

Abstract
Key points
The arterial baroreflex's operating point pressure is reset upwards and rightwards from rest in direct relation to the increases in dynamic exercise intensity.
The intraneural pathways and signalling mechanisms that lead to upwards and rightwards resetting of the operating point pressure, and hence the increases in central sympathetic outflow during exercise, remain to be identified.
We tested the hypothesis that the central production of angiotensin II during dynamic exercise mediates the increases in sympathetic outflow and, therefore, the arterial baroreflex operating point pressure resetting during acute and prolonged dynamic exercise.
The results identify that perindopril, a centrally acting angiotensin converting enzyme inhibitor, markedly attenuates the central sympathetic outflow during acute and prolonged dynamic exercise.
Abstract
We tested the hypothesis that the signalling mechanisms associated with the dynamic exercise intensity related increases in muscle sympathetic nerve activity (MSNA) and arterial baroreflex resetting during exercise are located within the central nervous system. Participants performed three randomly ordered trials of 70° upright back‐supported dynamic leg cycling after ingestion of placebo and two different lipid soluble angiotensin converting enzyme inhibitors (ACEi): perindopril (high lipid solubility), captopril (low lipid solubility). Repeated measurements of whole venous blood (n = 8), MSNA (n = 7) and arterial blood pressures (n = 14) were obtained at rest and during an acute (SS1) and prolonged (SS2) bout of steady state dynamic exercise. Arterial baroreflex function curves were modelled at rest and during exercise. Peripheral venous superoxide concentrations measured by electron spin resonance spectroscopy were elevated during exercise and were not altered by ACEi at rest (P ≥ 0.4) or during exercise (P ≥ 0.3). Baseline MSNA and mean arterial pressure were unchanged at rest (P ≥ 0.1; P ≥ 0.8, respectively). However, during both SS1 and SS2, the centrally acting ACEi perindopril attenuated MSNA compared to captopril and the placebo (P < 0.05). Arterial pressures at the operating point and threshold pressures were decreased with perindopril from baseline to SS1 with no further changes in the operating point pressure during SS2 under all three conditions. These data suggest that centrally acting ACEi is significantly more effective at attenuating the increase in the acute and prolonged exercise‐induced increases in MSNA.
Keywords: Muscle Sympathetic Nerve Activity, Angiotensin II, Electron Spin Resonance, Free Radicals, Superoxide, Nitric Oxide, Functional Sympatholysis
Key points
The arterial baroreflex's operating point pressure is reset upwards and rightwards from rest in direct relation to the increases in dynamic exercise intensity.
The intraneural pathways and signalling mechanisms that lead to upwards and rightwards resetting of the operating point pressure, and hence the increases in central sympathetic outflow during exercise, remain to be identified.
We tested the hypothesis that the central production of angiotensin II during dynamic exercise mediates the increases in sympathetic outflow and, therefore, the arterial baroreflex operating point pressure resetting during acute and prolonged dynamic exercise.
The results identify that perindopril, a centrally acting angiotensin converting enzyme inhibitor, markedly attenuates the central sympathetic outflow during acute and prolonged dynamic exercise.
Introduction
Short‐term adjustments of arterial blood pressure (ABP) in response to abrupt changes in blood volume, cardiac output and peripheral resistance, such as those that occur during exercise, are affectively buffered by the activation of the arterial baroreflex (ABR). During dynamic exercise and in contrast to the resting condition, a physiological hypertension coexists with tachycardia. These parallel increases in ABP and heart rate (HR) are essential to maintain increased cardiac output and to provide appropriate delivery of oxygen and nutrients to the working muscles (Mitchell, 1990; Rowell & O'Leary, 1990). It has been established by a number of investigators that activation of central command and/or the exercise pressor reflex is essential for the progressive physiological resetting of the reference ‘operating point’ pressures of the ABR in direct relation to the progressive increase in exercise intensity (Melcher & Donald, 1981; Walgenbach & Donald, 1983; Potts et al. 1993; Papelier et al. 1994; Norton et al. 1999; Fadel & Raven, 2012) . The resetting of the ABR enables the observed parallel increases in HR and ABP with progressive increases in exercise intensity. Furthermore, during dynamic exercise, the physiological production of angiotensin II (Ang II) and its related generation of free radicals are increased linearly in relation to increases in exercise intensity (Fallo, 1993; Bailey et al. 2004, 2011; Shim et al. 2008; Powers et al. 2016).
Exacerbated increases in central oxidative stress are implicated in impaired autonomic regulation of cardiovascular function associated with ageing (Monahan et al. 2004), chronic heart failure (Nightingale et al. 2003) and obstructive sleep apnoea (Yamauchi & Kimura, 2008). Furthermore, a body of evidence suggests that essential hypertension has a central neurogenic origin related to free radicals via Ang II linked free radical production (Zimmerman et al. 2002; Chan et al. 2005; Zucker, 2006). Physiologically central sympathetic outflow is inhibited by central nitric oxide (NO•) (Aslan et al. 2001) within the nucleus of the tractus solitarius (NTS) and the rostral ventral lateral medulla (Jiang et al. 1996). Animal data has implicated NO• as a modulator of central sympathetic nerve activity outflow. For example, introduction of central NO• dampens central sympathetic outflow (Patel et al. 2001; Gao et al. 2011). Recently, it has been established that central NO• is scavenged by centrally generated free radicals (Paton & Waki, 2009; Waki et al. 2011), thereby allowing greater central sympathetic outflow (Waki et al. 2008; Fisher & Fadel, 2010).
For the current study, we investigated the hypothesis that the central physiological production of Ang II during dynamic exercise within the brain was related to the increases in sympathetic outflow. We planned to compare radial muscle sympathetic nerve activity (MSNA) from rest to dynamic leg cycling exercise with a placebo or with a pharmacological inhibition of the angiotensin converting enzyme (ACEi) using effective equivalent doses (Agabiti‐Rosei et al. 1992): (i) a global 4 mg oral dose of perindopril, which is a highly lipid‐soluble ACEi that crosses the blood–brain barrier (Yamada et al. 2010, 2011; Dong et al. 2011); and (ii) a 25 mg oral dose of captopril, which has a low lipid solubility compared to perindopril. ACEi markedly limits Ang II binding with the AT1 receptor in the renin–angiotensin systems located in both the central nervous system (CNS) and the systemic neural system (Unger et al. 1982, 1984; Migdalof et al. 1984).
Methods
Ethical approval
This study conformed to guidelines set forth by the Declaration of Helsinki and was approved (IRB no. 2014‐062) by the Institutional Review Board for the Protection of Human Subjects in Research at the University of North Texas Health Science Center.
Subjects
Eleven males (24 ± 2 years, 181 ± 7 cm, 82 ± 5 kg) and three females (24 ± 1 years, 163 ± 5 cm, 67 ± 3 kg) volunteered for this study. Subjects were deemed healthy via a general health questionnaire, were not taking any medications, and did not smoke. Subjects were asked to abstain from caffeine, exercise and alcohol 24 h before each experimental session due to possible influences on cardiovascular regulatory mechanisms. Subjects also received familiarization training with the experimental protocol and procedures before any experiments were performed. Written informed consent was obtained from all subjects. Female subjects were screened with a urine over‐the‐counter pregnancy test to ensure that they were not pregnant on the day of testing. Due to the potential effect of oestrogen and progesterone on autonomic neural regulation of the cardiorespiratory systems, all female subjects not on hormonal contraception were tested in the early follicular (low hormone) phase of their menstrual cycle (i.e. days 1–4 post‐menses); female subjects on hormonal contraception were tested during the low hormone or placebo phase of their contraception medication schedule.
Experimental protocol
Subjects visited the laboratory on four separate occasions. The first visit was for an orientation day and the other three visits were randomized among the three experimental pharmacological protocols: placebo, perindopril and captopril. The subjects were assigned to one of three experimental groups for their first experiment (placebo, perindopril, captopril) in a partially randomized counterbalance design. The counterbalanced design was used because, in rare instances, severe anaphylaxis and angioedema have been observed following ACEi ingestion; therefore, our counterbalanced order always presented captopril ahead of perindopril. The pharmacokinetics of perindopril identifies ∼4 h post‐ingestion as achieving peak plasma concentrations (Bussien et al. 1986), whereas captopril takes only 1 h post‐ingestion (Hollenberg et al. 1981). Hence, subjects ingested perindopril 3 h prior to reporting to the laboratory while the captopril was ingested in the laboratory prior to the beginning of the experimental protocol, between 08.00 and 09.00 h. This counterbalanced design enabled supervised monitoring and screening of each subject during the faster acting captopril ingestion.
Experimental protocols were separated by at least 1 week, during which time subjects were instructed to maintain their normal exercise and diet regimen. Each subject performed an orientation session to familiarize themselves with the laboratory and its equipment in order to reduce any potential ‘white‐coat’ effects on measured variables. The subjects then underwent a graded exercise test on a 70° back‐supported semi‐recumbent cycle ergometer to determine their exercise tolerance and workload for steady state exercise at HRs of 120 beats min−1 (SS1, acute) and 150 beats min−1 (SS2, prolonged). These exercise work intensities are considered to be heavy and very heavy, respectively (Åstrand & Rodahl, 1986).
