

Supplemental Digital Content is available in the text
Keywords: checkpoint kinase 2, germline mutation, neurofibromatosis, single nucleotide variations
Abstract
Rationale:
Neurofibromatosis, including type 1 and type 2, is inherited dominant disease that causes serious consequences. The genetic mechanism of these diseases has been described, but germline mutation of checkpoint 2 kinase gene, together with other DNA repair related genes, has not been fully elucidated in the context of neurofibromatosis.
Patient concerns:
In this article, we reported identical germline mutation of CHEK2 gene (p.R180C) in a 7-year-old Tibetan boy with NF1, and in a 12-year-old Chinese girl with NF2.
Diagnoses:
Neurofibromatosis 1 and 2 with CHECK2 gene germline mutation.
Interventions:
Both patients underwent operation to obtain tumor tissue, and peripheral blood of their family was tested.
Outcomes:
Identical germline mutation of CHEK2 gene (p.R180C) was detected in both patients, and germline mutations of POLE, MUTYH and ATR were also detected.
Lessons:
This is the first article to describe CHEK2 mutation in both NF1 and NF2. This article highlights a possible role of CHEK2, in association with other germline genetic mutations, in tumorigenesis of NF1 and NF2.
1. Introduction
Neurofibromatosis is inherited dominant disease that is prone to a number of tumors. It can be divided into 2 types: neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). NF1 is caused by heterozygous mutation of tumor suppressor gene NF1, and NF2 is caused by mutations in the NF2 gene on chromosome 22. Checkpoint kinase 2 (CHEK2) is a serine/threonine kinase that is activated upon DNA damage, and is involved in DNA repair, cell cycle arrest, and apoptosis.[1] When activated, it phosphorylates a number of substrates, including tumor suppressor gene P53, and BRCA1, and CHEK2 gene thus acts as a tumor suppressor gene.[2] Mutation of CHEK2 was reported to be associated with elevated risk for breast cancer and other familiar cancers.[1,3]
Currently, there is no direct evidence supporting the role of CHEK2 in NF pathogenesis. Nevertheless, mutation of this gene is involved in impaired DNA damage repair, which can cause chromosomal instability in NF2.[4] In this article, we report for the first time an identical germline CHEK2 mutation, p.R180C, in a Tibetan boy with NF1 and in a Chinese girl with NF2, and discuss the significance of CHEK2 and other genetic mutations in tumorigenesis of this disease.
2. Patient and methods
2.1. Patient history
2.1.1. Patient 1
A 7-year-old Tibetan boy complained of painful subcutaneous mass in the right frontotemporoparietal region for 4 years. He underwent tumor resection on the same site 4 years before admission, and pathology showed plexiform neurofibroma. After that, he experienced slow regrowth of the tumor. On physical examination, the tumors were nodular, plexiform, and located under scalp on the right side. They were painful with moderate movability. He had multiple café-au-lait spots all over his body, and mass on his head was painful (Fig. 1). The patient underwent tumor resection under general anesthesia. His postoperative course was uneventful. Pathology confirmed previous diagnosis of plexiform neurofibroma and NF1. Because the boy is the only one with neurofibroma in his family, his parents expressed concern for heredity of the disease, and we discussed gene mutation test with patient's parents and obtained informed consent. Peripheral blood was drawn from the patient, his sister, and both his parents for subsequent tests.
Figure 1.
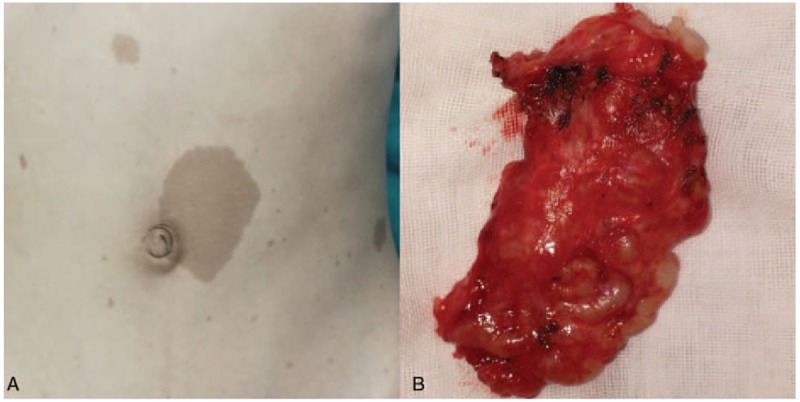
Clinical photograph of patient with neurofibromatosis type 1 (NF1). A, Multiple café-au-lait spots on the abdomen. B, Gross specimen of the plexus neurofibroma after resection.
