

Abstract
Tissue calcification is an important physiological process required for the normal structure and function of bone. However, ectopic or excessive calcification contributes to diseases such as chondrocalcinosis, to calcium deposits in the skin or to vascular calcification. SMOC2 is a member of the BM-40/osteonectin family of calcium-binding secreted matricellular proteins. Using osteoprogenitor MC3T3-E1 cells stably overexpressing SMOC2, we show that SMOC2 inhibits osteogenic differentiation and extracellular matrix mineralization. Stable Smoc2 knockdown in these cells had no effect on mineralization suggesting that endogenous SMOC2 is not essential for the mineralization process. Mineralization in MC3T3-E1 cells overexpressing mutant SMOC2 lacking the extracellular calcium-binding domain was significantly increased compared to cells overexpressing full length SMOC2. When SMOC2 overexpressing cells were cultured in the presence of extracellular calcium supplementation, SMOC2’s inhibitory effect on calcification was rescued. Our observations were translationally validated in primary human periosteal-derived cells. Furthermore, SMOC2 was able to impair mineralization in transdifferentiated human umbilical vein endothelial cells. Taken together, our data indicate that SMOC2 can act as an inhibitor of mineralization. We propose a possible role for SMOC2 to prevent calcification disorders.
Introduction
Tissue calcification is an important and physiological process required for the normal structure and function of bone [1]. Calcification of the bone extracellular matrix gives the bone and body structure, helps to protect the inner organs and is a storage site from which calcium can be mobilized when required. However, abnormal or excessive calcification of tissues contributes to symptoms or complications of different diseases. For instance, chondrocalcinosis is a skeletal disorder in which calcium pyrophosphate crystals are deposited in the joints and tendons, triggering acute and painful inflammation [2]. Moreover, calcium crystal deposits occur in the skin in patients suffering from systemic sclerosis. Also, calcium crystal deposits can be found in arteries, a feature associated with increased cardiovascular risk. Vascular calcification most often occurs in patients suffering from diabetes, renal insufficiency or atherosclerosis [3–5]. Thus, there is need for effective strategies that prevent pathological calcification.
SMOC2 (SPARC-related modular calcium-binding protein 2) is a secreted calcium-binding protein from the BM-40/SPARC/osteonectin family of secreted matricellular proteins. BM-40/SPARC/osteonectin family members all contain an extracellular calcium-binding (EC) domain, a follistatin-like (FS) domain and an acidic N-terminal domain. SMOC2 has a unique composition different from the other family members as 2 thyroglobulin domains and a SMOC-specific domain separate the EC domain and FS domain [6–8].
SMOC2 was originally identified from an extracellular extract of the articular cartilage [9–11], a tissue in which calcification must be avoided. Indeed, the uncalcified proteoglycan and water rich extracellular matrix of the articular cartilage allows efficient and low-friction mobility between the bones. This function must be preserved during aging to avoid the development of osteoarthritis, the most common chronic joint disease [12]. Based on its structure and its expression in the articular cartilage, we hypothesized that SMOC2 may have inhibitory effects on calcification. Thus, we investigated the effect of SMOC2 on mineralization and calcification. We demonstrate, in different in vitro models, that SMOC2 strongly inhibits calcification. Calcium sequestration by SMOC2’s calcium binding domain is proposed as part of the underlying mechanism.
Materials and methods
Materials and cells
All products used were purchased from Sigma unless otherwise stated. Human periosteum-derived cells (hPDC) and human umbilical vein endothelial cells (HUVEC) were a kind gift of the Tissue Engineering Unit, SBE center, KU Leuven. All procedures were approved by the ethical committee for clinical research (UZ Leuven), and informed consent was obtained from the patients.
