the manuscript. NPB was a major contributor to the study conceptualization, analyzed the data and drafted the manuscript. TIM supervised the study and drafted the manuscript. All authors read and approved the final manuscript. The researches were grant holders during the data collecting and analyzing. All authors read and approved the final manuscript.
Acknowledgements
The research has been carried out with support of the Grant #16-15-10131 from the Russian Science Foundation. The computations were performed at the Siberian Supercomputer Center of SB RAS (Novosibirsk, Russia).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All accession numbers and details on genome-wide data analysed during this study are included in this published article and its supplementary information files. The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
This manuscript reports studies involving human RNA-Seq datasets that were obtained from:
the International Cancer Genome Consortium (ICGC) Controlled Data (EGAD00001000215) by Seshagiri et al. [108] here the patient-matched fresh-frozen primary colon tumours and normal tissue samples with appropriate Institutional Review Board approval and patient-informed consent were obtained from commercial sources. The human tissue samples used in the study were de-identified (double-coded) before their use and hence the study using these samples is not considered human subject research under the US Department of Human and Health Services regulations and related guidance (45 CFR Part 46) and
from the NIH Short Read Archive (SRP035524) by Xiao et al. [27]. This reference study was approved by the Institutional Review Board of Texas Tech University Health Science Center, Texas, United States. All patients provided written informed consent. The human tissue samples were collected from the Southwest Brain Bank with consent from the next-of-kin (NOK). The NOK agreed to provide the donation and they read a State approved form. The authors called the NOK and recorded their agreement.
Ethics approval and consent to participate
The human cell data (Chip-Seq, ChlA-PET, RNA-Seq), were also used, all made available through public research projects {SRA archive and ENCODE). Hence the present study using available NGS data is not considered human subject research and said concerns and ethical statements are not applicable.
Funding
The study and the article-processing fee were funded by the Russian Science Foundation (Grant #16-15-10131).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- SNP(s)
single nucleotide polymorphism(s)
- rSNP
regulatory SNP
- TF
transcriptional factor
- OTFR
overlapping transcriptional factor binding region
- UTR(s)
untranslated region(s)
- TSS(s)
transcription start site(s)
- ChIP-Seq
chromatin immunoprecipitation followed by high-throughput DNA sequencing
- RNA-Seq
RNA sequencing
- ChIA-PET
chromatin interaction analysis by paired-end tag sequencing
- DE (G)
differentially expressed (differential gene expression)
- MAF
global minor allele frequency
- kbp
kilobase pairs
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12868-018-0414-3) contains supplementary material, which is available to authorized users.
Contributor Information
Leonid O. Bryzgalov, Email: leonolr14@gmail.com
Elena E. Korbolina, Phone: +7(383) 363-49-80, Email: lungry@bionet.nsc.ru
Ilja I. Brusentsov, Email: brusentsovi@gmail.com
Elena Y. Leberfarb, Email: leberfarb@mail.ru
Natalia P. Bondar, Email: nbondar@bionet.nsc.ru
Tatiana I. Merkulova, Email: merktativ@gmail.com
References
- 1.Naidoo N, Pawitan Y, Soong R, Cooper DN, Ku C-S. Human genetics and genomics a decade after the release of the draft sequence of the human genome. Hum Genomics. 2011;5:577. doi: 10.1186/1479-7364-5-6-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung CC, Chanock SJ. Current status of genome-wide association studies in cancer. Hum Genet. 2011;130:59–78. doi: 10.1007/s00439-011-1030-9. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Y, Clark WT, Mort M, Cooper DN, Radivojac P, Mooney SD. Prediction of functional regulatory SNPs in monogenic and complex disease. Hum Mutat. 2011;32:1183–1190. doi: 10.1002/humu.