Ultrasound and blood flow Doppler measurements were used to verify the location of the carotid sinus area and visualize flow of the carotid arteries within the neck to ensure that the bilateral vascularization of the neck was intact and free of atherosclerotic plaques. If no abnormalities were noted in the blood vessel anatomy or blood flow, the subject practiced wearing the neck collar and underwent two to three trials of neck pressure and neck suction at rest and during mild exercise. The collar provides an airtight chamber which encompasses the anatomical location of the carotid sinus area. When prompted, the computer software initiates a positive neck pressure (NP) or a negative neck suction (NS), pressure pulse for 5 s. The positive or negative chamber pressure within the collar transmits 89% of the NP and 83% of the NS to the extramural carotid tissues (Querry et al. 2001), which are sensed by the carotid baroreceptors as changes in arterial transmural pressure (Potts et al. 1993). The neck pressure simulates hypotension and the neck suction simulates hypertension and both simulations are sensed by the carotid baroreceptors.
ACE inhibitors
Comparison dosages for the ACE inhibitors were determined by our board‐certified cardiologist consultant and were set as the acute dose required for approximately equal changes in blood pressure in clinical applications based on previous studies (Lees & Reid, 1987; Agabiti‐Rosei et al. 1992; Jankowski et al. 1995; Chik et al. 2010). Furthermore, single dose studies in healthy human subjects have identified that plasma angiotensin II (Ang II) concentrations are reduced by 31% with both 25 mg captopril measured at 1 h post‐ingestion (Hollenberg et al. 1981) and 4 mg of perindopril measured at 4 h post‐ingestion (Bussien et al. 1986). Therefore, within the acute time frame in which we obtained the data and the similar peripheral reductions in Ang II with both ACE inhibitors, we contend that captopril and perindopril effects peripherally within that acute time frame are similar. Several animal studies have confirmed that an oral dosage of perindopril is a centrally active ACE inhibitor (Yamada et al. 2010, 2011; Dong et al. 2011). In one study the brain ACE activities in mice were decreased by more than 50% with an oral dosage of perindopril when compared to other non‐centrally active ACE inhibitors (Yamada et al. 2010). Furthermore, Dong et al. (2011) demonstrated that oral ingestion of perindopril significantly inhibited hippocampal ACE activity, which prevented cognitive impairment in an Alzheimer's disease mouse model. The authors attribute this beneficial effect to the suppression of microglia/astrocyte activation and the attenuation of oxidative stress caused by inducible nitric oxide synthase induction and extracellular superoxide dismutase down‐regulation (Dong et al. 2011). In contrast, when antihypertensive doses of captopril were given acutely orally or intravenously, captopril could scarcely be detected in the brain or cerebrospinal fluid (Unger et al. 1982, 1984). These findings suggested that following acute peripheral administration, captopril did not penetrate the blood–brain barrier in sufficient quantity to inhibit the renin–angiotensin system production of Ang II in the brain tissue or cerebrospinal fluid (Unger et al. 1982, 1984; Migdalof et al. 1984). We used these differences in the central effects of perindopril versus captopril, while each of the ACE inhibitors had similar peripheral effects on the AT1 receptors, as a means of selectively identifying central effects of ACEi on central sympathetic outflow.
Instrumentation and exercise protocol
Once subjects successfully completed the orientation day, they were invited back to participate in the experimental protocol. Each experimental protocol was performed at the same time of day for each subject to account for the influence of circadian rhythms on the measured variables. After ingestion of either the placebo or ACEi capsules, subjects were instrumented with a three‐lead ECG (Hewlett‐Packard, Inc.; Palo Alto, CA, USA) to monitor the beat‐to‐beat heart rate (HR), and arterial blood pressure (ABP) measurements were obtained using finger photoplethysmography (Finometer, Finapres Medical Systems, Amsterdam, The Netherlands).
Multiunit muscle sympathetic nerve activity (MSNA) was recorded directly from the radial nerve at the spiral groove in the upper arm using standardized (Hagbarth & Vallbo, 1968; Wallin et al. 1994; Vallbo et al. 2004) and ultrasound‐guided microneurography techniques (Curry & Charkoudian, 2011). Additionally, an intravenous catheter was inserted into an antecubital fossa vein to withdraw 5 mL samples of whole blood for the measurement of peripheral superoxide concentrations ([O2 •−]). In the present study, the workloads were increased incrementally from rest to the acute exercise SS1, and from SS1 to the prolonged SS2 workload, without randomization of the workloads in order to maintain the microneurography recording microelectrode at the initial site obtained at the start of the protocol. Even then, we only successfully obtained MSNA records on 7 of the 14 subjects recruited throughout the three repeated measures exercise protocols.
Figure 1 illustrates an overview of the experimental protocol procedures. Subjects were positioned on the same stationary bicycle ergometer where they performed the graded exercise test, in a 70° back‐supported semi‐recumbent position with the legs extended parallel with the floor for ∼60 min during instrumentation, which included obtaining MSNA from the radial nerve. The experimental protocol on each experimental day consisted of three stages: (1) baseline (resting); (2) SS1 (acute – the exercise workload determined at HRs of 120 beats min−1 during the orientation session); and (3) SS2 (prolonged – the exercise workload determined at HRs of 150 beats min−1 during the screening stress test). After instrumentation, subjects rested in the semi‐recumbent position for at least 5 min to establish pre‐exercise baseline values. Data were collected for an additional 5 min at baseline followed by the NP/NS protocol (∼20 min), which was performed to identify changes in HR and ABP that enabled our modelling of ABR function curves, as previously described (Potts et al. 1993; Norton et al. 1999). After collecting the resting data, the subject began to exercise on a computer‐controlled magnetically braked ergometer (Intellifit, Houston, TX, USA) with a zero load and a slow pedalling frequency exercise. Both the pedal frequency and the intensity of exercise were progressively increased to a maximum of 60 r.p.m. over ∼5 min to achieve a constant workload obtained at the HR of 120 beats min−1 (SS1) during the orientation screening exercise test. Once the subject achieved the steady state target workload for approximately 5 min, the HR and ABP data were collected for an additional 5 min during SS1 and followed by the 20 min NP/NS protocol, for a total of 30 min. This was identified as the ‘acute exercise’ period. After completion of the NP/NS protocol during the SS1 workload, the subject, without stopping, continued to exercise at 60 r.p.m. while the workload was slowly increased over 5 min to achieve the workload determined on their orientation screening exercise test to a steady state HR of 150 beats min−1 (SS2) for another 30 min. During the SS2, the steady state HR and ABP, and their responses to the NP/NS protocol, were obtained after a total exercise time of 60 min. The last 30 min of the total exercise time was identified as the ‘prolonged exercise’ protocol. Venous blood samples were obtained during each steady state stage: baseline, SS1 and SS2 (before the NP/NS protocol). The two exercise workloads, SS1 (acute and heavy) and SS2 (prolonged and very heavy) (Åstrand & Rodahl, 1986), were performed continuously for approximately 30 min each for a total of 60 min. Furthermore, to maintain the same metabolic demand across all three treatments, the workloads, once established during the orientation day, were not changed for each of the participants. Therefore, any changes in our dependent variables of interest were attributed to the intervention (i.e. ACEi) and not the manipulation of the workloads or heart rates.
Figure 1. Illustration of the experimental protocol over time.
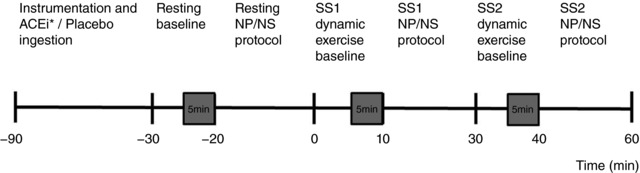
Shaded boxes represent measurement of haemodynamic, microneurographic and ESR data. Experimental data were averaged over the last 2 min of the 5 min period. Venous blood samples were obtained after each steady state stage during baseline, steady state exercise at workload HRs of 120 beats min−1 (SS1) and workload HRs of 150 beats min−1 (SS2) before the neck pressure/neck suction (NP/NS) protocol. * indicates the ingestion of the ACEi; Captopril (60 min before baseline) or the ingestion of Perindopril (180 min before coming into the laboratory for the instrumentation period).
ABR stimulus–response function and curves
We have previously identified baroreflex regulation of vasomotion as the primary means by which arterial blood pressure is regulated during dynamic exercise (Ogoh et al. 2003). Hence, we focused our attention on obtaining arterial baroreflex stimulus–response curves of vasomotion as measured by changes in MAP. We modelled the MAP change to each NP/NS pulse for each subject to a four‐parameter logistic function described by Kent et al. (1972) and is as follows:
where MAP is the dependent variable, ECSP is the estimated carotid sinus pressure, A 1 is the range (i.e. maximum – minimum), A 2 is the gain coefficient (average of the slope of the function), A 3 is the value at the centring point (i.e. point of maximum gain) and A 4 is the minimum response of MAP. Furthermore, the carotid sinus threshold pressures (the point where no further increases in MAP were elicited by reductions in ECSP) and saturation pressures (the point where no further decreases in MAP were elicited by increases in ECSP) were calculated using the following equations (the definitions of the variables A 1–A 4 are provided above) (Potts et al. 1993; McDowall & Dampney, 2006):
The operating point of each curve was defined as a stimulus‐estimated carotid sinus pressure and was averaged over the respective time point (baseline, SS1, SS2) after steady state circulatory responses were obtained. The operating point, saturation and carotid sinus threshold pressures were calculated for each subject and condition and then compared between and across all subjects.