Generation of stable gene overexpression or silencing cell lines
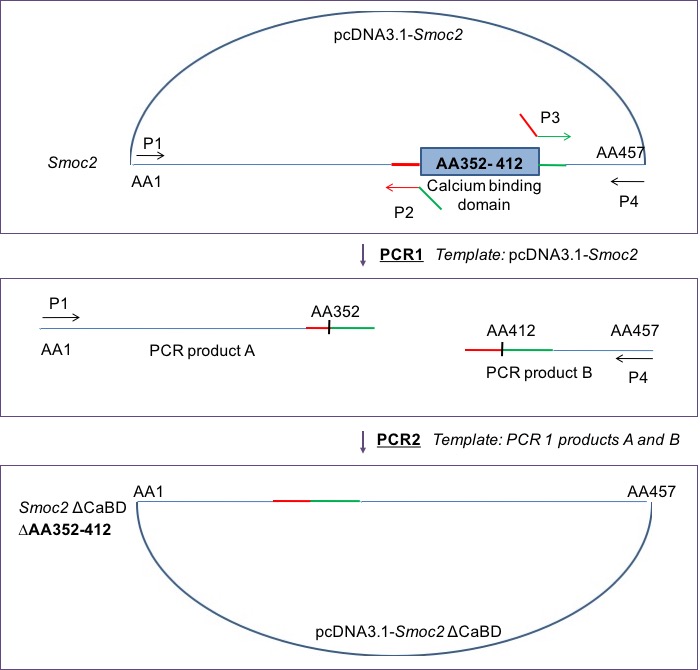
MC3T3-E1 cells were plated at a density of 2,600 cells/cm2 in a 6 well-plate and transfected with 2 μg of an empty pcDNA3.1+ vector (3.1) as a control, the pcDNA3.1-Smoc2 (Smoc2+), pcDNA3.1-Smoc2 lacking the calcium binding domain (ΔCaBD), non-interfering short hairpin micro (shmi)RNA (Gipz) or a shmiRNA against Smoc2 (ShSmoc2). pcDNA3.1-Smoc2 ΔCaBD was generated by performing PCR-directed mutagenesis using the pcDNA3.1-Smoc2 as a template as explained in the scheme in S1 Fig. Briefly, the calcium binding domain spans from aminoacid 352 to 412. For the first PCR reaction, we used the pcDNA3.1 plasmid containing wild type Smoc2 and primer pair A (P1 and P2) to obtain the PCR product A and primer pair B (P3 and P4) to obtain PCR product B. Primers were designed in such a way that the products had an overlap to bind to each other when used as templates in PCR reaction 2. The resulting product is the pcDNA3.1 plasmid containing mutant Smoc2 lacking the calcium binding domain (ΔAA352-412). Primers sets were manually designed using free internet software Primer3 (http://frodo.wi.mit.edu/primer3/). Primer set A consisted of P1 fwd, 5’-ATGCTGCCGCCACAGCTGT-3’; and P2 rev, 5’-GACACCCAAAACCCTCTCCTCCAGGGTGTG -3’. Primer set B consisted of P3 fwd, 5’-GAGAGGGTTTTGGGTGTCACCAGAGAGGAG-3’; and P4 rev, 5’-TCATCCTTGTTTCCTGGGCTGT-3’. ShmiRNA against Smoc2 was purchased from Open Biosystems (now Dharmacon; clone ID V2LMM_74468). The transfection reagent Arrest-In (Thermo Scientific) was used for the transfection reaction. To select stable lines, cells were treated for 10 days with the appropriate selection antibiotics (3.1, Smoc2+, Smoc2 ΔCaBD at 1 mg/ml of geneticin, (G418) Gibco; Gipz, ShSmoc2 at 0.5 μg/μl of puromycin (Life technologies/invitrogen)). Then, single colonies were created with a dilution method in which a single resistant cell was isolated by serial half-dilutions. The single cell was then cultured and expanded keeping the appropriate antibiotic pressure.
In vitro experiments
MC3T3-E1 cells
MC3T3-E1 cells stably transfected with either the empty pcDNA3.1+ vector or with the pcDNA3.1-Smoc2 (mouse) overexpression vector were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (2 mM Glutamax; 4.5 g/L high glucose, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% sodium pyruvate (Gibco), 1% antibiotics-antimycotic (penicillin-streptomycin/amphotericin B, Gibco). Cells were seeded at 2600 cells/cm2 in 6-well plates (D0) and induction of MC3T3-E1 mineralization started the day after (D1) by culturing cells for 21 days in Alpha Minimal Essential medium (αMEM) from Gibco, supplemented with 10% FBS, 1% sodium pyruvate, 1% antibiotics-antimycotic, 10 mM β-glycerophosphate (β-GP) and 50 μg/ml ascorbic acid-2-phosphate (AA). D1 samples represent undifferentiated cells whereas D21 samples are differentiated. Medium was changed every 2 days, but AA was added daily. The suitable antibiotic pressure (used to establish the stable clones) was maintained for the duration of the experiment using either 1 mg/ml of G418 or 0.5 μg /ml of puromycin.
Human periosteum-derived cells (hPDCs) and human umbilical vein endothelial cells (HUVECs)
Human periosteum-derived cells (hPDCs) were seeded at a density of 4,500 cells/cm2. Human umbilical vein endothelial cells (HUVECs) were seeded on gelatin-coated plastic at a density of 4,500 cells/cm2 and stimulated in basal endothelial cell growth (EGM)-2 medium (Lonza) in the presence of 200 ng/ml of Bone morphogenetic protein 6 (BMP6; R&D systems), known to trigger the endothelial to mesenchymal transition [13]. Both hPDCs and HUVECs were then cultured for 21 days as MC3T3-E1 cells but supplemented with 100 nM dexamethasone to trigger calcification. hPDCs and HUVECs were cultured in the presence of supernatants harvested from either 3.1, Smoc2+ or ΔCaBD MC3T3-E1 cells.
