

Abstract
The β-amyloid (Aβ) deposition is one of the major pathological hallmark of Alzheimer’s disease. Dysfunction in autophagy has been reported to lead to the Aβ deposition. The current study aimed to investigate the effects of treadmill exercise on autophagy activity and the Aβ deposition and to demonstrate whether exercise-induced reduction in the Aβ deposition was associated with changes in autophagy activity. APP/PS1 transgenic mice were divided into transgenic sedentary (TG-SED, n=12) and transgenic exercise (TG-EXE, n=12) groups. Wild-type mice were also divided into sedentary (WT-SED, n=12) and exercise (WT-EXE, n=12) groups. The WT-EXE and TG-EXE mice were subjected to treadmill exercise for 12 weeks. The levels of Aβ plaques and soluble forms of Aβ, autophagy markers light chain 3 and P62, and lysosomal marker lysosome-associated membrane protein 1 (Lamp1) were measured in the hippocampus. Both Aβ plaques and soluble forms of Aβ (Aβ40 and Aβ42) were significantly increased in TG-SED mice compared with WT-SED mice, whereas exercise reduced Aβ deposition in APP/PS1 transgenic mice. Coincidentally, TG-SED mice displayed a decrease in autophagy activity as evidenced by a significant increase in the levels of light chain 3-II and P62, as well as an accumulation of lysosome as evidenced by a significant over-expression of Lamp1. Interestingly, exercise increased autophagy activity as evidenced by a significant reduction in the levels of P62 and Lamp1 in TG-EXE mice. These findings suggest that treadmill exercise is efficient in decreasing Aβ deposition by enhancing autophagy–lysosomal activity in APP/PS1 transgenic mice, demonstrating a possible approach in Alzheimer’s disease prevention and treatment.
Keywords: Aβ, Alzheimer’s disease, autophagy, treadmill exercise
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disease. The major pathological features of AD include the accumulation of extracellular β-amyloid (Aβ) plaques and intracellular tangles of abnormally phosphorylated τ-proteins. The Aβ deposition is the key factor of AD, resulting in a series of pathological cascades and neuronal loss 1.
It has been found that Aβ deposition is closely related to dysfunction of autophagy 2. The autophagy is a self-destructive process by which damaged proteins and dysfunctional organelles are delivered to lysosomes for degradation 3. The deficits of autophagy significantly damage the healthy cell and lead to AD 4. Deficits in autophagy are associated with the increased Aβ deposition in the hippocampus, whereas increased autophagy activity can ameliorate Aβ deposition in APP/PS1 transgenic mice 5. The parallel between the Aβ deposition and deficits of autophagy in the brain suggests that autophagy activity might be involved in the clearance of Aβ, and alleviating the Aβ deposition 6.
Physical exercise has gained much attention as preventive and therapeutic approaches for AD. According to previous studies, exercise contributes to diminishing of Aβ deposition and prevention of AD, which might be associated with facilitated Aβ clearance across the blood–brain barrier 7, increased levels of Aβ-degrading enzymes 8, and reduced inflammatory activation of microglia and astrocytes 9. Interestingly, recent studies have shown that both acute (a single bout of) and long-term moderate exercise activate autophagy in the brain.
Regarding autophagy, Beclin1 is involved in autophagosome formation. Microtubule associated protein light chain 3 (LC3-II) controls the completion of autophagosome elongation and maturation. P62 is the autophagy substrate and reflects autophagy activity 10. Lysosome-associated membrane protein 1 (Lamp1) is a marker protein associated with lysosomes content 11. As He et al. 12 reported, acute exercise-induced higher LC3-II and reduced P62 in the brain of adult wild-type (WT) mice. Bayod and colleagues showed that long-term moderate exercise significantly increased LC3-II/LC3-I and Lamp1, as well as reduced P62 in the cortex of Sprague Dawley rats 13,14. We had previously shown that treadmill exercise slows cognitive deficits in AD model rats by inhibiting Aβ production 15. However, there is still a lack of evidence whether exercise-induced Aβ degradation is associated with enhanced autophagy activity. Thus, in the current study, we used APP/PS1 transgenic mice to investigate the effect of exercise on Aβ degradation and autophagy activity and to demonstrate whether exercise-induced reduction in the Aβ deposition was associated with changes in autophagy activity.