Measurement of free radicals
Electron spin resonance (ESR) spectroscopy was utilized to measure peripheral free radical concentrations (in particular, superoxide [O2 •−]) directly in venous blood samples. Triplicate 200 μL whole blood samples for each time point (baseline, SS1 and SS2) were incubated for 15 min in a buffer solution consisting of 3.5 mm deferoxamine methanesulfonate salt, 9.08 mm diethyldithiocarbamic acid sodium (DETC), and Krebs–Hepes buffer (Noxygen Science Transfer & Diagnostics GmbH, Elzach, Germany) containing methoxycarbonyl‐2,2,5,5‐tetramethyl‐pyrrolidine (CMH) as the spin probe, which preferentially detects O2 •− (Deo et al. 2012; Vianna et al. 2015). Whole blood samples of 50 μL for each time point in duplicate were then loaded into a 1 mL syringe and flash frozen using liquid nitrogen between buffer solutions to form a continuous frozen plug. Samples were then stored at −80°C and shipped to the University of Nebraska Medical Center's ESR Spectroscopy Core for analysis (Deo et al. 2012; Vianna et al. 2015). ESR amplitude was measured using a Bruker e‐Scan ESR Spectrometer (Bruker Corporation, Billerica, MA ,USA) and was averaged for all triplicate data to generate an individual subject average for each time point.
Data acquisition and analysis
Electronically recorded data were sampled at 500 Hz and recorded to a computer via data acquisition software (WINDAQ, DATAQ Instruments, Akron, OH, USA). The data were then analysed using a commercially available biomedical analysis software program (WinCPRS, Absolute Aliens, Turku, Finland). Data from the last 2 min of the 5 min baseline were analysed to establish baseline values. Additionally, exercise data were analysed in 2 min epochs during the first workload (SS1) and second workload (SS2) after 3 min of steady state exercise (depicted as grey boxes in Fig. 1). R‐waves generated from the ECG were detected and marked at their occurrence in time. Systolic and diastolic arterial pressures (SAPs, DAPs) were marked from the arterial pressure waveforms and MAP was automatically calculated as the area under the arterial pressure waveforms, via the WinCPRS software. Beat‐to‐beat stroke volume (SV) was estimated using the pulse contour method (Jansen et al. 1990). Cardiac output () was calculated from the product of SV and HR. Total vascular conductance (TVC) was calculated as divided by MAP. Multiunit muscle sympathetic nerve activity (MSNA) signals were band‐pass filtered (100–2000 Hz) and integrated (time constant, 0.1 s) to obtain mean voltage neurograms. Neurograms were subsequently imported into the aforementioned biomedical analysis software program which has the capability of detecting bursts of MSNA. The software detects bursts of MSNA based on two primary criteria: (1) pulse synchronous spontaneous bursts with signal‐to‐noise ratios of approximately 3:1; and (2) reflex latencies from preceding R‐waves of approximately 0.9 s (Wallin et al. 1994; Cooke et al. 2009). Furthermore, the same experienced microneurographer (G.M.) manually checked the computerized burst detection results. MSNA was expressed as bursts per minute (burst frequency). Due to the baseline fluctuations of the neurograms that occur during heavy intensity exercise and inherent to our repeated measures design, the amplitude and area of sympathetic bursts were not estimated. As the amplitude and area of sympathetic bursts varies within and between subjects due to electrode position, we cannot verify that the position of the electrode on a subject is the same on all three different occasions. Therefore, we concluded that it would be inappropriate to express the amplitude and area of sympathetic bursts for this particular protocol (White et al. 2015).
Statistical analysis
Subject recruitment and testing spanned over a period of 18 months. We screened and recruited 36 subjects (12 females, 24 males). Three subjects did not qualify for medical reasons. Ten subjects voluntarily withdrew after the initial graded exercise test session. Nine subjects voluntarily withdrew after the 1st experimental session. Therefore, 14 subjects (3 females, 11 males) completed both of the experimental steady state protocols. Acceptable repeated measures of plasma superoxide concentrations were obtained in 8 of the 14 subjects and repeated measurement of MSNA recordings in 7 of the 14 subjects across three different experimental days. Furthermore, carotid baroreflex stimulus–response curves were obtained from 9 of the 14 subjects. Due to the technical challenge of obtaining viable recordings of MSNA during heavy and/or very heavy intensity exercise, while performing the carotid baroreflex function curve protocol, the ABR‐MSNA responses were unable to be modelled by the Kent logistic model. All data were analysed with commercially available software (Sigma Plot, Systat Software Inc., San Jose, CA, USA). For assessment of differences among the mean values of interest between baseline and exercise, a three (treatment; placebo, captopril and perindopril) by three (time; baseline, SS1 and SS2) factorial ANOVA for repeated measures was implemented. A Student–Newman–Keuls post hoc test was conducted to determine differences between groups. Data are presented as means ± SEM. Statistical significance was determined as P < 0.05.
Results
MSNA responses to acute and prolonged dynamic exercise with and without ACEi
Figure 2 is a comparison summary of a representative subject's MSNA and arterial blood pressure recordings comparing baseline, SS1 and SS2 under all three conditions. ACEi did not result in a notable change in MSNA burst frequency at rest (n = 7; P ≥ 0.2, Fig. 3 A). However, the repeated measures ANOVA identified an interaction of MSNA burst frequency with placebo, captopril and perindopril at different exercise conditions of SS1 and SS2 (P < 0.05). Dynamic leg cycling exercise provoked significant increases in MSNA burst frequency from rest to SS1 under all conditions: 50 ± 12% increase for placebo, 66 ± 15% increase for captopril, and 24 ± 8% increase for perindopril (P < 0.01, Fig. 3 A). In addition, MSNA burst frequency at SS2 was markedly increased from baseline with placebo (93 ± 18%) and captopril (114 ± 11%) above the burst frequency in the SS1 with placebo and in the perindopril conditions (32 ± 12%) (P < 0.01). Compared to placebo and captopril, the perindopril burst frequency of MSNA during SS1 was decreased by an average of 6 bursts min−1 and decreased by an average of 11 bursts min−1 during SS2 (P ≤ 0.03; n = 7, Fig. 3 A). The attenuation of MSNA burst frequency is illustrated as percentage change from baseline in Fig. 3 B.
Figure 2. Repeated measurements of radial muscle sympathetic nerve activity during dynamic leg cycling exercise.
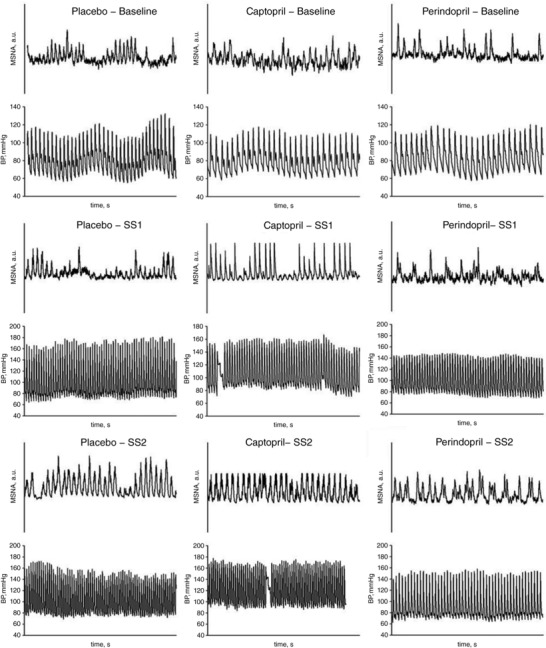
Muscle sympathetic activity (MSNA, arbitrary units (a.u.)) and arterial blood pressure (BP) 30 s tracings are shown for one representative subject during baseline and both steady state exercise workloads SS1 and SS2, and under all conditions: placebo, captopril and perindopril.
Figure 3. MSNA burst frequency (bursts min−1, A) and percentage change of MSNA burst frequency (B) at rest and during both steady state exercise workloads (SS1) and (SS2).
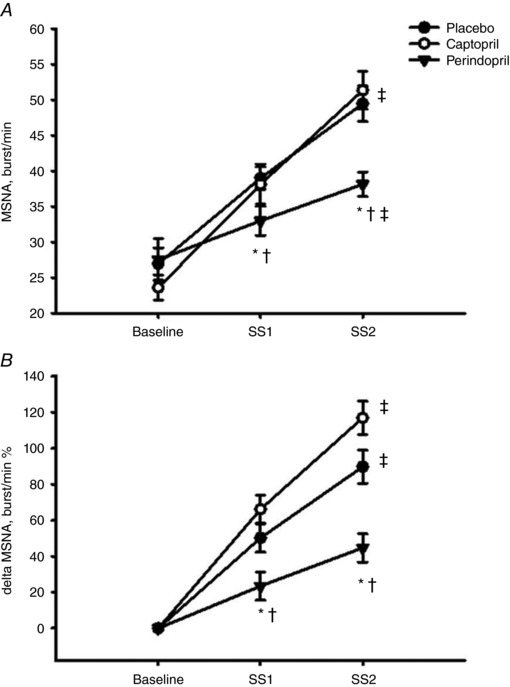
n = 7. Values are means ± SEM; * P ≤ 0.05 compared to placebo; † P ≤ 0.05 compared to captopril; ‡ P ≤ 0.05 compared to SS1 within condition.