Alizarin red staining
At D1 and D21, samples were washed with phosphate buffered saline (PBS) and fixed with ice-cold methanol for at least 30 min at 4°C. Cells were then washed again with PBS and stored in water at 4°C until further use. Plates were stained for 1 hour with 1% Alizarin red solution pH 4.1–4.3 and washed with water afterwards. The staining was then quantified using a protocol according to Gregory C.A. et al [14].
Gene expression analysis
At D1 and D21, samples were washed with PBS and stored dry at -80°C until further use. Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel). Gene expression was analyzed by RT-qPCR using Maxima SYBR Green qPCR Master Mix (Fermentas) in the rotor-gene 6000 (Corbett research). The ΔΔCt method was used to determine fold changes. 40S ribosomal protein S29 was used as internal control gene for normalization. ΔCt values are calculated as Ct (gene of interest)–Ct (S29). Fold change for each gene of interest compared to reference (control D1) was equal to 2-ΔΔCt with ΔΔCt = ΔCt (target)—ΔCt (reference). Mouse S29 was detected using the following primers: S29 fwd, 5’-CCAGCAGCTCTACTGGAGTCA-3’; and S29 rev, 5’-GCCTATGTCCTTCGCGTACT-3’. For endogenous mouse Smoc2, the following primers were used: Smoc2 fwd, 5’-CAAGTGCAAAGATCCACAGC-3’; and Smoc2 rev, 5’-GCCTATGTCCTTCGCGTACT-3’. Osteogenic markers mouse Osx, Opn and Col1a2 were detected by using the following primers: Osx fwd, 5’-ATGGCGTCCTCTCTGCTTGA-3’; and Osx rev, 5’-AGTCCCGCAGAGGGCTAGAG-3’; Opn fwd, 5’-TCCAATCGTCCCTACAGTCG-3’; and Opn Rev, 5’-AGGTCCTCATCTGTGGCATC-3’; Col1a2 fwd, 5’-GGCAGAGATGGTGTTGATGG-3’; and Col1a2 rev, 5’-AGGGCCAGATGAAACTCCTT-3’.
Western blot analysis
Total protein was extracted by adding 0.05% TritonX-100 to the samples followed by ultrasonication (Microson ultrasonic cell disruptor, Misonic). Afterwards, half of the samples were stored at -80°C for alkaline phosphatase (ALP) assay. Phosphatase inhibitors Na3VO4 2.3 mM and 5 mM and protease inhibitor phenylmethylsulfonyl fluoride (PMSF) 0.36 mM and the commercial protease inhibitor cocktail PIC (Sigma P2714)) were added to the other half of the samples. Proteins were quantified according to the Pierce bicinchoninic acid (BCA) Protein assay kit (Thermo scientific). Absorbance was measured with the micro plate reader Titertek plus MS212 at 570 nm. For Western blot analysis, supernatants from MC3T3-E1 cells were harvested and clarified by centrifugation at 12,000 × g for 5 min. The same volume of supernatants (10 μl for each sample) was placed in a final concentration of 1× Laemmli buffer. Proteins were separated by electrophoresis on a NuPAGE® Bis-Tris gel (Invitrogen) and transferred from the gel to an activated polyvinylidene difluoride (PVDF) membrane (Millipore) using the Trans-blot® SD semi-dry electrophoretic transfer cell system (Biorad). Ponceau red staining was performed to confirm equal protein loading. Membranes were blocked with 5% non-fat dry milk in Tris buffered saline with 0,1% Tween (TBS-T). Mouse SMOC2 was detected with anti-SMOC2 rabbit polyclonal (diluted 1:1000; Santa Cruz sc-67396 (no longer available)), followed by incubation with anti-rabbit horseradish peroxidase-linked secondary antibody (diluted 1:5000; Jackson ImmunoResearch). The target protein was visualized using the Supersignal West Femto maximum Sensitivity Substrate kit (Thermo Scientific) and the Las3000 mini (Fujifilm).
Alkaline phosphatase assay
An ALP assay was performed using the BluePhos Microwell Substrate kit (KPL). Briefly, 20 μl of proteins (stored at -80°C) were placed, in duplicate, in a 96-well plate. 50 μl of work solution (mix of ½ solution A and ½ solution B) was added to every sample followed by incubation at 37°C. Absorbance at 620 nm was measured after 15 minutes of incubation and results were expressed as absorbance values normalized to total protein content.
Statistical analysis
Results are presented as the mean ± standard deviation (SD) of three independent replicates, unless otherwise specified. Comparisons were made by 2- way ANOVA using GraphPad Prism 7 software. A value of P < 0.05 was considered significant.