2.1.2. Patient 2
A 12-year-old girl complained of decreased eyesight for 7 years. She had total loss of vision in her right eye about 4 months before admission, and before that, she did not tell her parents about her illness. On physical examination, there was bulging mass in her left frontal bone. Her right eye was completely blind, and visual acuity in her left eye was almost normal. Her bilateral hearing decreased, which was worse for her left ear. Her computed tomography (CT) scan showed choroidal plexus calcification in bilateral ventricles. Reconstructed skull CT scan showed expansion and erosion of her left frontal bone. On enhanced magnetic resonance imaging scan, there was thickening and enhancement in her left frontal bone and adjacent meninges. Also noticed was enhanced mass in her right optic nerve and bilateral vestibular masses. In addition, there were multiple enhanced masses in suprasellar region and lower posterior fossa (Fig. 2). An initial diagnosis of NF2 was made. Because she was not deaf in either of her ears and the chance of recovering her right eyesight was almost zero, we did not attempt to remove the vestibular schwannoma or right optic nerve tumor; instead, her main concern was the mass in her left frontal bone. She underwent skull and meningeal lesion removal with cranioplasty under general anesthesia. She recovered well and pathology confirmed World Health Organization grade I meningioma with invasion to skull and NF2. Her parents were concerned with heredity of the disease, and we performed gene mutation test for the girl and her parents using peripheral blood under informed consent. Both studies were reviewed and approved by Ethnic Committee of West China Hospital, and both patients’ guardians provided informed consent for publication of these cases.
Figure 2.
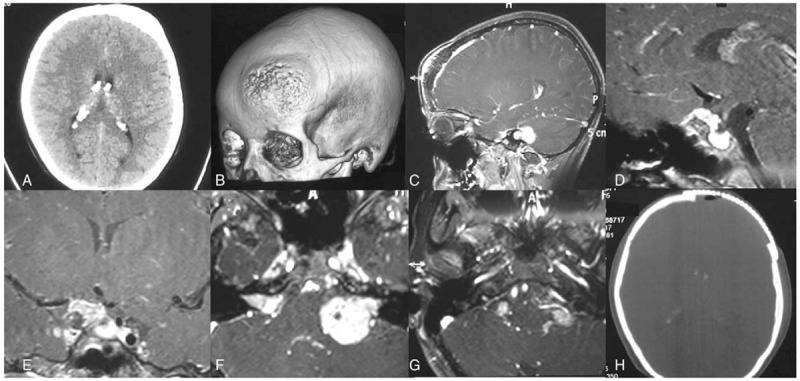
Pre- and postoperative images for patient with neurofibromatosis type 2 (NF2). A, Computed tomography (CT) scan showing thickening of left frontal bone and calcification of bilateral choroidal plexus. B, 3D-reconstructed CT scan showing bulging and erosion of left frontal bone. C, Sagittal enhanced magnetic resonance imaging (MRI) showing thickening of frontal bone and enhancement of underlying meninges. D, Sagittal enhanced MRI showing suprasellar mass. E, Coronal image showing enhanced mass on the right optic nerve, possibly optic glioma. F, Axial image showing bilateral vestibular schwannoma. G, Axial image on lower pons and medulla section showing multiple neuromas of cranial nerve in the posterior fossa. H, Postoperative CT scan showing removal of affected bone and cranioplasty.