Plasma superoxide concentration responses to ACEi during rest and exercise
Oral ingestion of the ACEi did not reduce plasma superoxide concentrations [O2 •−] at rest or during the exercise protocols (n = 8, P ≥ 0.3, Fig. 4). Superoxide did increase from baseline to SS1 in all three treatment groups (P ≤ 0.05) and remained elevated throughout SS2. There were no further increases in [O2 •−] during the SS2 workload for placebo and captopril (P ≥ 0.16, Fig. 4) but with perindopril, it increased from SS1 to SS2 (P = 0.06, Fig. 4).
Figure 4. Summary data of ESR spectra amplitude (arbitrary units, a.u) in response to ACEi during baseline and two workloads of steady state dynamic exercise (SS1) and (SS2).
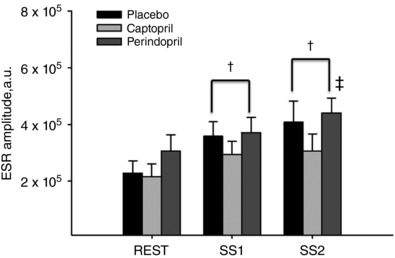
n = 8. Values are means ± SEM; † P ≤ 0.05 compared to baseline within condition; ‡ P ≤ 0.05 compared to SS1 within condition.
Arterial BP responses to ACEi during rest and exercise
Table 1 and Fig. 5 provide a summary of the changes in haemodynamic variables. No changes in arterial blood pressure were observed at rest with the oral administration of either of the ACE inhibitors (n = 14, P ≥ 0.3). However, systolic arterial pressures and diastolic arterial pressures were reduced with perindopril during SS1 when compared to placebo (P < 0.05). Steady state exercise MAPs (arterial baroreflex's operating point pressure) measured during the perindopril protocol were significantly lower during SS1 when compared to both placebo and captopril (P ≤ 0.05, Fig. 5). In addition, when expressed as a percentage change from baseline, perindopril attenuated diastolic arterial pressures compared to placebo and captopril (P ≤ 0.05, Fig. 5 B and C). The increase in work load during SS2 did not result in a significant rise in arterial blood pressure under all three protocols (P ≥ 0.1). However, similar reductions in diastolic and mean arterial pressures were observed with perindopril during SS2 when compared to placebo (P ≤ 0.05, Fig. 5 B and C). In addition, leg cycling exercise elicited significant increases in heart rate (HR), stroke volume (SV) and cardiac output under all conditions compared to baseline and progressively increased from SS1 to SS2 with the exception of SV during both ACEi protocols (captopril, P = 0.9; perindopril, P = 0.9, Table 1). Oral ingestion of the ACEi did not alter total vascular conductance (TVC) at rest (P > 0.1) nor during exercise (P = 0.6); however, TVC was progressively increased during SS1 and SS2 under all three protocols compared to baseline (P ≤ 0.01, Table 1).
Table 1.
Haemodynamic responses to angiotensin converting enzyme inhibitor and exercise
| Baseline | Exercise SS1 | Exercise SS2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | PL | CAP | PER | PL | CAP | PER | PL | CAP | PER |
| HR (beats min−1) | 66 ± 2 | 62 ± 1 | 64 ± 2 | 121 ± 2† | 119 ± 1† | 117 ± 2† | 151 ± 2†‡ | 144 ± 1†‡ | 145 ± 3†‡ |
| SV (mL) | 80 ± 3 | 72 ± 5 | 70 ± 4 | 124 ± 5† | 109 ± 6*† | 105 ± 6*† | 115 ± 5†‡ | 109 ± 6† | 105 ± 5† |
| (L min−1) | 5.3 ± 0.4 | 4.4 ± 0.3 | 4.5 ± 0.4 | 15.1 ± 0.2† | 12.9 ± 0.1*† | 12.2 ± 0.1*† | 17.3 ± 0.3†‡ | 15.6 ± 1*†‡ | 15.2 ± 1*†‡ |
| TVC (L min−1 mmHg−1) | 0.06 ± 0.004 | 0.05 ± 0.003 | 0.05 ± 0.005 | 0.13 ± 0.006† | 0.11 ± 0.003† | 0.11 ± 0.005† | 0.15 ± 0.004†‡ | 0.14 ± 0.003†‡ | 0.14 ± 0.007†‡ |
Values are means ± SEM; n = 14; PL, placebo; CAP, captopril; PER, perindopril; HR, heart rate; SV, stroke volume; , cardiac output; TVC, total vascular conductance. At rest and during steady state exercise at workload HRs of 120 beats min−1 (SS1) and workload HRs of 150 beats min−1 (SS2): * P ≤ 0.05 compared to PL; † P ≤ 0.05 compared to baseline within condition; ‡ P ≤ 0.05 compared to SS1 within condition.
Figure 5. Systolic arterial pressures (SAP, A), diastolic arterial pressures (DAP, B) and mean arterial pressure (MAP, C) at rest and during dynamic leg steady state exercise at workload HRs of 120 beats min−1 (SS1) and workload HRs of 150 beats min−1 (SS2).
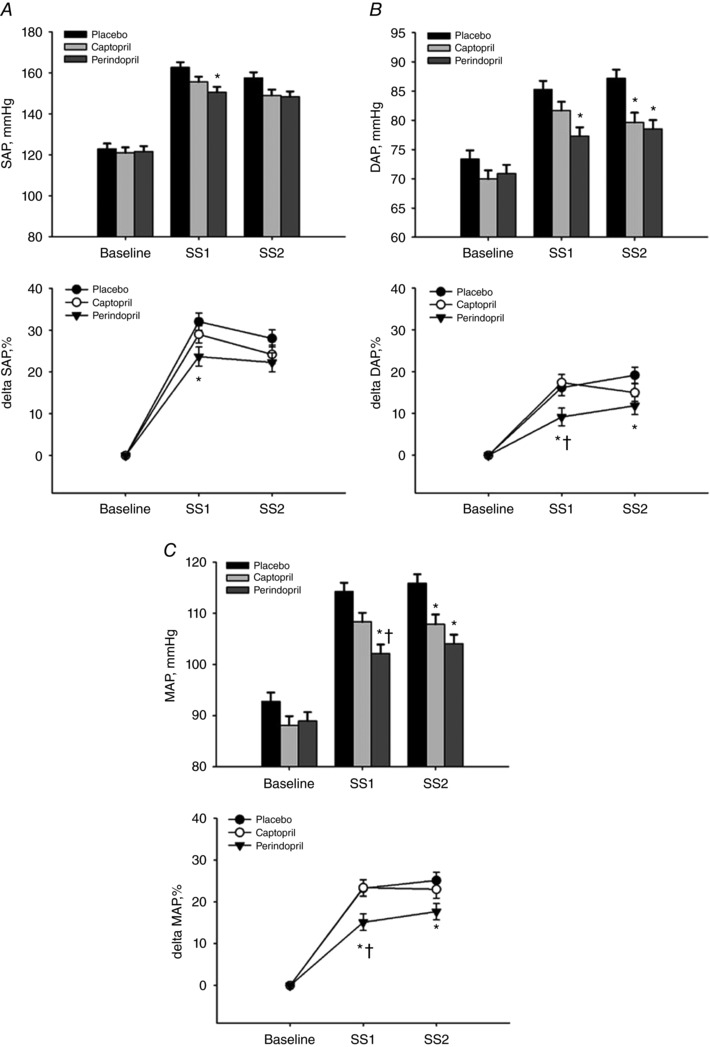
n = 14. Values are means ± SEM; * P ≤ 0.05 compared to placebo; † P ≤ 0.05 compared to captopril.
Arterial baroreflex control of mean arterial pressure with and without ACEi
The stimulus–response relationship for the arterial baroreflex control of MAP at rest and during both workloads of steady state dynamic leg cycling exercise is presented in Fig. 6 A, B and C. For a clearer interpretation of the data, each stimulus–response curve was plotted individually on each graph for each protocol: baseline (Fig. 6 A), workload SS1 (Fig. 6 B) and workload SS2 (Fig. 6 C). Furthermore, logistic parameters describing the ABR–MAP relationship are shown in Table 2. Oral ingestion of ACEi did not change the ABR–MAP parameters at baseline (P ≥ 0.10). However, ACEi did alter the ABR–MAP response range during SS1 (P ≤ 0.05) but not SS2 when compared to baseline. In addition, the point of maximal gain at the centring point pressure (where an equal pressor and depressor response occurs) of the ABR–MAP stimulus–response relationship was similar at rest and during both exercise workloads (SS1 and SS2) and was not affected by ACEi throughout the protocol (P ≥ 0.50; Table 2). In contrast, arterial pressures at the operating point and carotid sinus threshold pressures were decreased with perindopril from baseline to SS1 (i.e. downward resetting) with no further changes in the operating point pressure during SS2 under all three protocols. Moreover, perindopril significantly decreased the operating point pressures at SS1 and SS2 indicating that central Ang II was involved in establishing the operating point pressure of the ABR during dynamic exercise (Fig. 6 B and C).
Figure 6. Mean responses of blood pressure elected by perturbations of the carotid sinus baroreceptors at rest (A) and during both steady state dynamic exercise workloads SS1 (B) and SS2 (C).