Results
SMOC2 inhibits osteogenic differentiation and ECM mineralization
To study how SMOC2 affects osteogenic differentiation and mineralization, we overexpressed Smoc2 in MC3T3-E1 osteoblast precursor cells (Smoc2+) and differentiated these into mature osteoblasts that produce a calcified extracellular matrix (ECM). Following stable transfection, relative Smoc2 mRNA levels were determined in Smoc2+ cells compared to control cells expressing an empty vector (3.1) at baseline (day 1) and after 21 days in the differentiation culture. At day 1, Smoc2 mRNA levels were highly upregulated (20-fold increase) in Smoc2+ cells compared to 3.1 cells (Fig 1A). This upregulation was sustained during the differentiation process. In control cells, endogenous Smoc2 expression was increased after osteogenic differentiation but remained about 4 times lower than in Smoc2+ cells. We then assessed the secretion of SMOC2 protein in supernatants of differentiated Smoc2+ cells compared to 3.1+ cells by Western blot. These results confirm that secreted SMOC2 levels from the modified cells remain increased after 21 days of culture (Fig 1B).
Fig 1. Influence of full length Smoc2 overexpression on differentiation and mineralization of MC3T3-E1 cells.

Gene expression (A) and Western blot (B) analysis of MC3T3-E1 cells overexpressing Smoc2 show successful overexpression of Smoc2. At D21 Smoc2+ cells show smaller increase in ALP activity (C) and less Alizarin red staining (D-E) compared to 3.1. (F) Smoc2+ modifies mRNA expression of Opn, Osx and Col1a2 during osteogenic differentiation. Statistically significant differences vs. D1 3.1 (internal control) are indicated as *: P<0.05; vs. D21 3.1 as #: P<0.05). Figures are representative of two independent experiments with three different Smoc2+ overexpressing clones, each experiment performed in triplicates. mRNA levels were normalized to S29.
As a functional readout for osteogenic differentiation, ALP activity was measured (Fig 1C). At baseline, no difference in ALP activity could be detected between Smoc2+ and control cells. However, the expected increase in ALP levels after differentiation at day 21 was significantly lower in Smoc2+ cells compared to control cells. As a marker for matrix mineralization, calcium deposition was evaluated by alizarin red staining. At baseline, we did not detect mineralization in Smoc2+ or control cells, but after 21 days of differentiation, matrix mineralization was significantly lower in Smoc2+ cells than in control cells (Fig 1D and 1E). Messenger RNA levels of phenotypic markers of osteogenic differentiation, i.e. transcription factor osterix (Osx), ECM gene osteopontin (Opn) and bone-related collagen 1a2 (Col1a2) were assessed by RT-q-PCR. At day 1, no significant difference could be detected in mRNA expression level of Osx and Opn, whereas Col1a2 was significantly decreased in Smoc2+ cells compared to control cells. The increase in mRNA levels of Osx and Col1a2 over time after 21 days of differentiation was significantly lower in Smoc2+ cells compared to control cells. Smoc2+ cells showed significantly reduced Opn mRNA levels at D21 (Fig 1F). Altogether, these data support that SMOC2 acts as negative modulator of osteogenic differentiation and ECM mineralization in MC3T3-E1 cells.
SMOC2 is not essential for osteogenic differentiation and ECM mineralization
To further examine the role of SMOC2 in osteogenic differentiation and mineralization in vitro, we generated MC3T3-E1 cells in which endogenous Smoc2 was silenced. Relative Smoc2 mRNA levels were determined in control cells expressing a non-interfering short hairpin micro (shmi)RNA (Gipz) and cells with stable knock-down generated by shmiRNA against Smoc2 (ShSmoc2), after 1 and 21 days in culture. Fig 2A confirms lower Smoc2 mRNA level in ShSmoc2 cells compared to control cells both at D1 and D21 (6-fold and 16-fold respectively). In Gipz control cells, Smoc2 expression was induced 3- to 3.5-fold upon 21 days of differentiation. We confirmed the effective reduction of secreted SMOC2 protein by Western blot analysis on supernatants of differentiated ShSmoc2 cells (Fig 2B). We found no significant differences in ALP activity between Gipz and ShSmoc2 cells at D1 and D21 (Fig 2C). Likewise, alizarin red staining did not differ between Gipz and ShSmoc2 cells upon differentiation (Fig 2D and 2E). These findings suggest that endogenous SMOC2 is not essential for osteogenic differentiation and ECM mineralization.
Fig 2. Influence of full length Smoc2 knock down on differentiation and mineralization of MC3T3-E1 cells.

Gene expression (A) and western blot (B) analysis of MC3T3-E1 cells with Smoc2 knock down show successful knockdown of Smoc2. At D21 ShSmoc2 cells show no difference in ALP activity (C) and Alizarin red staining (D-E) compared to control cells (Gipz). Statistically significant differences vs. D1 Gipz (internal control) are indicated as *: P<0.05; vs. ShSmoc2 at D1 as ¶: P<0.05; vs. D21 Gipz as #: P<0.05). Figures are representative of 3 independent experiments with 3 different ShSmoc2 clones, each experiment performed in triplicate.