2.2. gDNA extraction
AllPrep DNA/RNA mini Kit (Qiagen 80204) was used for gDNA extraction from blood lymphocytes according to the instructions. The gDNA was qualified with a 2200 Bioanalyzer (Agilent Technologies) in fragment size, quality and total concentration.
2.3. Library construction
The library of gDNA was constructed according to the manufacturer's protocols by a KAPA Hyper Prep kit (Kapa Biosystems). The quantities of the library were all measured with Qbit 3 (Thermo Fisher).
2.4. Panel design and targeted sequencing
A panel of 139 were used in our research (gene are listed in supplementary files), gDNA libraries were enriched for regions of this custom designed captured probe manufactured by Agilent. A total of 750 ng prepared libraries were hybridized with 2 different hybridization reagents and blocking agents in SureSelectXT Target Enrichment System (Agilent Technologies). The enriched libraries were amplified with P5/P7primer. After qualified by the 2200 Bioanalyzer and quantified with Qbit3 and a qPCR NGS library quantification kit (Agilent Technologies), then the libraries were sequenced on a Hiseq X10 platform (Illumina, San Diego, CA).
2.5. Data analysis
Sequencing reads were mapped to a human reference genome (hg19) using the Burrows–Wheeler Aligner.[5] Duplicate removal, local realignment, and base quality recalibration were performed using PICARD (http://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (GATK).[6] Somatic single nucleotide variations and small indels were called using GATK.[6] Effects of variants were annotated using Oncotator[7] and Variant Effect Predictor,[8] as well as an in-house database (GnentronDB). A mutation was filtered if the depth was smaller than 5, the mutational frequency was less than 20%, or was recurrently found in healthy individuals. Each candidate mutation was reviewed visually by integrative genomics viewer.[9]
3. Results
In patient 1, he inherited CHEK2 mutation from his mother, and polymerase epsilon (POLE) mutation from his father (Fig. 3). In patient 2, she inherited CHEK2 mutation from her mother, but not MutL homolog 1 (MLH1), colon cancer, nonpolyposis type 2 (E coli), and she inherited both mutY DNA glycosylase, earlier mutY Homolog (MUTYH), and ataxia telangiectasia mutated and rad-related kinase (ATR) mutations from her father. NF2 mutation was also detected (Fig. 4).
Figure 3.
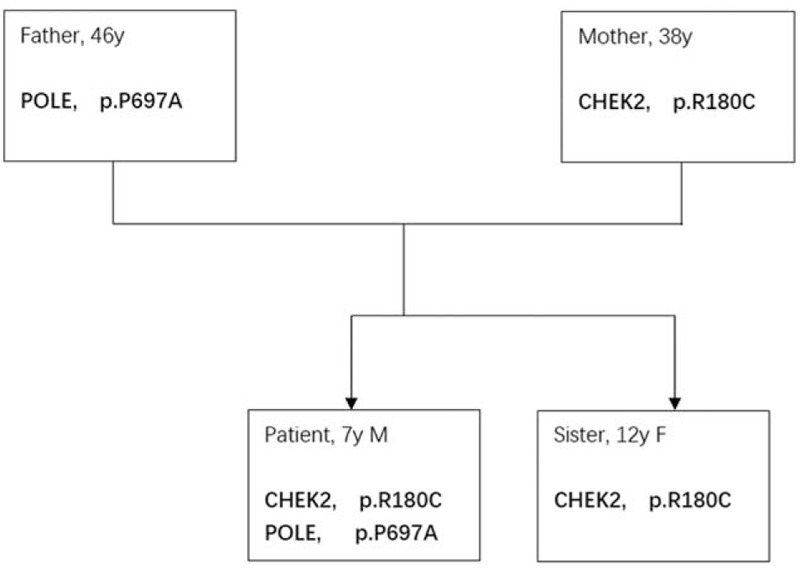
Pattern of germline mutation of patient with neurofibromatosis type 1 (NF1). CHEK2 = checkpoint kinase 2, POLE = polymerase epsilon.
Figure 4.