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–860. doi: 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- 6.Sneha P, George Priya Doss C. Molecular dynamics: new frontier in personalized medicine. Adv Protein Chem Struct Biol. 2016;102:181–224. doi: 10.1016/bs.apcsb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 7.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards D, Batley J, Snowdon RJ. Accessing complex crop genomes with next-generation sequencing. Theor Appl Genet. 2013;126:1–11. doi: 10.1007/s00122-012-1964-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet. 2010;86(5):730–742. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lappalainen T. Functional genomics bridges the gap between quantitative genetics and molecular biology. Genome Res. 2015;25:1427–1431. doi: 10.1101/gr.190983.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy TE, Gertz J, Pauli F, Kucera KS, Varley KE, Newberry KM, et al. Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res. 2012;22:860–869. doi: 10.1101/gr.131201.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 2014;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavalli M, Pan G, Nord H, Wallen Arzt E, Wallerman O, Wadelius C. Allele-specific transcription factor binding in liver and cervix cells unveils many likely drivers of GWAS signals. Genomics. 2016;107:248–254. doi: 10.1016/j.ygeno.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Ogletree BT, Morrow-Odom KL, Westling D. Understanding the brain-behaviour relationship in persons with ASD: implications for PECS as a treatment choice. Dev Neurorehabil. 2015;18:88–96. doi: 10.3109/17518423.2013.833995. [DOI] [PubMed] [Google Scholar]
- 19.Fagan ES, Pihlstrøm L. Genetic risk factors for cognitive decline in Parkinson’s disease: a review of the literature. Eur J Neurol. 2017;24:561-e20. doi: 10.1111/ene.13258. [DOI] [PubMed] [Google Scholar]
- 20.de Geus EJ, Wright MJ, Martin NG, Boomsma DI. Genetics of brain function and cognition. Behav Genet. 2001;31:489–495. doi: 10.1023/A:1013360909048. [DOI] [PubMed] [Google Scholar]
- 21.Kremen WS, Panizzon MS, Cannon TD. Genetics and neuropsychology: a merger whose time has come. Neuropsychology. 2016;30:1–5. doi: 10.1037/neu0000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni Y, Weber Hall A, Battenhouse A, Iyer VR. Simultaneous SNP identification and assessment of allele-specific bias from ChIP-seq data. BMC Genet. 2012;13:46. doi: 10.1186/1471-2156-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryzgalov LO, Antontseva EV, Matveeva MY, Shilov AG, Kashina EV, Mordvinov VA, Merkulova TI. Detection of regulatory SNPs in human genome using ChIP-seq ENCODE data. PLoS One. 2013;8(10):e78833. doi: 10.1371/journal.pone.0078833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, et al. International cancer genome consortium data portal-a one-stop shop for cancer genomics data. Database (Oxford) 2011;2011:bar026. doi: 10.1093/database/bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevenson KR, Coolon JD, Wittkopp PJ. Sources of bias in measures of allele-specific expression derived from RNA-seq data aligned to a single reference genome. BMC Genom. 2013;14(536):6. doi: 10.1186/1471-2164-14-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vijaya Satya R, Zavaljevski N, Reifman J. A new strategy to reduce allelic bias in RNA-Seq readmapping. Nucleic Acids Res. 2012;40:e127. doi: 10.1093/nar/gks425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao Y, Camarillo C, Ping Y, Arana TB, Zhao H, Thompson PM, et al. The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS One. 2014;9:e95875. doi: 10.1371/journal.pone.0095875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montojo J, Zuberi K, Rodriguez H, et al. GeneMANIA: fast gene network construction and function prediction for Cytoscape [version 1; referees: 2 approved] F1000Research. 2014;3:153. doi: 10.12688/f1000research.4572.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31(3):241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 32.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–20. [DOI] [PMC free article] [PubMed]