The calcium binding domain partially mediates the inhibitory effects of SMOC2 on osteogenic differentiation and ECM mineralization
Calcium signaling plays an important role in osteogenic differentiation. We therefore studied whether the effects of SMOC2 on MC3T3-E1 cells were mediated by its calcium binding domain. To address this question, we generated MC3T3-E1 cells overexpressing mutant Smoc2 lacking the calcium binding domain (ΔCaBD) and we compared them to MC3T3-E1 cells overexpressing full length Smoc2 (Smoc2+). Fig 3A demonstrates successful and comparable overexpression of Smoc2 ΔCaBD and full length Smoc2 based on their relative mRNA levels. ALP activity and Alizarin red staining (Fig 3B–3D) of the cells did not differ at baseline. Following the 21 days differentiation protocol we detected a significant increase in ALP activity (2-fold) in differentiated ΔCaBD cells compared to Smoc2+ cells (Fig 3B). Likewise, after 21 days of differentiation, ΔCaBD cells showed stronger mineralization compared to Smoc2+ cells (Fig 3C and 3D). Gene expression analysis of osteoblast phenotype markers Opn, Osx, and Col1a2 is depicted in Fig 3E. At D1, no significant differences could be detected in mRNA expression levels of Osx and Col1a2 whereas Opn was significantly increased in ΔCaBD cells compared to Smoc2+ cells. The increase in mRNA levels of Opn, Osx and Col1a2 after 21 days of differentiation was significantly higher in ΔCaBD cells compared to Smoc2+ cells. Taken together, these results indicate that the calcium binding domain of SMOC2 is playing a role in the inhibitory effects on osteogenic differentiation and ECM mineralization in MC3T3-E1 cells.
Fig 3. Influence of Smoc2 ΔCaBD on differentiation and mineralization of MC3T3-E1 cells.

Gene expression analysis of MC3T3-E1 cells overexpressing Smoc2 lacking the calcium binding domain shows successful overexpression (A). At D21 increase in ALP activity (B) is higher and Alizarin red staining (C-D) is stronger in ΔCaBD cells compared to Smoc2+. (E) mRNA expression of Osx, Opn, Col1a2 shows that ΔCaBD can decrease or suppress the influence of Smoc2 overexpression on MC3T3-E1 differentiation. mRNA levels were normalized to S29. Statistically significant differences vs. D1 Smoc2+ (internal control) are indicated as *: P<0.05; vs. ΔCaBD at D1 as ¶: P<0.05; vs. D21 Smoc2+ as #: P<0.05). Figures are representative for one experiment with 3 different Smoc2+ overexpressing clones performed in triplicate.
Extracellular calcium supplementation diminishes the inhibitory effect of SMOC2 on osteogenic differentiation and ECM mineralization
We hypothesized that high levels of SMOC2 could titer part of the calcium out of the extracellular environment, by capturing it through its calcium binding domain. To test this hypothesis, we evaluated whether the addition of extracellular calcium (1 mM of CaCl2) to the medium (2mM of CaCl2) during differentiation could rescue the impaired mineralization observed in Smoc2+ cells. Therefore MC3T3-E1 Smoc2+ cells were differentiated for 21 days in the absence or presence of extracellular calcium supplementation. Fig 4A shows no difference in alizarin red staining between undifferentiated Smoc2+ and Smoc2+ + 1mM CaCl2. However, at D21, Smoc2+ + 1mM CaCl2 cells exhibited a stronger alizarin red staining than Smoc2+ cells. This result suggests a rescue of mineralization in Smoc2+ cells when extracellular calcium is added. Extracellular calcium addition also enhanced ALP activity in Smoc2+ cells (Fig 4B). These data, in agreement with the results obtained with the calcium binding domain deletion, support that calcium plays a role in the impairment of SMOC2 on osteogenic differentiation and mineralization.
Fig 4. Influence of calcium supplementation to Smoc2+ MC3T3-E1 cells on differentiation and mineralization.

Addition of extracellular calcium to Smoc2+ cells shows increased Alizarin Red staining (A-B) and a higher increase in ALP activity (C) at D21. Statistically significant differences vs. D1 Smoc2+ (internal control) are indicated as *: P<0.05; vs. Smoc2+ + 1mM Ca at D1 as ¶: P<0.05; vs. D21 Smoc2+ as #: P<0.05). Figures are representative for one experiment with 2 different Smoc2+ overexpressing clones performed in triplicate.