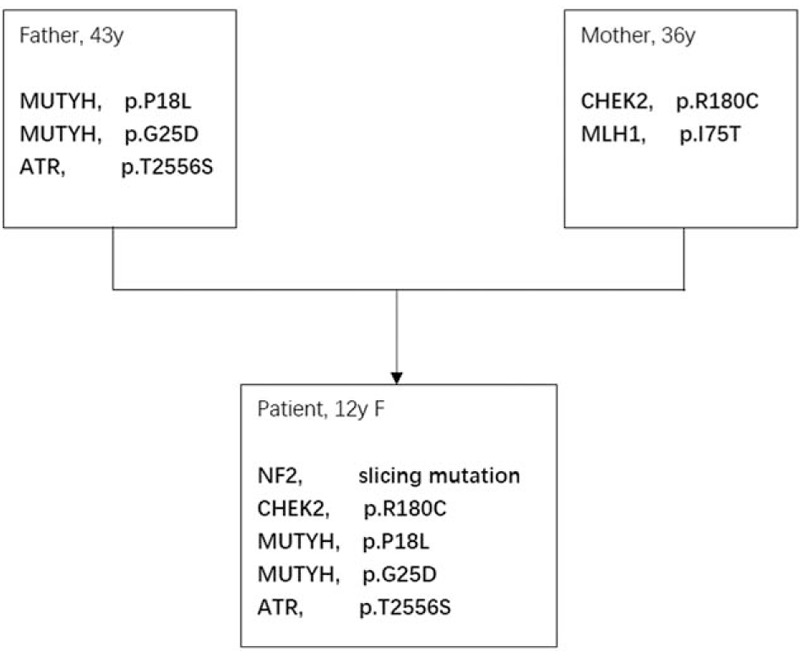
Pattern of germline mutation of patient 2. ATR = ataxia telangiectasia mutated and rad-related kinase, CHEK2 = checkpoint kinase 2, MUTYH = mutY DNA glycosylase, earlier mutY homolog.
4. Discussion
This is the first article to report that identical germline mutation of CHEK2 gene, p.R180C, exists in both NF1 and NF2 patients, and the first article to show coexistence of germline mutation of CHEK2 and POLE in a Tibetan child. These genes are involved in DNA synthesis, repair of mutation, and proofreading, all of which are critical steps in normal cell division and errors could cause tumorigenesis. CHEK2 as a tumor suppressor gene is activated in response to DNA damage, and can prevent cell from entering mitosis or arrest cell cycle. It was reported that CHEK2 mutation was associated with elevated risk of breast cancer, colorectal cancer, Li-Fraumei syndrome and other early-onset familiar cancers.[1–3,10,11] Our findings demonstrate that CHEK2 mutation could also be associated with both NF1 and NF2.
CHEK2∗1100delC allele is the most widely studied mutation point and is associated with Li-Fraumei syndrome. The risk of breast cancer in its carriers varies in different ethnic groups, with the highest in Russian and Brazilian populations, but in Southeast Asia, CHEK2∗1100delC allele is rarely detected.[1] The rate of p.R180C mutation was reported to be 0.6% in Ashkenazi Jewish breast cancer patients and 0.38% in Chinese patients.[1,12] Although this mutation is not located in a functional domain, it may affect protein folding and is deemed intolerant, and further study in yeast Rad53 gene, an equivalent to human CHEK2, confirms that it is pathogenic.[13] In Chinese population, the CHEK2 mutation p.H371Y has only been recently reported to be associated with Li-Fraumei–like syndrome.[11] Otherwise, there is no report concerning the p.R180C mutation in either NF1 or NF2 patients.