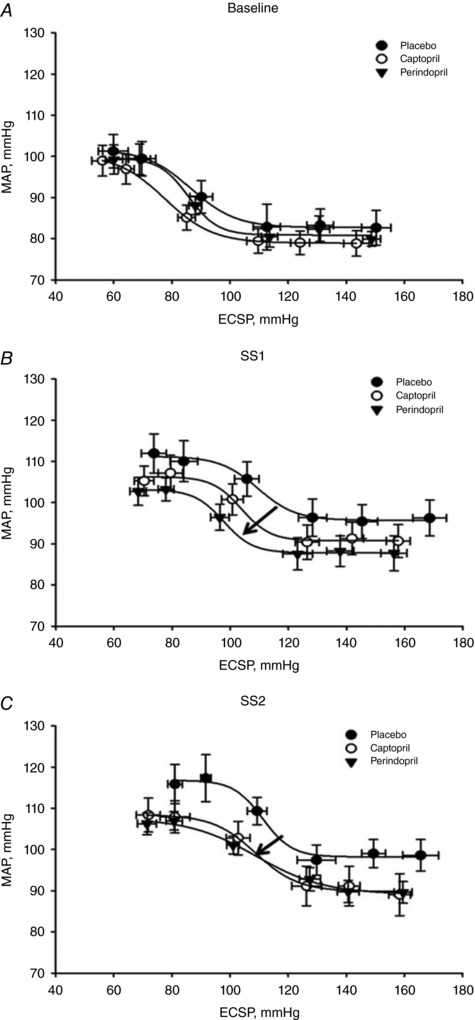
ECSP, estimated carotid sinus pressure. Arrows indicate the directional effect of ACEi on baroreflex resetting compared with placebo. Data were fitted with the logistic function to represent the mean response across each condition. Values are means ± SEM.
Table 2.
Carotid–MAP baroreflex function curve parameter responses to ACEi and exercise
| Baseline | Exercise SS1 | Exercise SS2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | PL | CAP | PER | PL | CAP | PER | PL | CAP | PER |
| Response range (mmHg) | 20 ± 2 | 23 ± 3 | 22 ± 2 | 18 ± 3 | 16 ± 2† | 17 ± 1† | 18 ± 2 | 19 ± 3 | 20 ± 1 |
| Max. gain (mmHg mmHg−1) | −0.7 ± 0.1 | −0.8 ± 0.1 | −1.1 ± 0.1 | −0.8 ± 0.1 | −0.8 ± 0.1 | −0.7 ± 0.1 | −0.5 ± 0.1 | −0.4 ± 0.1 | −0.3 ± 0.1 |
| Operating point (mmHg) | 94 ± 2 | 87 ± 3 | 89 ± 2 | 109 ± 2† | 102 ± 3† | 97 ± 3 * | 107 ± 3 | 103 ± 2 | 100 ± 2* |
| Threshold (mmHg) | 65 ± 7 | 54 ± 7 | 71 ± 6 | 90 ± 5† | 89 ± 6† | 79 ± 2 | 99 ± 7† | 93 ± 5† | 69 ± 6 *§ |
| Saturation (mmHg) | 111 ± 4 | 104 ± 5 | 101 ± 5 | 131 ± 3† | 120 ± 5† | 117 ± 4† | 129 ± 10 | 140 ± 7 | 147 ± 7‡ |
| Min. MAP response (mmHg) | 85 ± 3 | 80 ± 2 | 81 ± 3 | 99 ± 3† | 92 ± 2† | 88 ± 2 * | 96 ± 2 | 90 ± 4† | 88 ± 2† |
Values are means ± SEM; n = 9; PL, placebo; CAP, captopril; PER, perindopril; response range equals maximum minus minimum mean arterial pressure (MAP) response. At rest and during steady state exercise at workload HRs of 120 beats min−1 (SS1) and workload HRs of 150 beats min−1 (SS2): * P ≤ 0.05 compared to PL; § P ≤ 0.05 compared to CAP; † P ≤ 0.05 compared to baseline within condition; ‡ P ≤ 0.05 compared to SS1 within condition. Bold text highlight notable values of interest.
Discussion
The data obtained in this investigation provide new insights into the role of central Ang II production and its influence on the acute arterial baroreflex resetting of MSNA and the operating point pressure during acute and prolonged dynamic leg cycling exercise in humans. These findings identified that a centrally acting ACEi, perindopril, attenuated the steady state central outflow of MSNA by 26% during SS1 and 61% during SS2 compared to placebo and captopril (Fig. 3). The perindopril‐induced 26% reduction of MSNA bursts min−1 translated to an 11% reduction in the upward resetting of the ABR‐operating point pressure during the acute heavy exercise SS1 period. However, the translation of the 61% reduction in MSNA bursts min−1 during the prolonged very heavy exercise SS2 period resulted in a similar ABR‐operating point pressure to that observed in SS1.
The contribution of key inputs to ABR resetting have been established (Smith et al. 1976; Gallagher et al. 2001a, b; Ogoh et al. 2007) and include increased input to the nucleus of the tractus solitarius (NTS) from central command and exercise pressor reflex to enhance the sympathetic outflow and increase blood pressure (Victor et al. 1987; Potts, 2006). In contrast, increases in baroreceptor afferent input to the NTS will restrain sympathetic outflow and decrease blood pressure (Sheriff et al. 1990). Recent animal data have implicated NO• as a modulator of central sympathetic outflow. For example, introduction of central NO• dampens central sympathetic outflow (Patel et al. 2001; Gao et al. 2011) and there is a growing body of evidence that indicates that central NO• is scavenged by centrally generated [O2 •−] (Paton & Waki, 2009; Waki et al. 2011; Leal et al. 2012, 2013). Hence, during dynamic exercise, increases in centrally generated [O2 •−], formed by increased exercise workload‐related oxidative metabolism and central Ang II (Fallo, 1993; Bailey et al. 2004, 2011; Shim et al. 2008), suggest that increased production of central free radicals are the likely candidate signalling molecules involved in allowing the ABR to progressively reset with increasing exercise intensity. For example, NO• can be depleted by its reaction with [O2 •−] to generate peroxynitrite resulting in disinhibition within the NTS and rostral ventral lateral medulla and, subsequently, increased central sympathetic outflow. Studies performed in conscious animals support a general consensus that central NO• restrains sympathetic outflow and facilitates parasympathetic output, particularly when feedback from the baroreceptor reflex is controlled (Patel et al. 2001; Zucker et al. 2001). Zimmerman et al. demonstrated that [O2 •−] is a requirement for the central sympathetic response to Ang II in the mouse (Zimmerman et al. 2002, 2004). Furthermore, several groups have now established a link among Ang II, the AT1 receptor and [O2 •−] signalling in the central nervous system relating to the regulation of sympathetic nerve activity and arterial baroreflex function (Campese et al. 2005; Gao et al. 2005; Han et al. 2005; Fahim et al. 2012).
In this investigation, our data, comparing the resting baseline to the SS1 exercise conditions, suggest that central Ang II production is a prerequisite for the increase in central MSNA outflow and directly related to the increase in exercise workload. Subsequently, the increases in MSNA result in vasoconstriction of the blood vessels and the upward and rightward resetting of the ABR‐operating point pressure associated with exercise. By utilizing the permeability kinetics of a centrally acting ACEi, perindopril, we demonstrate that its central inhibition of Ang II production resulted in the greatest attenuation of the exercise‐induced resetting of ABR control of MSNA in SS1, when compared to placebo and captopril (Figs 2 and 3). Furthermore, even though MAP in the SS1 exercise was decreased with captopril compared to placebo, we suggest that this can be attributed to systemic ACE's inhibition of Ang II production, as there was no decrease in MSNA during exercise with captopril (Fig. 3). In fact, MSNA was slightly elevated with captopril compared to placebo, suggesting that the systemic effect of captopril elicited hypotension that is sensed by the ABR mediated response resulting in an increase in MSNA. However, this ABR response was not observed with perindopril, indicating that it was the central blocking and not the peripheral effects of perindopril that resulted in a manifested reduction of MSNA and the ABR‐operating point pressure (Fig. 6).
However, even though during the prolonged very heavy exercise period (SS2) perindopril reduced MSNA by 61% compared with placebo and 82% compared with captopril (Fig. 3 B), the expected significant greater blunting of the upward resetting of the steady state ABR‐operating point pressure was not manifested (Fig. 6 C). In addition, our hypothesized upward and rightward resetting of the ABR‐operating point pressure between SS1 and SS2 in the placebo condition was absent.
What explains the lack of resetting of the ABR‐operating point pressure during the transition from the SS1 to the SS2 workloads in the placebo condition (Fig. 6 C), despite the 43% increase in MSNA above the MSNA observed in SS1 (Fig. 3)? As noted in the Methods section, we decided to increase the exercise workloads from rest to SS1 and on to SS2 without the previously used 45 min recovery rest in between the change in workloads (Potts et al. 1993; Norton et al. 1999). In the current study, the continuous exercise protocol was performed in order to maintain the microneurography electrode's location established at the beginning of the experimental protocol. An unintended consequence of using the continuous exercise protocol was that the translation of the increased MSNA to an increased vasoconstriction between SS1 and SS2 was most likely blunted by the mechanisms of functional sympatholysis (Mitchell, 2017) associated with the cardiovascular drift of prolonged exercise (Ekelund, 1967; Norton et al. 1999). In the investigation by Norton et al. of cardiovascular drift, the downward drift of MAP was observed to be −10 to −12 mmHg resulting in a baroreflex‐mediated increase in HR of +10 to +12 beats min−1 at 40–60 min of continuous cycling exercise at 65% compared to the steady state MAP at 10 min (Norton et al. 1999). During the present investigation, the steady state measurement of MAP during SS1 was made at 10 min and during SS2 was made at 40 min of the continuous exercise. Hence, we conclude that the MAP of SS2 (workloads eliciting HRs of 150 beats min−1) was not significantly greater than the MAP at SS1 (workloads eliciting HRs of 120 beats min−1) and was the result of the downward drift in MAP associated with continuous steady state exercise (Ekelund, 1967; Norton et al. 1999).