Materials and methods
Subjects
Male APPswe/PS1dE9 (APP/PS1) double transgenic mice [B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/Mmjax] were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China), and their age-matched and background-matched WT mice (C57BL/6) were purchased from the East China Normal University Experimental Animal Center. All mice were housed individually in standard plastic cages under conventional laboratory conditions (22–24°C, 40–60% relative humidity), with ad-libitum access to food and water, and maintained on a standard 12/12 h light/dark cycle with lights on at 6:00 a.m. All experimental procedures were approved by the Experimental Animal Care and Use Committee at East China Normal University. All efforts were made to minimize the number and suffering of animals in these experiments. At the age of 3 months, both male APP/PS1 transgenic mice and male WT littermate controls were randomly assigned to four groups: transgenic control group (TG-SED, n=12), transgenic exercise group (TG-EXE, n=12), wild-type control group (WT-SED, n=12), and wild-type exercise group (WT-EXE, n=12).
Treadmill exercise protocols
Treadmill exercise training protocols were adapted from those previously described 15. Mice in exercise groups received a 6-day familiarization period to adapt to their new environment followed by a 12-week exercise training. The familiarization period (15 min/day) was designed for the mice to learn how to run on a horizontal treadmill (Xinruan Technology Co. Ltd, Hangzhou, China). On the first 2 days (days 1–2), the treadmill was engaged to a walking speed of 5 m/min, and then the speed was increased to 8 m/min on days 3–4 and 12 m/min on days 5–6. The mice were then subjected to an exercise protocol during 6–8 p.m., for 45 min/day, 5 days/week for 12 weeks. Each day, mice were exercised at a sequence of speed in the following order: 5 m/min for 5 min, 8 m/min for 5 min, 12 m/min for 30 min, and 5 m/min for 5 min. Mice in sedentary control groups were placed on the static treadmill for the same amount of time.
Tissue preparation
After 12 weeks of treadmill exercise, all mice were anesthetized with pentobarbital, and the brains were removed after decapitation. Half of the brains (n=6) from each group were fixed overnight in 4% buffered formaldehyde, paraffin imbedded, and cut into 6-µm coronal section for immunohistochemistry. The hippocampi of the other six mice from each group were isolated and collected. The hippocampi were snap-frozen and stored at –80°C until use for protein extraction. The hippocampi from the extracted brain tissues were homogenized in radioimmunoprecipitation assay buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) using a homogenizer (polytron PT3100, KINEMATIC, Luzern, Switzerland). In summary, the radioimmunoprecipitation assay buffer (1 ml) containing hippocampal tissue (20 mg) and four to five magnetic beads were loaded in the homogenate tube and mechanically homogenized at 3.55 m/s for 30 s, and placed on ice for 5 min. The procedure was repeated three times, and the final homogenate was centrifuged (12 000g) for 30 min at 4°C. The supernatant was collected, and protein concentration was measured using the Bradford method 14.
Western blot
Western blot analyses were conducted as previously described 16 using antibodies against Beclin1 (#3788; Cell Signaling Technology, Boston, Massachusetts, USA), P62 (#5114; Cell Signaling Technology), LC3 (sc16755; SantaCruz, California, USA), and Lamp1 (ab24170; Abcam, Cambridge, UK). The protein bands on the membrane were scanned densitometrically and quantified with a gel image processing system (GIS-2008; Tanon, Shanghai, China). β-Actin was used to normalize the amounts of protein loaded.
Enzyme-linked immunosorbent assay
Commercial sandwich enzyme-linked immunosorbent assay (ELISA) kits were used to quantify the levels of Aβ40 (KHB3482; Invitrogen, Carlsbad, California, USA) and Aβ42 (KHB3442; Invitrogen) in the hippocampal tissue following the manufacturer’s protocols. In short, hippocampal samples were homogenized in PBS (pH: 8.0), and centrifuged at 3000g for 1 h at 4°C to remove insoluble material. The supernatant fractions were applied to ELISA microplate reader for Aβ quantification. The plate was read in an ELISA (TECAN Infinite M200, TECAN, Männedorf, Switzerland) with an absorbance wavelength of 450 nm. Standard curves were obtained from the values generated from known concentrations of Aβ40 and Aβ42 provided by the kits.