- 33.Kingeter LM, Lin X. C-type lectin receptor-induced NF-κB activation in innate immune and inflammatory responses. Cell Mol Immunol. 2012;9(2):105–112. doi: 10.1038/cmi.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010;22:1397–1405. doi: 10.1016/j.cellsig.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coe BP, Girirajan S, Eichler EE. The genetic variability and commonality of neurodevelopmental disease. Am J Med Genet Part C Semin Med Genet. 2012;160C(2):118–129. doi: 10.1002/ajmg.c.31327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forstner AJ, Hecker J, Hofmann A, Maaser A, Reinbold CS, Muhleisen TW, et al. Identification of shared risk loci and pathways for bipolar disorder and schizophrenia. PLoS One. 2017;12(2):e0171595. doi: 10.1371/journal.pone.0171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arslan A. Imaging genetics of schizophrenia in the post-GWAS era. Prog Neuro-Psychopharmacol Biol Psychiatry. 2018;80(Pt B):155–165. doi: 10.1016/j.pnpbp.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 38.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–1036. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budde M, Forstner AJ, Adorjan K, Schaupp SK, Nöthen MM, Schulze TG. Genetics of bipolar disorder. Nervenarzt. 2017;88(7):755–759. doi: 10.1007/s00115-017-0336-9. [DOI] [PubMed] [Google Scholar]
- 40.Ferreira MAR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cichon S, Mühleisen TW, Degenhardt FA, Mattheisen M, Miró X, Strohmaier J, et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. 2011;88:372–381. doi: 10.1016/j.ajhg.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Takumi T. Genomic and genetic aspects of autism spectrum disorder. Biochem Biophys Res Commun. 2014;452:244–253. doi: 10.1016/j.bbrc.2014.08.108. [DOI] [PubMed] [Google Scholar]
- 44.Veenstra-VanderWeele J, Cook EH. Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9:819–832. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- 45.Bralten J, van Hulzen KJ, Martens MB, Galesloot TE, Arias Vasquez A, Kiemeney LA, et al. Autism spectrum disorders and autistic traits share genetics and biology. Mol Psychiatry. 2017. 10.1038/mp.2017.98. [DOI] [PubMed]
- 46.Li X, Zou H, Brown WT. Genes associated with autism spectrum disorder. Brain Res Bull. 2012;88:543–552. doi: 10.1016/j.brainresbull.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry. 2015;77(1):66–74. doi: 10.1016/j.biopsych.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim Y, Zerwas S, Trace SE, Sullivan PF. Schizophrenia genetics: where next? Schizophr Bull. 2011;37(3):456–463. doi: 10.1093/schbul/sbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smeland OB, Wang Y, Lo M-T, Li W, Frei O, Witoelar A, et al. Identification of genetic loci shared between schizophrenia and the big five personality traits. Sci Rep. 2017;7:2222. doi: 10.1038/s41598-017-02346-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oey NE, Leung HW, Ezhilarasan R, Zhou L, Beuerman RW, VanDongen HM, VanDongen AM. A neuronal activity-dependent dual function chromatin-modifying complex regulates arc expression. eNeuro 2015;2(1). pii: ENEURO.0020-14.2015. [DOI] [PMC free article] [PubMed]
- 51.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 53.Li X, Bao X, Wang R. Neurogenesis-based epigenetic therapeutics for Alzheimer’s disease (review) Mol Med Rep. 2016;14:1043–1053. doi: 10.3892/mmr.2016.5390. [DOI] [PubMed] [Google Scholar]
- 54.Grigorenko EL, Kornilov SA, Naumova OY. Epigenetic regulation of cognition: a circumscribed review of the field. Dev Psychopathol. 2016;28:1285–1304. doi: 10.1017/S0954579416000857. [DOI] [PubMed] [Google Scholar]
- 55.Parkel S, Lopez-Atalaya JP, Barco A. Histone H3 lysine methylation in cognition and intellectual disability disorders. Learn Mem. 2013;20:570–579. doi: 10.1101/lm.029363.112. [DOI] [PubMed] [Google Scholar]
- 56.Jarome TJ, Lubin FD. Histone lysine methylation: critical regulator of memory and behavior. Rev Neurosci. 2013;24:375–387. doi: 10.1515/revneuro-2013-0008. [DOI] [PubMed] [Google Scholar]
- 57.Franklin TB, Mansuy IM. The prevalence of epigenetic mechanisms in the regulation of cognitive functions and behaviour. Curr Opin Neurobiol. 2010;20:441–449. doi: 10.1016/j.conb.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 58.Whitton L, Cosgrove D, Clarkson C, Harold D, Kendall K, Richards A, et al. Cognitive analysis of schizophrenia risk genes that function as epigenetic regulators of gene expression. Am J Med Genet Part B Neuropsychiatr Genet. 2016;171(8):1170–1179. doi: 10.1002/ajmg.b.32503. [DOI] [PubMed] [Google Scholar]