SMOC2 impairs mineralization in primary human osteoblast precursors and of transdifferentiated HUVECs
To translationally validate the data obtained in a murine cell line, we evaluated the influence of SMOC2 in a human in vitro model of osteogenesis. As SMOC2 is a secreted protein, we differentiated human periosteum derived mesenchymal cells for 21 days in the absence or presence of supernatants from MC3T3-E1 3.1, Smoc2+ or ΔCaBD cells. Fig 5A confirms equal protein levels of secreted full length SMOC2 and SMOC2 ΔCaBD. At D21, Fig 5B and 5C and 5D show reduced alizarin red staining and ALP activity, respectively in the presence of Smoc2+ supernatants compared to 3.1 controls. Notably, hPDCs in the presence of ΔCaBD supernatants exhibited a smaller reduction in ALP activity and alizarin red staining compared to Smoc2+. To further investigate the potential clinical implications of our findings, we explored whether SMOC2 could also have an impact on vascular calcification, which is a common complication of vascular diseases such as atherosclerosis, predicting cardiovascular morbidity and mortality [15–16]. To this end, we differentiated HUVECs for 21 days in the absence or presence of supernatants from MC3T3-E1 3.1, Smoc2+ or ΔCaBD cells. After 21 days of differentiation, HUVECs cultured in the presence of Smoc2+ supernatants exhibited a reduced alizarin red staining as compared to 3.1 control cells. Moreover, HUVECs in the presence of ΔCaBD supernatants exhibited a smaller decrease in Alizarin red staining compared to Smoc2+ (Fig 5E and 5F). These results show that, consistent with our results in MC3T3-E1 cells, SMOC2 negatively modulates osteogenic differentiation and mineralization in primary human osteoblast precursors and in human endothelial cells. This suggests that SMOC2 could have a potential therapeutic benefit in diseases which involve vascular calcification such as atherosclerosis.
Fig 5. Influence of Smoc2+ and of Smoc2 ΔCaBD on differentiation and mineralization of hPDCs and HUVECs.

(A) Confirmation of equal protein levels of secreted full length SMOC2 and SMOC2 ΔCaBD. (B-C) hPDCs treated with Smoc2+ supernatant show less Alizarin staining compared to 3.1, while ΔCaBD produces an intermediate response. Similar results were obtained for ALP activity (D). (E) HUVECs treated with Smoc2+ supernatant show less Alizarin staining compared to 3.1, ΔCaBD being intermediate as demonstrated by quantification of absorbance at 405 nm (F). Statistically significant differences vs. D1 (within same condition) are indicated as *: P<0.05; vs. 3.1 at D21 as ¶: P<0.05; vs. D21 Smoc2+ as #: P<0.05). Figures are representative of two independent experiments, performed in triplicate in hPDCs and of one experiment, performed in triplicate in HUVECs.
Discussion
In this study, we investigated the influence of SMOC2 on mineralization and calcification in different model systems. First, we assessed the influence of SMOC2 on mineralization in the MC3T3-E1 cell line. Therefore, we used MC3T3-E1 cells overexpressing Smoc2 or with Smoc2 knockdown. Measurement of endogenous Smoc2 level in control cells (both 3.1 and Gipz) shows Smoc2 upregulation during the differentiation and mineralization process, which is however significantly lower than Smoc2 levels in Smoc2 overexpression cells. Cells overexpressing Smoc2 show a severely reduced mineralization in this model. On the other hand, Smoc2 knockdown did not affect mineralization. As endogenous Smoc2 knockdown did not affect the mineralization process, we suggest the endogenous Smoc2 upregulation in the differentiating cells might be a consequence of mineralization rather than a regulator and that the levels are too low to effectively inhibit mineralization. It has been described however that SMOC2 deficiency is associated with human and zebrafish dental and craniofacial defects [17–18]. Bloch-Zupan et al identified a homozygous SMOC2 mutation in patients suffering from dentin dysplasia phenotype with major microdontia, oligodontia, and shape abnormalities. In a zebrafish study using the morpholino technique to knockdown Smoc2, they demonstrated abnormalities in the pharyngeal teeth similar to the human phenotype [17]. Melvin et al at the other hand identified Smoc2 as a gene involved in craniofacial morphogenesis as Smoc2 knockdown in Zebrafish led to craniofacial abnormalities [18]. A possible explanation for the contrast with our data is that MC3T3-E1 cells are derived from neonatal mouse calvaria, when cells are already committed to osteogenic differentiation [19]. Thus, the presence of Smoc2 may be critical in earlier developmental stages.