Human studies utilizing selective anaesthetic (fentanyl) blockade of respiratory muscle afferents (Secher & Amann, 2012) and epidural anaesthesia (lidocaine/lignocaine) of leg skeletal muscle afferents (Smith et al. 2003) during exercise have yielded similar results to those reported in the current investigation. For example, epidural blockade causes the carotid baroreflex control of MAP function curves to reset downward to a lower operating point pressure (Smith et al. 2003) similar to the effect perindopril produced between SS1 and SS2 in the present investigation (Fig. 6). In the present study, analyses of the carotid baroreflex MAP reflex function curves during exercise, particularly SS1 (Fig. 6 B, Table 2), identified an attenuation of the MAP‐operating point and threshold pressures from rest to exercise with perindopril when compared to placebo and captopril. In Figs 3 A and 6 B, the central and peripheral acting ACEi, perindopril, selectively reduces the operating point pressure of MAP and MSNA during exercise compared to both placebo and the peripherally acting ACEi, captopril.
The magnitude of the reduction in the operating point pressure for ACEi at SS2 illustrates that perindopril and captopril were equally effective at attenuating the exercise‐induced resetting of the ABR (Fig. 6 C). However, the presence of a greater decrease in MSNA burst frequency with perindopril compared to captopril and placebo at SS1 and SS2 (Fig. 3) suggests that perindopril is acting both centrally and peripherally, whereas captopril is acting only peripherally on the vascular system (Fig. 3). Despite these marked differences in the amount of blockade of MSNA by perindopril compared with captopril and placebo, the reduction in the operating point pressure between SS1 and SS2 amounted to only a 10–12% reduction for perindopril compared with captopril and placebo (Fig. 6 C). Furthermore, there were no differences in the operating point pressures between SS1 and SS2 across either condition.
Perspectives
In humans, we proposed to identify the role of central and peripheral Ang II production on exercise‐induced arterial baroreflex resetting by using the differential lipid solubility of effective dose equivalents of ACE inhibitors. To our knowledge, the use of different lipid soluble ACEs on ABR resetting during exercise has not been demonstrated previously. Neither ACE inhibitor significantly decreased peripheral venous superoxide concentrations compared to placebo; however, perindopril significantly lowered the MSNA response to exercise. This may be explained by perindopril crossing the blood–brain barrier and decreasing central outflow of MSNA (Fig. 3) without altering systemic redox balance (Fig. 4). The findings of the present investigation suggest that central Ang II has a physiological role in ABR‐operating point pressure resetting and steady state MSNA during exercise.
Clinically, impaired autonomic control of sympathetic nerve activity leads to poor prognosis in cardiovascular morbidities such as hypertension, left ventricular hypertrophy, chronic heart failure, obstructive sleep apnoea, diabetes, obesity and normal ageing (Narkiewicz et al. 1998; Zucker et al. 2001; Nightingale et al. 2003; Monahan et al. 2004; Osborn et al. 2005; Xue et al. 2005; Paton et al. 2008). This investigation not only advances our understanding regarding the physiological mechanisms governing reflex cardiovascular control during exercise in healthy humans but also may provide insight into the patho‐physiological mechanisms of impaired autonomic regulation of cardiovascular responses such as exercise‐induced hypertension (EIHt). EIHt is a clinical disorder that has a 5 year prognosis for humans to develop essential hypertension. EIHt is caused by an impairment of the arterial baroreflex and is defined as a systolic arterial blood pressure (SAP) value exceeding the 90th percentile, equating to a SAP of greater than 210 mmHg for males and greater than 190 mmHg for females. In addition, EIHt has been linked to augmented exercise‐induced increases in Ang II (Shim et al. 2008). EIHt, at moderate exercise intensity, is an independent risk factor for cardiovascular events and mortality (Schultz et al. 2013). The results of the present investigation may provide physicians with a clear choice of treatment options dependent upon the primary central or peripheral cause of exercise‐induced hypertension. Hence, it is logical to investigate inhibition of central Ang II as an adjunctive therapy for the exercise‐induced augmented increase in blood pressure of EIHt patients.
Limitations
In this current investigation, circulating Ang II was not measured during exercise, before or after ACEi ingestion. It is well known that circulating Ang II inhibits baroreflex activity through interactions with AT1 receptors in the circumventricular organs (Sanderford & Bishop, 2002; Tan et al. 2007). The subfornical organ and organum vasculosum of the lamina terminalis are circumventricular organs which lack a functional blood–brain barrier and are sensitive to circulating Ang II (Osborn et al. 2007). The subfornical organ and organum vasculosum of the lamina terminalis innervate the median preoptic nucleus (Johnson & Gross, 1993), which projects to the paraventricular nucleus in the hypothalamus (Stocker & Toney, 2005) and modulates sympathetic outflow through the NTS and rostral ventral lateral medulla. In this current investigation, the doses of ACEi that we utilized have been reported to reduce plasma Ang II concentration by 31% with both 25 mg captopril measured at 1 h post‐ingestion (Hollenberg et al. 1981) and 4 mg of perindopril measured at 4 h post‐ingestion (Bussien et al. 1986). Furthermore, several animal studies have confirmed that an oral dosage of perindopril is a centrally active ACE inhibitor (Yamada et al. 2010, 2011; Dong et al. 2011). In one study, the brain ACE activities in mice were decreased by more than 50% with an oral dosage of perindopril when compared to other non‐centrally active ACE inhibitors (Yamada et al. 2010). Another limitation is not knowing central Ang II or free radical concentrations. In human subjects, these measurements would require arterial–venous differences across the brain. As an initial step, we chose to identify the involvement of the CNS by having captopril act as a peripheral control for perindopril and attenuating the circulating Ang II peripherally to the same extent before exercise. Hence, any changes in central sympathetic outflow can be attributed to the centrally acting ACEi acting on the central renin–angiotensin system.
Another limitation of this study is only having three female participants and only one in the MSNA data. A body of literature has identified sex differences in neural vascular control in men and women. Hart & Charkoudian identify that in young women, the influence of the β‐adrenergic receptors significantly offsets the relationship between MSNA and vasoconstriction (Hart & Charkoudian, 2014). The current study utilized both male and female volunteers, but the ratio of male to female subjects was large (11:3) for the haemodynamic data and for the MSNA data (6:1), so we did not anticipate significant differences in the overall physiological conclusions of the present study.
Conclusions
The question addressed in this investigation was whether generation of central Ang II during exercise mediates the increases in sympathetic outflow and, therefore, the arterial baroreflex operating point pressure resetting during acute and prolonged dynamic exercise. The main finding identifies that, indeed, central Ang II production plays a role in the reflex control of central sympathetic nerve activity during acute and prolonged dynamic exercise. While the peripheral free radical concentrations did not change with perindopril or captopril from these data, we conclude that the centrally acting ACEi, perindopril, attenuated the exercise related increase in central Ang II resulting in a dampened response of sympathetic nerve activity outflow during dynamic exercise.
Additional information
Competing interests
None declared.
Author contributions
Conception and design: G.M., M.C.Z., P.B. and P.B.R.; analysis and interpretation of data: G.M., N.P.J., J.T., M.C.Z. and P.B.R.; drafting the article or revising it critically for important intellectual content: G.M., N.P.J., J.T., M.C.Z., P.B. and P.B.R. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. All experiments were carried out in Dr. Peter Raven's laboratory at the University of North Texas Health Science Center in Fort Worth, TX, USA.
Funding
ESR spectroscopy was performed by the University of Nebraska Medical Center's ESR Spectroscopy Core, which is supported, in part, by a grant from the National Institute of General Medical Sciences of the National Institutes of Health (P30GM103335) awarded to the University of Nebraska's Redox Biology Center. Intramural Funding was provided by the University of North Texas Health Science Center's Office of Research and the Institute of Cardiovascular and Metabolic Diseases. G.M. (2014) was awarded the Texas ACSM 2014 Student Research Award and partially supported the completion of this project. Currently, G.M. is a postdoctoral fellow at The Institute of Exercise and Environmental Medicine at the University of Texas Southwestern Medical Center and Presbyterian Hospital in Dallas.
Acknowledgements
The investigators wish to acknowledge the subjects that volunteered their time and effort in participating and adhering to the stringent requirements necessary to complete the investigation.
Biography
Gilbert Moralez received his PhD in Biomedical Sciences with an emphasis on Integrative Physiology (2016) from the University of North Texas Health Science Centre, Fort Worth, Texas under the supervision of Professor Peter B. Raven. Subsequently, Gilbert has been working as a postdoctoral fellow at the University of Texas Southwestern Medical Centre and the Institute for Exercise and Environmental Medicine, Dallas, Texas under the guidance of Professor Craig G. Crandall. His research focuses on the signalling mechanisms regulating neural control of cardiovascular responses to exercise and environmental stressors in healthy and clinical populations.