Human POLE is the catalytic subunit of POLE complex. It is responsible for synthesis of leading strand during DNA replication, and is capable of proofreading, which is essential for replication fidelity.[14] Mutation of this gene is involved in impaired DNA damage repair,[4] which is a key mechanism in DNA mismatch repair deficiency (MMRD) syndrome, characterized by early onset of hematological and brain tumors, as well as cutaneous features similar to café-au-lait spots in NF1 patients.[15,16] Recently, the phenotype overlap between NF1 and MMRD has been reported, raising the hypothesis that NF1 and MMRD may share some common pathogenic mechanism.[17–19]
MUTYH gene is located in the short arm of chromosome 1 (1p34.1) and encodes human DNA glycosylase that is associated with DNA repair, specifically base excision repair pathway.[20] It removes adenine from oxidized form of guanine, and mutation of MUTYH is related to a condition vulnerable to colorectal cancer called MUTYH-associated polyposis (MAP). Most MAP patients harbor somatic G:C to T:A transversion mutation in adenomatous polyposis coli and Kras genes.[20] ATR, together with ataxia telangiectasia mutated kinase (ATM), are critical mediators in response to DNA damage. ATR mainly activates Chek1 in response to ultraviolet damage, whereas ATM activates p53 and Chek2 in response to ionizing radiation damage. Recently, ATM has been shown to be required for ATR recruitment and later activation of Chek1, which subsequently activates a number of substrates that function in cell-cycle arrest, replication forks stabilization and DNA repair.[21]
More than 2600 inherited mutations leading to NF1 have been reported. Although germline mutation rate of NF1 is approximately 10 times higher than other inherited disease genes, more than half of NF1 is caused by de novo mutations.[22] We did not perform somatic gene mutation test in the neurofibroma tissue from our first case, because it is usually very difficult to identify the causative NF1 mutation due to its large size (350 kbp), structural complexity and lack of mutation hotspot and clusters. No germline mutation of NF1 was detected in the Tibetan boy. It is noteworthy that only the boy who harbored both CHEK2 and POLE mutation was inflicted by NF1, and not those who harbored either one mutation, such as his sister or parents. Therefore, it is possible that CHEK2 and POLE germline mutation would work synergistically to evoke tumorigenesis of NF1 in the child. In addition, some MMRD patients present symptoms and signs indistinguishable from NF1 patients before the early onset of malignant tumors, and although the role of POLE in MMRD has been confirmed,[16] our case suggests that in addition to POLE mutation, CHEK2 mutation might also contribute to development of MMRD. However, the role of CHEK2 germline mutation in MMRD needs further study.
In the second case, the girl inherited mutation of MUTYH and ATR from her father and CHEK2 from her mother. It was reported that ATR-Chek1 pathway suppressed tumorigenesis, whereas ATM-Chek2 pathway might promote it.[23] Germline mutations of both pathways coexisted in the second patient, but we do not know how these mutations affect function of the respective pathways and their interplay. Mutation of MUTYH, ATR, and CHEK2 might have caused failure to correct de novo NF2 gene mutation during early embryonic stage before she was born, as germline mutation of NF2 was detected only in the girl, not her parents. This case suggests that de novo germline mutation of NF2 can occur when DNA repair mechanism is incompetent, and causes symptomatic NF2 phenotype.
It is interesting that germline mutation of CHEK2 with the exact identical location (p.R180C) exists in both NF1 and NF2 patients in the present study, irrespective of ethnic differences. This suggests p.R180C mutation may not be rare for Asian population. Furthermore, since p.R180C is pathogenic, and inactivation of ATM-Chek2 and ATR-Chek1 pathway has been reported to sensitize human tumors to radio-/chemo-therapy based on cisplatin, temozolomide, topoisomerase inhibitor, and poly(ADP-ribose) polymerase inhibitor,[23] our findings implicate possible role of these agents in systemic or adjuvant treatment of NF1 and NF2 patients.
Author contributions
Conceptualization: Qiang Li, Yan Ju.
Data curation: Qiang Li.
Funding acquisition: Qiang Li.
Investigation: Qiang Li, Feilong Zhao, Yan Ju.
Methodology: Feilong Zhao, Yan Ju.
Resources: Yan Ju.
Software: Feilong Zhao.
Supervision: Yan Ju.
Validation: Feilong Zhao.
Visualization: Qiang Li.
Writing – original draft: Qiang Li.
Writing – review and editing: Qiang Li, Yan Ju.