Since calcium signaling plays an important role in osteogenic differentiation, we investigated the role of the EC domain in the inhibitory effects of SMOC2 [20–21]. Our results suggest that the inhibitory effect of SMOC2 is, at least in part, dependent on the EC domain, as a deletion of this domain partially rescues the inhibitory effects of wild type SMOC2 overexpression. We hypothesized that high levels of SMOC2 could titer part of the calcium out of the extracellular environment, by sequestering it through its calcium-binding domain. To test this hypothesis, we tried to measure calcium levels in conditioned medium of control cells, Smoc2+ cells and ΔCaBD cells but could not demonstrate consistent differences. Different factors may explain this issue: (1) binding of SMOC2 and calcium is likely reversible as described by Novinec M et al [22], and the processing of the samples may affect this process. (2) Effects of SMOC2 may be linked to the actions close to the cell surface where matrix molecules are produced. Nevertheless, our hypothesis was supported by the phenotypic partial rescue upon extracellular calcium addition to SMOC2 overexpressing cells. Our observations on the overexpression of wild-type and truncated SMOC2 forms were replicated in a human model system, using hPDCs.
In this study, we focused on the calcium signaling as contributing factor to the inhibitory effects of SMOC2 on mineralization that we observed. However, as we only observe partial rescue effects with the calcium studies and based on literature, we acknowledge that the other domains of SMOC2 and thus other signaling pathways might also play a role in the observed effects. Moos et al showed in gain-of-function assays that XSMOC-1, the orthologue of human SMOC1, inhibited the BMP signaling as potently as known BMP antagonists. In the gain-of-function studies, they found that unlike known BMP antagonists such as noggin, XSMOC-1 antagonized BMP activity by acting downstream of the receptor. Loss-of-function studies using morpholinos, led to developmental failure indicating that XSMOC-1 is necessary for postgastrulation development [11]. Moos et al demonstrated moreover, using SMOC deletion constructs, that SMOC can both act as an expander or antagonist of BMP signaling, as SMOC-ΔEC, lacking the calcium-binding (EC) domain, inhibited BMP2 signaling whereas SMOC-EC (EC domain only) enhanced BMP signaling by expanding the range of signaling [23]. Mommaerts et al also demonstrated modulation of BMP target genes by SMOC2 in Zebrafish [24]. Altogether, their results suggest that the SMOC pathway could be a potential therapeutic target in diseases such as osteoarthritis, which are associated with an altered BMP signaling and altered calcification.
To investigate whether our results could have potential clinical implications, we explored the possible impact of SMOC2 on vascular calcification, which is a common complication of frequent vascular diseases such as atherosclerosis, predicting cardiovascular morbidity and mortality [15–16]. Therefore, we studied the effect of SMOC2 on HUVECs. Culturing HUVECs in the presence of Smoc2+ supernatant caused decreased mineralization partially rescued when cultured in the presence of the EC-domain deletion mutant supernatant. These results suggest that SMOC2 could have beneficial effects in vascular calcification disorders.
In conclusion, we showed that SMOC2 overexpression inhibits mineralization in different model systems and identified the importance of the EC domain in this process.
Supporting information
The calcium-binding domain spans from aminoacid 352 to 412. For the first PCR reaction, the pcDNA3.1 plasmid containing wild type Smoc2 and primer pair A (P1 and P2) to obtain the PCR product A and primer pair B (P3 and P4) to obtain PCR product B was used. PCR product A and B were used as templates and primers P1 and P4 were used in PCR reaction 2. The resulting product was the pcDNA3.1 plasmid containing mutant Smoc2 lacking the calcium binding domain (ΔAA352-412).
(JPG)
{kind=link}
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The study was supported by FWO Vaanderen grant G.0717.09 to RL and by FP7 Health grant TREATOA to RL, FPL and PT. FC was the recipient of a Marie Curie Fellowship "SMOOS". The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet 2011;377(9773):1276–87. doi: 10.1016/S0140-6736(10)62349-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivory D, Velazquez CR. The forgotten crystal arthritis: calcium pyrophosphate deposition. Mo Med 2012;109(1):64–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Molloy ES, McCarthy GM. Calcium crystal deposition diseases: update on pathogenesis and manifestations. Rheum Dis Clin North Am 2006;32(2):383–400, vii. doi: 10.1016/j.rdc.2006.02.001 [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez A Jr., Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther 2012;25(2):195–206. doi: 10.1111/j.1529-8019.2012.01492.x [DOI] [PubMed] [Google Scholar]