Edited by: Harold Schultz & Julie Chan
References
- Agabiti‐Rosei E, Ambrosioni E, Finardi G, Folino P, Gambassi G, Malini P, Marchesi E, Muiesan ML, Semplicini A & Pessina AC (1992). Perindopril versus captopril: efficacy and acceptability in an Italian multicenter trial. Am J Med 92, 79S–83S. [DOI] [PubMed] [Google Scholar]
- Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic‐Haberle I, White CR & Freeman BA (2001). Oxygen radical inhibition of nitric oxide‐dependent vascular function in sickle cell disease. Proc Natl Acad Sci USA 98, 15215–15220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åstrand P‐O & Rodahl K (1986). Textbook of Work Physiology: Physiological Bases of Exercise. McGraw‐Hill, New York, London. [Google Scholar]
- Bailey DM, Evans KA, McEneny J, Young IS, Hullin DA, James PE, Ogoh S, Ainslie PN, Lucchesi C, Rockenbauer A, Culcasi M & Pietri S (2011). Exercise‐induced oxidative‐nitrosative stress is associated with impaired dynamic cerebral autoregulation and blood‐brain barrier leakage. Exp Physiol 96, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Young IS, McEneny J, Lawrenson L, Kim J, Barden J & Richardson RS (2004). Regulation of free radical outflow from an isolated muscle bed in exercising humans. Am J Physiol Heart Circ Physiol 287, H1689–H1699. [DOI] [PubMed] [Google Scholar]
- Bussien JP, d'Amore TF, Perret L, Porchet M, Nussberger J, Waeber B & Brunner HR (1986). Single and repeated dosing of the converting enzyme inhibitor perindopril to normal subjects. Clin Pharmacol Ther 39, 554–558. [DOI] [PubMed] [Google Scholar]
- Campese VM, Shaohua Y & Huiquin Z (2005). Oxidative stress mediates angiotensin II‐dependent stimulation of sympathetic nerve activity. Hypertension 46, 533–539. [DOI] [PubMed] [Google Scholar]
- Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC & Chan JY (2005). NADPH oxidase‐derived superoxide anion mediates angiotensin II‐induced pressor effect via activation of p38 mitogen‐activated protein kinase in the rostral ventrolateral medulla. Circ Res 97, 772–780. [DOI] [PubMed] [Google Scholar]
- Chik Z, Basu RC, Pendek R, Lee TC & Mohamed Z (2010). A bioequivalence comparison of two formulations of rifampicin (300‐ vs 150‐mg capsules): An open‐label, randomized, two‐treatment, two‐way crossover study in healthy volunteers. Clin Ther 32, 1822–1831. [DOI] [PubMed] [Google Scholar]
- Cooke WH, Rickards CA, Ryan KL, Kuusela TA & Convertino VA (2009). Muscle sympathetic nerve activity during intense lower body negative pressure to presyncope in humans. J Physiol 587, 4987–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry TB & Charkoudian N (2011). The use of real‐time ultrasound in microneurography. Auton Neurosci 162, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo SH, Fisher JP, Vianna LC, Kim A, Chockalingam A, Zimmerman MC, Zucker IH & Fadel PJ (2012). Statin therapy lowers muscle sympathetic nerve activity and oxidative stress in patients with heart failure. Am J Physiol Heart Circ Physiol 303, H377–H385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YF, Kataoka K, Tokutomi Y, Nako H, Nakamura T, Toyama K, Sueta D, Koibuchi N, Yamamoto E, Ogawa H & Kim‐Mitsuyama S (2011). Perindopril, a centrally active angiotensin‐converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer's disease. FASEB J 25, 2911–2920. [DOI] [PubMed] [Google Scholar]
- Ekelund LG (1967). Circulatory and respiratory adaptation during prolonged exercise of moderate intensity in the sitting position. Acta Physiol Scand 69, 327–340. [DOI] [PubMed] [Google Scholar]
- Fadel PJ & Raven PB (2012). Human investigations into the arterial and cardiopulmonary baroreflexes during exercise. Exp Physiol 97, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahim M, Gao L, Mousa TM, Liu D, Cornish KG & Zucker IH (2012). Abnormal baroreflex function is dissociated from central angiotensin II receptor expression in chronic heart failure. Shock 37, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallo F (1993). Renin‐angiotensin‐aldosterone system and physical exercise. J Sports Med Phys Fitness 33, 306–312. [PubMed] [Google Scholar]
- Fisher JP & Fadel PJ (2010). Therapeutic strategies for targeting excessive central sympathetic activation in human hypertension. Exp Physiol 95, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KM, Fadel PJ, Stromstad M, Ide K, Smith SA, Querry RG, Raven PB & Secher NH (2001a). Effects of exercise pressor reflex activation on carotid baroreflex function during exercise in humans. J Physiol 533, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KM, Fadel PJ, Stromstad M, Ide K, Smith SA, Querry RG, Raven PB & Secher NH (2001b). Effects of partial neuromuscular blockade on carotid baroreflex function during exercise in humans. J Physiol 533, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Zhang H, Le KD, Chao J & Gao L (2011). Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am J Hypertens 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG & Zucker IH (2005). Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol 288, H2271–H2279. [DOI] [PubMed] [Google Scholar]
- Hagbarth KE & Vallbo AB (1968). Discharge characteristics of human muscle afferents during muscle stretch and contraction. Exp Neurol 22, 674–694. [DOI] [PubMed] [Google Scholar]
- Han Y, Zhang Y, Wang HJ, Gao XY, Wang W & Zhu GQ (2005). Reactive oxygen species in paraventricular nucleus modulates cardiac sympathetic afferent reflex in rats. Brain Res 1058, 82–90. [DOI] [PubMed] [Google Scholar]
- Hart EC & Charkoudian N (2014). Sympathetic neural regulation of blood pressure: influences of sex and aging. Physiology (Bethesda) 29, 8–15. [DOI] [PubMed] [Google Scholar]
- Hollenberg NK, Meggs LG, Williams GH, Katz J, Garnic JD & Harrington DP (1981). Sodium intake and renal responses to captopril in normal man and in essential hypertension. Kidney Int 20, 240–245. [DOI] [PubMed] [Google Scholar]
- Jankowski A, Skorek A, Krzysko K, Zarzycki PK, Ochocka RJ & Lamparczyk H (1995). Captopril: determination in blood and pharmacokinetics after single oral dose. J Pharm Biomed Anal 13, 655–660. [DOI] [PubMed] [Google Scholar]
- Jansen JR, Wesseling KH, Settels JJ & Schreuder JJ (1990). Continuous cardiac output monitoring by pulse contour during cardiac surgery. Eur Heart J 11 (Suppl. I), 26–32. [DOI] [PubMed] [Google Scholar]
- Jiang H, Rummage JA, Stewart CA, Herriott MJ, Kolosova I, Kolosov M & Leu RW (1996). Evidence for endogenous C1q modulates TNF‐α receptor synthesis and autocrine binding of TNF‐α associated with lipid A activation of murine macrophages for nitric oxide production. Cell Immunol 170, 34–40. [DOI] [PubMed] [Google Scholar]
- Johnson AK & Gross PM (1993). Sensory circumventricular organs and brain homeostatic pathways. FASEB J 7, 678–686. [DOI] [PubMed] [Google Scholar]
- Kent BB, Drane JW, Blumenstein B & Manning JW (1972). A mathematical model to assess changes in the baroreceptor reflex. Cardiology 57, 295–310. [DOI] [PubMed] [Google Scholar]
- Leal AK, Mitchell JH & Smith SA (2013). Treatment of muscle mechanoreflex dysfunction in hypertension: effects of l‐arginine dialysis in the nucleus tractus solitarii. Exp Physiol 98, 1337–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal AK, Murphy MN, Iwamoto GA, Mitchell JH & Smith SA (2012). A role for nitric oxide within the nucleus tractus solitarii in the development of muscle mechanoreflex dysfunction in hypertension. Exp Physiol 97, 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR & Reid JL (1987). Haemodynamic and humoral effects of oral perindopril, an angiotensin converting enzyme inhibitor, in man. Br J Clin Pharmacol 23, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowall LM & Dampney RA (2006). Calculation of threshold and saturation points of sigmoidal baroreflex function curves. Am J Physiol Heart Circ Physiol 291, H2003–H2007. [DOI] [PubMed] [Google Scholar]
- Melcher A & Donald DE (1981). Maintained ability of carotid baroreflex to regulate arterial pressure during exercise. Am J Physiol 241, H838–H849. [DOI] [PubMed] [Google Scholar]
- Migdalof BH, Antonaccio MJ, McKinstry DN, Singhvi SM, Lan SJ, Egli P & Kripalani KJ (1984). Captopril: pharmacology, metabolism and disposition. Drug Metab Rev 15, 841–869. [DOI] [PubMed] [Google Scholar]
- Mitchell JH (1990). J.B. Wolffe memorial lecture. Neural control of the circulation during exercise. Med Sci Sports Exerc 22, 141–154. [PubMed] [Google Scholar]
- Mitchell JH (2017). Abnormal cardiovascular response to exercise in hypertension: contribution of neural factors. Am J Physiol Regul Integr Comp Physiol 312, R851–R863. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Eskurza I & Seals DR (2004). Ascorbic acid increases cardiovagal baroreflex sensitivity in healthy older men. Am J Physiol Heart Circ Physiol 286, H2113–H2117. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, Kato M, Phillips BG, Davison DE & Somers VK (1998). Baroreflex control of sympathetic nerve activity and heart rate in obstructive sleep apnea. Hypertension 32, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Nightingale AK, Blackman DJ, Field R, Glover NJ, Pegge N, Mumford C, Schmitt M, Ellis GR, Morris‐Thurgood JA & Frenneaux MP (2003). Role of nitric oxide and oxidative stress in baroreceptor dysfunction in patients with chronic heart failure. Clin Sci (Lond) 104, 529–535. [DOI] [PubMed] [Google Scholar]