Supplementary Material
Footnotes
Abbreviations: ATM = ataxia telangiectasia mutated kinase, ATR = ataxia telangiectasia mutated and rad-related kinase, CHEK2 = checkpoint kinase 2, CT = computed tomography, GATK = Genome Analysis Toolkit, MAP = MUTYH-associated polyposis, MLH1 = MutL homolog 1, colon cancer, nonpolyposis type 2 (E coli), MMRD = mismatch repair deficiency, MRI = magnetic resonance imaging, MUTYH = mutY DNA glycosylase, earlier mutY homolog, NF1 = neurofibromatosis type 1, NF2 = neurofibromatosis type 2, POLE = polymerase epsilon.
Funding Source: This study is supported by National Natural Science Foundation Grant No. 81602190.
The authors have no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Apostolou P, Papasotiriou I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer (Dove Med Press) 2017;9:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jalilvand M, Oloomi M, Najafipour R, et al. An association study between CHEK2 gene mutations and susceptibility to breast cancer. Comp Clin Path 2017;26:837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dominguez-Valentin M, Nakken S, Tubeuf H, et al. Potentially pathogenic germline CHEK2 c.319+2T>A among multiple early-onset cancer families. Fam Cancer 2018;17:141–53. [DOI] [PubMed] [Google Scholar]
- [4].Yang HW, Kim TM, Song SS, et al. Alternative splicing of CHEK2 and codeletion with NF2 promote chromosomal instability in meningioma. Neoplasia 2012;14:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ramos AH, Lichtenstein L, Gupta M, et al. Oncotator: cancer variant annotation tool. Hum Mutat 2015;36:E2423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hallamies S, Pelttari LM, Poikonen-Saksela P, et al. CHEK2 c.1100delC mutation is associated with an increased risk for male breast cancer in Finnish patient population. BMC cancer 2017;17:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhuang X, Li Y, Cao H, et al. Case report of a Li-Fraumeni syndrome-like phenotype with a de novo mutation in CHEK2. Medicine (Baltimore) 2016;95:e4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mohamad S, Isa NM, Muhammad R, et al. Low prevalence of CHEK2 gene mutations in multiethnic cohorts of breast cancer patients in Malaysia. PLos One 2015;10:e0117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hill J, John E, Lichten M, et al. Novel pathogenic CHEK2 mutations in African Americans with breast cancer: results of a Breast Cancer Family Registry study. Cancer Epidemiol Biomarkers Prev 2007;16: Supplement A13. [Google Scholar]
- [14].Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013;45:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010;95:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Andrianova MA, Chetan GK, Sibin MK, et al. Germline PMS2 and somatic POLE exonuclease mutations cause hypermutability of the leading DNA strand in biallelic mismatch repair deficiency syndrome brain tumours. J Pathol 2017;243:331–41. [DOI] [PubMed] [Google Scholar]
- [17].Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet 2017;91:507–19. [DOI] [PubMed] [Google Scholar]
- [18].Suerink M, Potjer TP, Versluijs AB, et al. Constitutional mismatch repair deficiency in a healthy child: on the spot diagnosis? Clin Genet 2018;93:134–7. [DOI] [PubMed] [Google Scholar]
- [19].Yeung JT, Pollack IF, Shah S, et al. Optic pathway glioma as part of a constitutional mismatch-repair deficiency syndrome in a patient meeting the criteria for neurofibromatosis type 1. Pediatr Blood Cancer 2013;60:137–9. [DOI] [PubMed] [Google Scholar]
- [20].Kashfi SM, Golmohammadi M, Behboudi F, et al. MUTYH the base excision repair gene family member associated with colorectal cancer polyposis. Gastroenterol Hepatol Bed Bench 2013;6(suppl 1):S1–0. [PMC free article] [PubMed] [Google Scholar]
- [21].Smits VA, Warmerdam DO, Martin Y, et al. Mechanisms of ATR-mediated checkpoint signalling. Front Biosci (Landmark Ed) 2010;15:840–53. [DOI] [PubMed] [Google Scholar]
- [22].Philpott C, Tovell H, Frayling IM, et al. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics 2017;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Manic G, Obrist F, Sistigu A, et al. Trial watch: targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol 2015;2:e1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.