Immunohistochemistry and quantification
Immunohistochemistry was performed to analyze the distribution of Aβ plaques in the APP/PS1 mouse brain. In summary, paraffin sections (6 µm) were deparaffinized in xylene and rehydrated through graded series of alcohol, and then treated in 0.1 M Tris–HCl buffer saline (pH: 7.4) containing 3% (H2O2) for 10 min to inhibit endogenous peroxidase activity. Paraffin sections were washed with PBS for 20 min at 4°C. After washing in PBS, paraffin sections were blocked with 2% BSA in PBS for 1 h at room temperature, and incubated with mouse anti-Aβ antibody (anti-Aβ, 1 : 2000, ab2539; Abcam) in a blocking solution containing 0.1% Tween 20 overnight at 4°C. The next day, after rinsing, sections were incubated with biotinylated goat anti-mouse IgG (Goat anti-Mouse IgG, 1 : 500, 131224; Jackson ImmunoResearch, West Grove, Pennsylvania, USA) for 1 h at room temperature. Signal amplification was accomplished with the avidin–biotin complex (ABC Reagent; Vector Laboratories, Burlingame, California, USA) and visualized with a 3,3′-diaminobenzidine brownish reaction product (DAB Kit; Vector Laboratories). The stained sections were dehydrated through graded alcohols, cleared in xylene, and covered with neutral balsam. The slices were imaged under an upright microscope (Leica, Germany). The area of plaques was counted using the optical fractionator technique. The data were analyzed with Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, Maryland, USA).
Statistical analysis
All statistical analyses were performed using IBM SPSS statistics software (version 19.0, Chicago, Illinois, USA) and GraphPad Prism (version 7.0, La Jolla, California, USA). All values are reported as means±SEM. Statistical significance was assumed at P value of less than 0.05. Two-way analysis of variance (ANOVA) (with exercise treatment and genotype as factors) was used for evaluation of differences among groups. Post-hoc t-tests were conducted to further interrogate statistically significant ANOVA results.
Results
Treadmill exercise decreased Aβ deposition in the hippocampus
As shown in Fig. 1, two-way ANOVA revealed that there were significant main effects of genotype [F(1,20)=89.09, P<0.01] and exercise [F(1,20)=7.412, P<0.01], as well as a significant interaction [F(1,20)=3.665, P<0.01]. Post-hoc analysis test revealed that the area of Aβ plaques was significantly higher in TG-SED mice than that in WT-SED mice (P<0.01). The area of Aβ plaques was significantly reduced in TG-EXE mice compared with TG-SED mice (P<0.001).
Fig. 1.
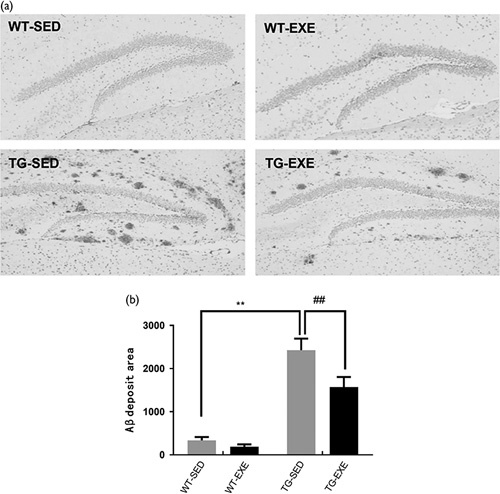
Treadmill exercise reduces the amount of β-amyloid (Aβ) plaque deposition in the hippocampus of APP/PSI transgenic mice. (a) Immunohistochemistry images showing Aβ plaque deposition; (b) quantitative analysis of Aβ deposition. N=6 for each group. **Aβ area was statistically different between WT-SED and TG-SED (P<0.01); ##Aβ area was statistically different between TG-SED and TG-EXE (P<0.01). TG-EXE, transgenic exercise; TG-SED, transgenic sedentary; WT-EXE, wild-type exercise; WT-SED, wild-type sedentary.