- 59.Ernst P, Vakoc CR. WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics. 2012;11:217–226. doi: 10.1093/bfgp/els017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ali A, Tyagi S. Diverse roles of WDR5-RbBP5-ASH2L-DPY30 (WRAD) complex in the functions of the SET1 histone methyltransferase family. J Biosci. 2017;42:155–159. doi: 10.1007/s12038-017-9666-9. [DOI] [PubMed] [Google Scholar]
- 61.Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–577. doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89:940–947. doi: 10.1016/j.neuron.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jezek M, Gast A, Choi G, Kulkarni R, Quijote J, Graham-Yooll A, et al. The histone methyltransferases Set5 and Set1 have overlapping functions in gene silencing and telomere maintenance. Epigenetics. 2017;12:93–104. doi: 10.1080/15592294.2016.1265712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;518:249–253. doi: 10.1038/nature13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Baysac KC, Yee L-F, Saporita AJ, Weber JD. Elevated DDX21 regulates c-Jun activity and rRNA processing in human breast cancers. Breast Cancer Res. 2014;16:449. doi: 10.1186/s13058-014-0449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qin Y, Ouyang H, Liu J, Xie Y. Proteome identification of proteins interacting with histone methyltransferase SET8. Acta Biochim Biophys Sin (Shanghai) 2013;45:303–308. doi: 10.1093/abbs/gmt011. [DOI] [PubMed] [Google Scholar]
- 67.Sloan KE, Leisegang MS, Doebele C, Ramírez AS, Simm S, Safferthal C, et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015;43:553–564. doi: 10.1093/nar/gku1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milite C, Feoli A, Viviano M, Rescigno D, Mai A, Castellano S, et al. Progress in the development of lysine methyltransferase SETD8 inhibitors. ChemMedChem. 2016;11:1680–1685. doi: 10.1002/cmdc.201600272. [DOI] [PubMed] [Google Scholar]
- 69.Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siddiqui MF, Elwell C, Johnson MH. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism Open Access 2016;6(5). pii: 1000190. [DOI] [PMC free article] [PubMed]
- 71.Griffiths KK, Levy RJ. Evidence of mitochondrial dysfunction in autism: biochemical links, genetic-based associations, and non-energy-related mechanisms. Oxid Med Cell Longev. 2017;2017:4314025. doi: 10.1155/2017/4314025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scaini G, Rezin GT, Carvalho AF, Streck EL, Berk M, Quevedo J. Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci Biobehav Rev. 2016;68:694–713. doi: 10.1016/j.neubiorev.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 73.Kato T. Neurobiological basis of bipolar disorder: mitochondrial dysfunction hypothesis and beyond. Schizophr Res. 2016;187:62–66. doi: 10.1016/j.schres.2016.10.037. [DOI] [PubMed] [Google Scholar]
- 74.Khacho M, Clark A, Svoboda DS, MacLaurin JG, Lagace DC, Park DS, et al. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum Mol Genet. 2017;26(17):3327–3341. doi: 10.1093/hmg/ddx217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Devaraju P, Zakharenko SS. Mitochondria in complex psychiatric disorders: lessons from mouse models of 22q11.2 deletion syndrome: hemizygous deletion of several mitochondrial genes in the 22q11.2 genomic region can lead to symptoms associated with neuropsychiatric disease. Bioessays. 2017 doi: 10.1002/bies.201600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ben-Shachar D. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr Res. 2017;187:3–10. doi: 10.1016/j.schres.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 77.Mohanasundaram P, Shanmugam MM. Role of syntaxin 4 in activity-dependent exocytosis and synaptic plasticity in hippocampal neurons. Sci Signal. 2010;3(144):jc7. doi: 10.1126/scisignal.3144jc7. [DOI] [PubMed] [Google Scholar]