- Norton KH, Boushel R, Strange S, Saltin B & Raven PB (1999). Resetting of the carotid arterial baroreflex during dynamic exercise in humans. J Appl Physiol (1985) 87, 332–338. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Fadel PJ, Nissen P, Jans O, Selmer C, Secher NH & Raven PB (2003). Baroreflex‐mediated changes in cardiac output and vascular conductance in response to alterations in carotid sinus pressure during exercise in humans. J Physiol 550, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoh S, Fisher JP, Fadel PJ & Raven PB (2007). Increases in central blood volume modulate carotid baroreflex resetting during dynamic exercise in humans. J Physiol 581, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn JW, Fink GD, Sved AF, Toney GM & Raizada MK (2007). Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9, 228–235. [DOI] [PubMed] [Google Scholar]
- Osborn JW, Jacob F & Guzman P (2005). A neural set point for the long‐term control of arterial pressure: beyond the arterial baroreceptor reflex. Am J Physiol Regul Integr Comp Physiol 288, R846–R855. [DOI] [PubMed] [Google Scholar]
- Papelier Y, Escourrou P, Gauthier JP & Rowell LB (1994). Carotid baroreflex control of blood pressure and heart rate in men during dynamic exercise. J Appl Physiol (1985) 77, 502–506. [DOI] [PubMed] [Google Scholar]
- Patel KP, Li YF & Hirooka Y (2001). Role of nitric oxide in central sympathetic outflow. Exp Biol Med (Maywood) 226, 814–824. [DOI] [PubMed] [Google Scholar]
- Paton JF & Waki H (2009). Is neurogenic hypertension related to vascular inflammation of the brainstem? Neurosci Biobehav Rev 33, 89–94. [DOI] [PubMed] [Google Scholar]
- Paton JF, Wang S, Polson JW & Kasparov S (2008). Signalling across the blood brain barrier by angiotensin II: novel implications for neurogenic hypertension. J Mol Med 86, 705–710. [DOI] [PubMed] [Google Scholar]
- Potts JT ( 2006). Inhibitory neurotransmission in the nucleus tractus solitarii: implications for baroreflex resetting during exercise. Exp Physiol 91, 59–72. [DOI] [PubMed] [Google Scholar]
- Potts JT, Shi XR & Raven PB (1993). Carotid baroreflex responsiveness during dynamic exercise in humans. Am J Physiol 265, H1928–H1938. [DOI] [PubMed] [Google Scholar]
- Powers SK, Radak Z & Ji LL (2016). Exercise‐induced oxidative stress: past, present and future. J Physiol 594, 5081–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querry RG, Smith SA, Stromstad M, Ide K, Secher NH & Raven PB (2001). Anatomical and functional characteristics of carotid sinus stimulation in humans. Am J Physiol Heart Circ Physiol 280, H2390–H2398. [DOI] [PubMed] [Google Scholar]
- Rowell LB & O'Leary DS (1990). Reflex control of the circulation during exercise: chemoreflexes and mechanoreflexes. J Appl Physiol (1985) 69, 407–418. [DOI] [PubMed] [Google Scholar]
- Sanderford MG & Bishop VS (2002). Central mechanisms of acute ANG II modulation of arterial baroreflex control of renal sympathetic nerve activity. Am J Physiol Heart Circ Physiol 282, H1592–H1602. [DOI] [PubMed] [Google Scholar]
- Schultz MG, Otahal P, Cleland VJ, Blizzard L, Marwick TH & Sharman JE (2013). Exercise‐induced hypertension, cardiovascular events, and mortality in patients undergoing exercise stress testing: a systematic review and meta‐analysis. Am J Hypertens 26, 357–366. [DOI] [PubMed] [Google Scholar]
- Secher NH & Amann M (2012). Human investigations into the exercise pressor reflex. Exp Physiol 97, 59–69. [DOI] [PubMed] [Google Scholar]
- Sheriff DD, O'Leary DS, Scher AM & Rowell LB (1990). Baroreflex attenuates pressor response to graded muscle ischemia in exercising dogs. Am J Physiol 258, H305–H310. [DOI] [PubMed] [Google Scholar]
- Shim CY, Ha JW, Park S, Choi EY, Choi D, Rim SJ & Chung N (2008). Exaggerated blood pressure response to exercise is associated with augmented rise of angiotensin II during exercise. J Am Coll Cardiol 52, 287–292. [DOI] [PubMed] [Google Scholar]
- Smith EE, Guyton AC, Manning RD & White RJ (1976). Integrated mechanisms of cardiovascular response and control during exercise in the normal human. Prog Cardiovasc Dis 18, 421–444. [DOI] [PubMed] [Google Scholar]
- Smith SA, Querry RG, Fadel PJ, Gallagher KM, Stromstad M, Ide K, Raven PB & Secher NH (2003). Partial blockade of skeletal muscle somatosensory afferents attenuates baroreflex resetting during exercise in humans. J Physiol 551, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD & Toney GM (2005). Median preoptic neurones projecting to the hypothalamic paraventricular nucleus respond to osmotic, circulating Ang II and baroreceptor input in the rat. J Physiol 568, 599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PS, Killinger S, Horiuchi J & Dampney RA (2007). Baroreceptor reflex modulation by circulating angiotensin II is mediated by AT1 receptors in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 293, R2267–R2278. [DOI] [PubMed] [Google Scholar]
- Unger T, Fleck T, Ganten D, Lang RE & Rettig F (1984). 2‐[N‐[(S)‐1‐ethoxycarbonyl‐3‐phenylpropyl]‐L‐alanyl]‐(1S,3S,5S)‐2‐ azabicyclo[3.3.0]octane‐3‐carboxylic acid (Hoe 498): antihypertensive action and persistent inhibition of tissue converting enzyme activity in spontaneously hypertensive rats. Arzneimittelforschung 34, 1426–1430. [PubMed] [Google Scholar]
- Unger T, Yukimura T, Marin‐Grez M, Lang RE, Rascher W & Ganten D (1982). SA446, a new orally active converting enzyme inhibitor: antihypertensive action and comparison with captopril in stroke‐prone spontaneously hypersensitive rats. Eur J Pharmacol 78, 411–420. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE & Wallin BG (2004). Microneurography: how the technique developed and its role in the investigation of the sympathetic nervous system. J Appl Physiol (1985) 96, 1262–1269. [DOI] [PubMed] [Google Scholar]
- Vianna LC, Deo SH, Jensen AK, Holwerda SW, Zimmerman MC & Fadel PJ (2015). Impaired dynamic cerebral autoregulation at rest and during isometric exercise in type 2 diabetes patients. Am J Physiology Heart Circ Physiol 308, H681–H687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor RG, Seals DR & Mark AL (1987). Differential control of heart rate and sympathetic nerve activity during dynamic exercise. Insight from intraneural recordings in humans. J Clin Invest 79, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M & Paton JF (2008). Specific inflammatory condition in nucleus tractus solitarii of the SHR: novel insight for neurogenic hypertension? Auton Neurosci 142, 25–31. [DOI] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M, Raizada MK & Paton JF (2011). Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol 178, 422–428. [DOI] [PubMed] [Google Scholar]
- Walgenbach SC & Donald DE (1983). Cardiopulmonary reflexes and arterial pressure during rest and exercise in dogs. Am J Physiol 244, H362–H369. [DOI] [PubMed] [Google Scholar]
- Wallin BG, Burke D & Gandevia S (1994). Coupling between variations in strength and baroreflex latency of sympathetic discharges in human muscle nerves. J Physiol 474, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DW, Shoemaker JK & Raven PB (2015). Methods and considerations for the analysis and standardization of assessing muscle sympathetic nerve activity in humans. Auton Neurosci 193, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Pamidimukkala J & Hay M (2005). Sex differences in the development of angiotensin II‐induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol 288, H2177–H2184. [DOI] [PubMed] [Google Scholar]
- Yamada K, Horita T, Takayama M, Takahashi S, Takaba K, Nagata Y, Suzuki N & Kanda T (2011). Effect of a centrally active angiotensin converting enzyme inhibitor, perindopril, on cognitive performance in chronic cerebral hypo‐perfusion rats. Brain Res 1421, 110–120. [DOI] [PubMed] [Google Scholar]
- Yamada K, Uchida S, Takahashi S, Takayama M, Nagata Y, Suzuki N, Shirakura S & Kanda T (2010). Effect of a centrally active angiotensin‐converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer's disease. Brain Res 1352, 176–186. [DOI] [PubMed] [Google Scholar]
- Yamauchi M & Kimura H (2008). Oxidative stress in obstructive sleep apnea: putative pathways to the cardiovascular complications. Antioxid Redox Signal 10, 755–768. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR & Davisson RL (2002). Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91, 1038–1045. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV & Davisson RL (2004). Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95, 210–216. [DOI] [PubMed] [Google Scholar]
- Zucker IH (2006). Novel mechanisms of sympathetic regulation in chronic heart failure. Hypertension 48, 1005–1011. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Wang W, Pliquett RU, Liu JL & Patel KP (2001). The regulation of sympathetic outflow in heart failure. The roles of angiotensin II, nitric oxide, and exercise training. Ann N Y Acad Sci 940, 431–443. [PubMed] [Google Scholar]