Treadmill exercise decreased the level of Aβ40 and Aβ42 in the hippocampus
Figure 2a and b shows the level of Aβ40 and Aβ42 in the hippocampus. Two-way ANOVA revealed main effects of genotype [F(1,20)=75.08, P<0.05; and F(1,20)=23.19, P<0.01 for Aβ40 and Aβ42, respectively]. There were also main effects of exercise [F(1,20)=6.08, P<0.05; and F(1,20)=10.81, P<0.01 for Aβ40 and Aβ42, respectively], but no significant interaction [F(1,20)=0.99, P=0.33; and F(1,20)=0.54, P=0.47 for Aβ40 and Aβ42, respectively]. Post-hoc analysis revealed that the levels of Aβ40 and Aβ42 were significantly increased in TG-SED mice compared with WT-SED mice (TG-SED vs. WT-SED; P<0.01 for both values). In APP/PS1 transgenic mice, exercise significantly reduced the level of Aβ40 and Aβ42 compared with sedentary mice (TG-EXE vs. TG-SED; P<0.05 for both values).
Fig. 2.

Effects of treadmill exercise on the levels of soluble Aβ40 and Aβ42 in the hippocampus (n=6 for each group). (a) Aβ40 expression level. (b) Aβ42 expression levels. **The level of Aβ40 was statistically different between WT-SED and TG-SED (P<0.01); #the level of Aβ42 was statistically different between TG-SED and TG-EXE (P<0.05). TG-EXE, transgenic exercise; TG-SED, transgenic sedentary; WT-EXE, wild-type exercise; WT-SED, wild-type sedentary.
Treadmill exercise increased autophagy activity in the hippocampus
To investigate whether exercise-induced reduction in the Aβ deposition was associated with changes in autophagy activity, we quantified the expression levels of several proteins that are critical for autophagy activity. Figure 3a showed the level of Beclin1 in the hippocampus. Analysis with two-way ANOVA revealed significant main effects of genotype [F(1,20)=8.16, P=0.01] and exercise [F(1,20)=8.18, P=0.01], but no significant effect of interaction [F(1,20)=1.05, P=0.32]. Post-hoc analysis revealed that the level of Beclin1 was significantly higher in WT-EXE mice than that in WT-SED mice (P<0.05). However, the effect of exercise on Beclin1 in the hippocampus was not statistically significant in APP/PS1 transgenic mice (P=0.19).
Fig. 3.
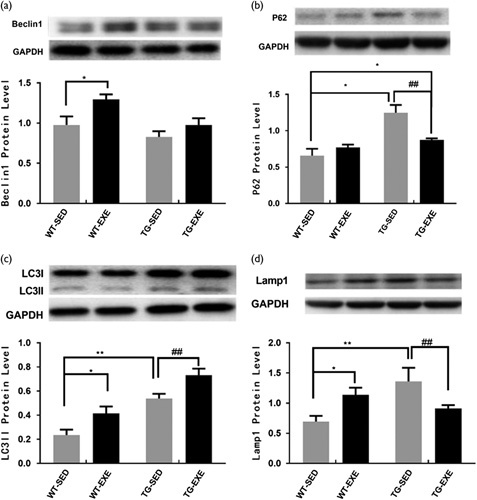
Effects of treadmill exercise on autophagy in hippocampus (n=6 for each group). (a) Beclin 1 expression level; (b) P62 expression levels; (c) LC3 expression level; (d) Lampl expression level. Statistically different from WT-SED. *P<0.05; **P<0.01. Statistically different from TG-SED. ##P<0.01. TG-EXE, transgenic exercise; TG-SED, transgenic sedentary; WT-EXE, wild-type exercise; WT-SED, wild-type sedentary.
An analysis of P62 expression with two-way ANOVA revealed that there was a significant main effect of genotype [F(1,20)=24.79, P<0.0001] but not exercise [F(1,20)=3.46, P=0.08]. There was a significant interaction of genotype×exercise [F(1,20)=12.06, P=0.003]. Post-hoc analysis revealed that the level of P62 was significantly increased in TG-SED mice compared with WT-SED mice (P<0.01). The level of P62 in TG-EXE mice was significantly decreased than that in TG-SED mice (P<0.01), but comparable with WT-SED mice (P=0.04) (Fig. 3b).
Figure 3c shows the level of LC3-II in the hippocampus. An analysis with two-way ANOVA revealed an significant main effects of genotype [F(1,20)=30.21, P=0.0001] and exercise [F(1,20)=11.65, P=0.001], but no significant effect of interaction [F(1,20)=0.08, P=0.78]. Post-hoc analysis revealed that the level of LC3-II was significantly higher in TG-SED mice compared with WT-SED mice (P<0.01). However, exercise significantly increased the levels of LC3-II in the hippocampus of both WT mice (P<0.05) and APP/PS1 transgenic mice (P<0.01).