- 78.Xu T, Wang S, Lalchandani RR, Ding JB. Motor learning in animal models of Parkinson’s disease: aberrant synaptic plasticity in the motor cortex. Mov Disord. 2017;32(4):487–497. doi: 10.1002/mds.26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qiu B, Shi X, Wong E, Lim J, Bezzi M, Low D, et al. NUCKS is a positive transcriptional regulator of insulin signaling. Cell Rep. 2014;7:1876–1886. doi: 10.1016/j.celrep.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 80.Jajodia A, Kaur H, Kumari K, Gupta M, Baghel R, Srivastava A, et al. Evidence for schizophrenia susceptibility alleles in the Indian population: an association of neurodevelopmental genes in case-control and familial samples. Schizophr Res. 2015;162(1–3):112–117. doi: 10.1016/j.schres.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 81.Shrimal S, Ng BG, Losfeld M-E, Gilmore R, Freeze HH. Mutations in STT3A and STT3B cause two congenital disorders of glycosylation. Hum Mol Genet. 2013;22:4638–4645. doi: 10.1093/hmg/ddt312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Medinas DB, Valenzuela V, Hetz C. Proteostasis disturbance in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26(R2):R91–R104. doi: 10.1093/hmg/ddx274. [DOI] [PubMed] [Google Scholar]
- 84.Chandel TI, Zaman M, Khan MV, Ali M, Rabbani G, Ishtikhar M, et al. A mechanistic insight into protein-ligand interaction, folding, misfolding, aggregation and inhibition of protein aggregates: an overview. Int J Biol Macromol. 2018;106:1115–1129. doi: 10.1016/j.ijbiomac.2017.07.185. [DOI] [PubMed] [Google Scholar]
- 85.Shrivastava AN, Aperia A, Melki R, Triller A. Physico-pathologic mechanisms involved in neurodegeneration: misfolded protein-plasma membrane interactions. Neuron. 2017;95:33–50. doi: 10.1016/j.neuron.2017.05.026. [DOI] [PubMed] [Google Scholar]
- 86.Klein ME, Monday H, Jordan BA. Proteostasis and RNA binding proteins in synaptic plasticity and in the pathogenesis of neuropsychiatric disorders. Neural Plast. 2016;2016:3857934. doi: 10.1155/2016/3857934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Louros SR, Osterweil EK. Perturbed proteostasis in autism spectrum disorders. J Neurochem. 2016;139:1081–1092. doi: 10.1111/jnc.13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wortham NC, Proud CG. eIF2B: recent structural and functional insights into a key regulator of translation. Biochem Soc Trans. 2015;43(6):1234–1240. doi: 10.1042/BST20150164. [DOI] [PubMed] [Google Scholar]
- 89.Han Y-G, Liu H-L, Zheng H-J, Li S-G, Bi R-C. Purification and refolding of human alpha5-subunit (PSMA5) of the 20S proteasome, expressed as inclusion bodies in Escherichia coli. Protein Expr Purif. 2004;35:360–365. doi: 10.1016/j.pep.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 90.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: Back to the future for anti-ras drug discovery? Clin Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ansel A, Rosenzweig JP, Zisman PD, Melamed M, Gesundheit B. Variation in gene expression in autism spectrum disorders: an extensive review of transcriptomic studies. Front Neurosci. 2016;10:601. doi: 10.3389/fnins.2016.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bjorklund G, Saad K, Chirumbolo S, Kern JK, Geier DA, Geier MR, et al. Immune dysfunction and neuroinflammation in autism spectrum disorder. Acta Neurobiol Exp (Wars) 2016;76(4):257–268. doi: 10.21307/ane-2017-025. [DOI] [PubMed] [Google Scholar]
- 93.Xia K, Guo H, Hu Z, Xun G, Zuo L, Peng Y, et al. Common genetic variants on 1p13.2 associate with risk of autism. Mol Psychiatry. 2014;19(11):1212–1219. doi: 10.1038/mp.2013.146. [DOI] [PubMed] [Google Scholar]
- 94.Ray S, Catnaigh PÓ, Anderson EC. Post-transcriptional regulation of gene expression by Unr. Biochem Soc Trans. 2015;43(3):323–327. doi: 10.1042/BST20140271. [DOI] [PubMed] [Google Scholar]
- 95.Ray S, Anderson EC. Stimulation of translation by human Unr requires cold shock domains 2 and 4, and correlates with poly(A) binding protein interaction. Sci Rep. 2016;6:22461. doi: 10.1038/srep22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wurth L, Papasaikas P, Olmeda D, Bley N, Calvo GT, Guerrero S, et al. UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell. 2016;30:694–707. doi: 10.1016/j.ccell.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 97.Kamenska A, Simpson C, Vindry C, Broomhead H, Benard M, Ernoult-Lange M, et al. The DDX6-4E-T interaction mediates translational repression and P-body assembly. Nucleic Acids Res. 2016;44:6318–6334. doi: 10.1093/nar/gkw565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stevens FL, Hurley RA, Taber KH. Anterior cingulate cortex: unique role in cognition and emotion. J Neuropsychiatry Clin Neurosci. 2011;23(2):121–125. doi: 10.1176/jnp.23.2.jnp121. [DOI] [PubMed] [Google Scholar]