- 5.Rennenberg RJ, Schurgers LJ, Kroon AA, Stehouwer CD. Arterial calcifications. J Cell Mol Med 2010;14(9):2203–10. doi: 10.1111/j.1582-4934.2010.01139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vannahme C, Gosling S, Paulsson M, Maurer P, Hartmann U. Characterization of SMOC- 2, a modular extracellular calcium-binding protein. Biochem J. 2003. August 1;373(Pt 3):805–14. doi: 10.1042/BJ20030532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pazin DE, Albrecht KH. Developmental expression of Smoc1 and Smoc2 suggests potential roles in fetal gonad and reproductive tract differentiation. Dev Dyn. 2009. November;238(11):2877–90. doi: 10.1002/dvdy.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maier S, Paulsson M, Hartmann U. The widely expressed extracellular matrix protein SMOC-2 promotes keratinocyte attachment and migration. Exp Cell Res. 2008. August 1;314(13):2477–87. doi: 10.1016/j.yexcr.2008.05.020 [DOI] [PubMed] [Google Scholar]
- 9.Chang SC, Hoang B, Thomas JT, Vukicevic S, Luyten FP, Ryba NJ, et al. Cartilage-derived morphogenetic proteins. New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J Biol Chem. 1994. November 11;269(45):28227–34. [PubMed] [Google Scholar]
- 10.Hoang B, o M Jr, Vukicevic S, Luyten FP. Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J Biol Chem. 1996. October 18;271(42):26131–7. [DOI] [PubMed] [Google Scholar]
- 11.Thomas JT, Canelos P, Luyten FP, Moos M. Jr. Xenopus SMOC-1 Inhibits bone morphogenetic protein signaling downstream of receptor binding and is essential for postgastrulation development in Xenopus. J Biol Chem. 2009. July 10;284(28):18994–9005. doi: 10.1074/jbc.M807759200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lories RJ, Luyten FP. The bone-cartilage unit in osteoarthritis. Nat Rev Rheumatol. 2011. January;7(1):43–9. doi: 10.1038/nrrheum.2010.197 Epub 2010 Dec 7. [DOI] [PubMed] [Google Scholar]
- 13.Valdimarsdottir G, Goumans MJ, Rosendahl A, Brugman M, Itoh S, Lebrin F, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002. October 22;106(17):2263–70. [DOI] [PubMed] [Google Scholar]
- 14.Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004. June 1;329(1):77–84. doi: 10.1016/j.ab.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 15.Pugliese G, Iacobini C, Blasetti Fantauzzi C, Menini S. The dark and bright side of atherosclerotic calcification. Atherosclerosis. 2015;238(2):220–30. doi: 10.1016/j.atherosclerosis.2014.12.011 [DOI] [PubMed] [Google Scholar]
- 16.Bostrom KI. Where do we stand on vascular calcification? Vascul Pharmacol. 2016; 84:8–14. doi: 10.1016/j.vph.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bloch-Zupan A, Jamet X, Etard C, Laugel V, Muller J, Geoffroy V, et al. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Am J Hum Genet. 2011. December 9;89(6):773–81. doi: 10.1016/j.ajhg.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melvin VS, Feng W, Hernandez-Lagunas L, Artinger KB, Williams T. A morpholino-based screen to identify novel genes involved in craniofacial morphogenesis. Dev Dyn. 2013. July;242(7):817–31. doi: 10.1002/dvdy.23969 Epub 2013 Jun 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sudo H, Kodama HA, Amagai Y, Yamamoto S, Kasai S. In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J Cell Biol. 1983. January;96(1):191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamauchi M, Yamaguchi T, Kaji H, Sugimoto T, Chihara K. Involvement of calcium-sensing receptor in osteoblastic differentiation of mouse MC3T3-E1 cells. Am J Physiol Endocrinol Metab. 2005. March;288(3): E608–16. Epub 2004 Nov 16. doi: 10.1152/ajpendo.00229.2004 [DOI] [PubMed] [Google Scholar]
- 21.Habel B, Glaser R. Human osteoblast-like cells respond not only to the extracellular calcium concentration but also to its changing rate. Eur Biophys J. 1998;27(4):411–6. [DOI] [PubMed] [Google Scholar]
- 22.Novinec M, Kovacic L, Skrlj N, Turk V, Lenarcic B. Recombinant human SMOCs produced by in vitro refolding: calcium-binding properties and interactions with serum proteins. Protein Expr Purif. 2008. November;62(1):75–82. doi: 10.1016/j.pep.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 23.Thomas JT, Eric Dollins D, Andrykovich KR, Chu T, Stultz BG, Hursh DA, et al. SMOC can act as both an antagonist and an expander of BMP signaling. Elife. 2017. March 21;6 pii: e17935. doi: 10.7554/eLife.17935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mommaerts H, Esguerra CV, Hartmann U, Luyten FP, Tylzanowski P. Smoc2 modulates embryonic myelopoiesis during zebrafish development. Dev Dyn. 2014. November;243(11):1375–90. doi: 10.1002/dvdy.24164 Epub 2014 Jul 30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The calcium-binding domain spans from aminoacid 352 to 412. For the first PCR reaction, the pcDNA3.1 plasmid containing wild type Smoc2 and primer pair A (P1 and P2) to obtain the PCR product A and primer pair B (P3 and P4) to obtain PCR product B was used. PCR product A and B were used as templates and primers P1 and P4 were used in PCR reaction 2. The resulting product was the pcDNA3.1 plasmid containing mutant Smoc2 lacking the calcium binding domain (ΔAA352-412).
(JPG)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.