Lastly, Fig. 3d shows the level of Lamp1 in the hippocampus. Two-way ANOVA revealed that neither the main effect of genotype [F(1,20)=3.36, P=0.09] nor exercise [F(1,20)=5.81, P=0.99] was statistically significant. However, there was a significant interaction of genotype×exercise [F(1,20)=13.51, P=0.002]. Post-hoc analysis revealed that the level of Lamp1 was significantly increased in TG-SED mice compared with WT-SED mice (P<0.01). Moreover, the level of Lamp1 was significantly increased in WT-EXE mice compared with WT-SED mice (P<0.01). However, Lamp1 was reduced in TG-EXE mice compared with TG-SED mice (P<0.05).
Discussion
The present study demonstrates that the area of Aβ plaques was significantly increased in the hippocampus of APP/PS1 transgenic mice at the age of 6 months, and this was accompanied by an increase in Aβ40 and Aβ42 levels in the hippocampus. Interestingly, long-term treadmill exercise (12 weeks) significantly reduced the area of Aβ plaques, as well as Aβ40 and Aβ42 levels in these transgenic mice. Furthermore, an analysis of the proteins that regulate autophagy activity suggests that the exercise-induced Aβ reduction in APP/PS1 transgenic mice was associated with enhanced autophagy activity.
Both Aβ plaques and higher soluble Aβ40 and Aβ42 were evident in the hippocampus of APP/PS1 transgenic mice, which may be the cause of observed cognitive deficits. Interestingly, the current study found that treadmill exercise significantly reduced the area of Aβ plaques (Fig. 1) and the level of soluble Aβ40 and Aβ42 (Fig. 2), which are consistent with previous studies from others 17 and our own 15. These data suggest that exercise can effectively reduce the Aβ deposition in the hippocampus.
Autophagy is a degradation system that is induced in response to various intracellular stressors (nutrient depletion, a growth factor deficiency, endoparasitic reticulum stress, etc.). The entire process of autophagy includes autophagosome formation, elongation and maturation, autophagosome–lysosome fusion, the delivery of cargo to lysosomes, as well as the subsequent degradation 18. The autophagy–lysosomal can clear Aβ from the cell, if these molecules are trafficked through to the lysosome. Aβ deposition might be highly associated with decreased autophagy activity 19. Therefore, we measured the effects of treadmill exercise on Beclin1, P62, LC3-II, and Lamp1 in APP/PS1 transgenic mice, which are related to autophagy.
Within autophagy signaling pathway, LC3 is known to control the completion stage of autophagosome elongation and maturation. During autophagy process, the cytosolic form of LC3 (LC3-I) is converted to the membrane-bound form (LC3-II), which promotes the formation of autophagosome and is recycled after being fused with lysosomes 20. In the current study, we found that LC3-II was reduced in WT but significantly increased in APP/PS1 transgenic mice (Fig. 3c), indicating autophagosomes were excessively accumulated in APP/PS1 transgenic mice. This is consistent with recent reports that the expression of LC3-II was significantly increased in Aβ-treated cell cultures 21 and in the postmortem human brain tissue from patients with AD 22. In addition, we found that exercise caused a significant increase in the level of LC3-II in WT mice (Fig. 3c). Meanwhile, we also found that exercise caused an additional increase of LC3-II in APP/PS1 transgenic mice (Fig. 3c). It is known that an accumulation of autophagosomes (increased LC3-II) reflects either increased autophagosome formation (increased Beclin1) owing to increase in autophagy activity (P62 reduction), or reduced turnover of autophagosomes (increased P62, reduced lysosomal activity). Thus, to understand whether the increase in LC3-II level stands for functional upregulation or downregulation, measuring the levels of Beclin1 (involved in autophagosome formation), P62 (autophagy substrate, reflecting autophagy activity), and lysosomal activity is important.