- 99.Ramanan VK, Saykin AJ. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am J Neurodegener Dis. 2013;2(3):145–175. [PMC free article] [PubMed] [Google Scholar]
- 100.Milaneschi Y, Lamers F, Peyrot WJ, Abdellaoui A, Willemsen G, Hottenga J-J, et al. Polygenic dissection of major depression clinical heterogeneity. Mol Psychiatry. 2016;21:516–522. doi: 10.1038/mp.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Charney AW, Ruderfer DM, Stahl EA, Moran JL, Chambert K, Belliveau RA, et al. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl Psychiatry. 2017;7(1):e993. doi: 10.1038/tp.2016.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cicenas J, Tamosaitis L, Kvederaviciute K, Tarvydas R, Staniute G, Kalyan K, et al. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med Oncol. 2017;34:26. doi: 10.1007/s12032-016-0879-9. [DOI] [PubMed] [Google Scholar]
- 103.Johnson DB, Smalley KSM, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res. 2014;20:4186–4192. doi: 10.1158/1078-0432.CCR-13-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ryu H-H, Lee Y-S. Cell type-specific roles of RAS-MAPK signaling in learning and memory: implications in neurodevelopmental disorders. Neurobiol Learn Mem. 2016;135:13–21. doi: 10.1016/j.nlm.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 105.Singh L, Pushker N, Sen S, Singh MK, Bakhshi S, Chawla B, Kashyap S. Expression of CDC25A and CDC25B phosphatase proteins in human retinoblastoma and its correlation with clinicopathological parameters. Br J Ophthalmol. 2015;99(4):457–463. doi: 10.1136/bjophthalmol-2014-305830. [DOI] [PubMed] [Google Scholar]
- 106.Evain-Bana E, Schiavo L, Bour C, Lanfranchi DA, Berardozzi S, Ghirga F, et al. Synthesis, biological evaluation and molecular modeling studies on novel quinonoid inhibitors of CDC25 phosphatases. J Enzyme Inhib Med Chem. 2017;32(1):113–118. doi: 10.1080/14756366.2016.1238364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sequence Read Archive. https://trace.ncbi.nlm.nih.gov/Traces/sra. Accessed 29 May 2015.
- 108.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.The UCSC Genome Center Archive. ftp://hgdownload.cse.ucsc.edu/goldenPath/. Accessed 4 Sept 2015.
- 111.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Human Annotation FTP Download. ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz. Accessed 29 May 2015.
- 115.Group TR core. The R project for statistical computing. https://www.r-project.org. Accessed 3 Apr 2014.
- 116.Human Annotation Repeat Regions. http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/simpleRepeat.txt. Accessed 29 May 2015.
- 117.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Contains the accession list 1 with the accession numbers for utilized ChIP-Seq datasets (by NCBI archive).
Additional file 2. Contains the Tables S1–S5, as listed below: Table S1. (The identified rSNPs) contains the IDs and information on the totalled regulatory variants identified in the study. Table S2. (The rSNPs that were found associated with cognitive disorders) contains the data on the identified regulatory variants that fell within the − 10,000 to + 10,000 bp window around GWAS-implicated SNPs for analyzed traits related to cognition and cognitive disorders. Table S3. (The list of genes in GeneMANIA query) contains the totalled annotation of the targeted genes and their interactors by GeneMANIA. Table S4. (The interactions within the targeted gene list by GeneMANIA) contains the information on the composite functional gene–gene interactions between the targeted genes and the genes most related to the original targeted list by GeneMANIA. Table S5. (Functional annotation of targeted genes) contains DAVID results for targeted genes and their potential interaction partners by GeneMANIA.
Data Availability Statement
All accession numbers and details on genome-wide data analysed during this study are included in this published article and its supplementary information files. The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.