Beclin1 is thought to control the onset of autophagosome formation. Interestingly, the current study found that the level of Beclin1 was not significantly altered in the hippocampus of APP/PS1 transgenic mice. This is consistent with recent reports that the level of Beclin1 was not significantly changed in the brain of APP/PS1 transgenic mice 23. This suggests that although Aβ was significantly increased in the brain, it does not affect the level of Beclin1. The current result states that exercise increased the level of Beclin1 in WT mice (Fig. 3a), suggesting that treadmill exercise may promote the process of autophagy by increasing autophagosome formation. This is also consistent with previous studies showing that the level of Beclin1 was increased in rat brain after treadmill exercise 14. However, the level of Beclin1 was not significantly changed in TG-EXE mice (Fig. 3a). Although there is a lack of evidence, we speculate that this is because that the accumulation of autophagosomes (increased LC3-II in APP/PS1 transgenic mice following exercise) causes reverse inhibition to the formation of Beclin1 24. Supporting this is the evidence that Bcl-2 sequesters Beclin1 and prevents its association with PI3KCIII (class III PI3K), thus suppresses autophagy initiation through the negative feedback produced by the accumulation of autophagosomes 25.
P62 is one of the best characterized substrates of selective autophagy. P62 interacts with LC3 and is subsequently incorporated into the autophagosomal membrane where they are degraded. Therefore, higher P62 level is associated with autophagy inhibition because of the incomplete degradation 26. Thus, combined with the increased in LC3-II, the current finding that P62 was significantly increased in the APP/PS1 transgenic mice (Fig. 3b) further suggests that autophagic substrates were accumulated in the hippocampus of APP/PS1 transgenic mice. Interestingly, we found that exercise decreased P62 in APP/PS1 transgenic mice, suggesting that exercise may facilitate autophagy activity through accelerating waste (including Aβ protein) degradation. It is worth noting that the data showed that the increase of LC3-II in TG-SED was paralleled with an increase in P62, whereas exercise further increased LC3-II level but reduced P62. These data suggested that in APP/PS1 mice the accumulation of autophagosome and Aβ occurred when Aβ plaques were over-loading the autophagosome processing ability, which was then able to be restored upon exercise. This is consistent with recent reports that exercise, as an extra stimulus, can further increase autophagy activity 27.
Lysosome plays a key role in the autophagy activity. The current finding that lysosome marker Lamp1 was over-expressed in the TG-SED mice compared with WT-SED mice (Fig. 3d) suggests that lysosomes were accumulated in APP/PS1 transgenic mice. This is consistent with a previous study by Joshi et al. 28, who found that there was a significant increase in lysosomal protein Lamp1 in APP/PS1 transgenic mice, which is associated with a deficient clearance of autophagy substrate. As Lee et al. 29 reported, the accumulation of late endosomes and lysosomes may be associated with dystrophic axonal swellings and induces Alzheimer’s-like axonal dystrophy.
Furthermore, the current study found that exercise significantly increased Lamp1 in WT mice (Fig. 3d). Combined with the increased LC3-II and Beclin1 levels, the increase of Lamp1 in WT mice following exercise suggests that exercise increased the lysosomal content, which might be associated with an increase in autophagosome formation (increased Beclin1 and LC3-II), and thus exercising facilitates the degradation of waste products. This is consistent with previous studies showing that both acute and chronic exercise-induced promote autophagy, whereas disruption of normal autophagy pathway impairs chronic exercise-mediated protection against high-fat-diet-induced glucose intolerance 14,30.
Interestingly, we found that treadmill exercise reduced Lamp1 level in TG-SED mice suggesting that exercise reversed the lysosome accumulation in APP/PS1 transgenic mice back to WT mice level (Fig. 3d). This contrasts with the WT mice, where exercise increased Lamp1 and seems contradictory. This is likely owing to the fact that, the increase of autophagosome in APP/PS1 transgenic mice facilitated production of empty lysosomes, whereas exercise promoted autophagosome–lysosome fusion and the degradation of extra Lamp1 and returned to WT mice level. However, this needs to be clarified in our future study. Nevertheless, this has been demonstrated in a previous study by Luo et al. 31, which found that long-term exercise training can improve autophagy/mitophagy in aged rats, and lysosomal was required for the effects.
Conclusion
The study demonstrates that APP/PS1 transgenic mice developed a significant area of Aβ plaques and soluble Aβ peptides in the hippocampus, which is associated with impaired autophagy activity. The area of Aβ plaques and soluble form of Aβ peptides were significantly reduced following a 12-week treadmill exercise training, which were accompanied by improving autophagy–lysosomal activity in the hippocampus. Therefore, our study suggested that treadmill exercise is efficient in decreasing Aβ deposition by improving autophagy-lysosomal activity in APP/PS1 transgenic mice, demonstrating a possible approach in AD prevention and treatment.
Acknowledgements
This work was supported by grants from the Project supported by the National Natural Science Foundation of China (Grant No: 31571225).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Small DH, Mok SS, Bornstein JC. Alzheimer’s disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci 2001; 2:595–598. [DOI] [PubMed] [Google Scholar]
- 2.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:1845–1846. [DOI] [PubMed] [Google Scholar]
- 3.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta 2009; 1793:664–673. [DOI] [PubMed] [Google Scholar]
- 4.Lei Y, Liu K, Hou L, Ding L, Li Y, Liu L. Small chaperons and autophagy protected neurons from necrotic cell death. Sci Rep 2017; 7:5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng M, Huang L, Ning B, Wang N, Zhang Q, Zhu C, et al. beta-asarone improves learning and memory and reduces Acetyl Cholinesterase and Beta-amyloid 42 levels in APP/PS1 transgenic mice by regulating Beclin-1-dependent autophagy. Brain Res 2016; 1652:188–194. [DOI] [PubMed] [Google Scholar]
- 6.Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res 2014; 81:54–63. [DOI] [PubMed] [Google Scholar]
- 7.Herring A, Munster Y, Metzdorf J, Bolczek B, Krussel S, Krieter D, et al. Late running is not too late against Alzheimer’s pathology. Neurobiol Dis 2016; 94:44–54. [DOI] [PubMed] [Google Scholar]
- 8.Liu HL, Zhao G, Zhang H, Shi LD. Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res 2013; 256:261–272. [DOI] [PubMed] [Google Scholar]
- 9.He XF, Liu DX, Zhang Q, Liang FY, Dai GY, Zeng JS, et al. Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci 2017; 10:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 2011; 14:2201–2214. [DOI] [PubMed] [Google Scholar]
- 11.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 2007; 26:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He C, Sumpter RJ, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012; 8:1548–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marques-Aleixo I, Santos-Alves E, Balca MM, Rizo-Roca D, Moreira PI, Oliveira PJ, et al. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015; 301:480–495. [DOI] [PubMed] [Google Scholar]
- 14.Bayod S, Del VJ, Pelegri C, Vilaplana J, Canudas AM, Camins A, et al. Macroautophagic process was differentially modulated by long-term moderate exercise in rat brain and peripheral tissues. J Physiol Pharmacol 2014; 65:229–239. [PubMed] [Google Scholar]
- 15.Yu F, Xu B, Song C, Ji L, Zhang X. Treadmill exercise slows cognitive deficits in aging rats by antioxidation and inhibition of amyloid production. Neuroreport 2013; 24:342–347. [DOI] [PubMed] [Google Scholar]
- 16.Kang EB, Cho JY. Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J Exerc Nutrition Biochem 2015; 19:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koo JH, Kang EB, Oh YS, Yang DS, Cho JY. Treadmill exercise decreases amyloid-beta burden possibly via activation of SIRT-1 signaling in a mouse model of Alzheimer’s disease. Exp Neurol 2017; 288:142–152. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol 2012; 123:53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YD, Zhao JJ. TFEB participates in the Aβ-induced pathogenesis of Alzheimer’s disease by regulating the autophagy-lysosome pathway. DNA Cell Biol 2015; 34:661–668. [DOI] [PubMed] [Google Scholar]
- 21.Singh AK, Bissoyi A, Kashyap MP, Patra PK, Rizvi SI. Autophagy activation alleviates amyloid-beta-induced oxidative stress, apoptosis and neurotoxicity in human neuroblastoma SH-SY5Y cells. Neurotox Res 2017. [DOI] [PubMed] [Google Scholar]
- 22.Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016; 12:2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, et al. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J Exp Med 2014; 211:1551–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927–939. [DOI] [PubMed] [Google Scholar]
- 26.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rocchi A, Yamamoto S, Ting T, Fan Y, Sadleir K, Wang Y, et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet 2017; 13:e1006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer’s disease-like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging 2015; 36:664–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 2011; 31:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo L, Dai JR, Guo SS, Lu AM, Gao XF, Gu YR, et al. Lysosomal proteolysis is associated with exercise-induced improvement of mitochondrial quality control in aged hippocampus. J Gerontol A Biol Sci Med Sci 2017; 72:1342–1351. [DOI] [PubMed] [Google Scholar]