

Abstract
Pierisin-1 is a potent apoptosis-inducing protein derived from the cabbage butterfly, Pieris rapae. It has been shown that pierisin-1 has an A⋅B structure–function organization like cholera or diphtheria toxin, where the “A” domain (N-terminal) exhibits ADP-ribosyltransferase activity. The present studies were designed to identify the target molecule for ADP-ribosylation by pierisin-1 in the presence of β-[adenylate-32P]NAD, and we found DNA as the acceptor, but not protein as is the case with other bacteria-derived ADP-ribosylating toxins. ADP-ribosylation of tRNAs from yeast was also catalyzed by pierisin-1, but the efficiency was around 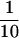 of that for calf thymus DNA. Pierisin-1 efficiently catalyzed the ADP-ribosylation of double-stranded DNA containing dG⋅dC, but not dA⋅dT pairs. The ADP-ribose moiety of NAD was transferred to the amino group at N2 of 2′-deoxyguanosine to yield N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form, which were determined by several spectral analyses including 1H- and 13C-NMR and mass spectrometry. The chemical structures were also ascertained by the independent synthesis of N2-(D-ribos-1-yl)-2′-deoxyguanosine, which is the characteristic moiety of ADP-ribosylated dG. Using the 32P-postlabeling method, ADP-ribosylated dG could be detected in DNA from pierisin-1-treated HeLa cells, in which apoptosis was easily induced. Thus, the targets for ADP-ribosylation by pierisin-1 were concluded to be 2′-deoxyguanosine residues in DNA. This finding may open a new field regarding the biological significance of ADP-ribosylation.
of that for calf thymus DNA. Pierisin-1 efficiently catalyzed the ADP-ribosylation of double-stranded DNA containing dG⋅dC, but not dA⋅dT pairs. The ADP-ribose moiety of NAD was transferred to the amino group at N2 of 2′-deoxyguanosine to yield N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form, which were determined by several spectral analyses including 1H- and 13C-NMR and mass spectrometry. The chemical structures were also ascertained by the independent synthesis of N2-(D-ribos-1-yl)-2′-deoxyguanosine, which is the characteristic moiety of ADP-ribosylated dG. Using the 32P-postlabeling method, ADP-ribosylated dG could be detected in DNA from pierisin-1-treated HeLa cells, in which apoptosis was easily induced. Thus, the targets for ADP-ribosylation by pierisin-1 were concluded to be 2′-deoxyguanosine residues in DNA. This finding may open a new field regarding the biological significance of ADP-ribosylation.
Pierisin-1, a 98-kDa protein, is abundantly present in the fifth instar larvae and the early phase of pupae in the cabbage butterfly, Pieris rapae (1, 2). This protein has potent cytotoxic activity against various human cancer cell lines with a wide range of IC50 values, inducing typical apoptotic cell death with characteristic morphological features, DNA fragmentation, and cleavage of poly(ADP-ribose) polymerase (2, 3). The N-terminal region of pierisin-1 has a partial regional sequence similarity with ADP-ribosylating toxins such as the A-subunit of cholera toxin, and disruption of this possible NAD-binding site by site-directed mutagenesis abolishes its apoptosis-inducing activity (4). Inhibitors of ADP-ribosyltransferase also significantly reduce its ability to induce apoptosis of cancer cells. Recently, we found that pierisin-1 is likely to have an A⋅B structure–function organization similar to that of cholera and diphtheria toxins, the “A” domain featuring the ADP-ribosyltransferase activity and the “B” domain binding to receptors in cell membranes, thereby incorporating the A domain into cells for ADP-ribosylation of intracellular target molecules. It is also suggested that the glycolipid in the membrane was a possible candidate for the receptor of pierisin-1, and the sensitivity of cancer cells to pierisin-1 depended on the presence of the receptor in cell membrane. Moreover, treatment using either the N-terminal polypeptide (amino acids 1–233) or the C-terminal polypeptide (amino acids 234–850) of pierisin-1 did not show any cytotoxicity. On the other hand, the combined use of both N- and C-terminal polypeptides or direct incorporation of the N-terminal alone by electroporation induced cell death in HeLa cells (5). Thus, pierisin-1 appears to belong to ADP-ribosylating toxins that exert their action by ADP-ribosyltransferase.
ADP-ribosylation is generally known to be a posttranslational modification where the ADP-ribose moiety of β-NAD is transferred to specific proteins (6). Several types of bacteria have been shown to produce mono(ADP-ribosyl)transferase the acceptors of which are usually specific amino acid residues in proteins in eukaryotic cells (7). Cholera toxin ADP-ribosylates arginine residues in G proteins, whereas pertussis toxin ADP-ribosylates a cysteine residue. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A use diphthamide, a modified histidine, as the acceptor amino acid. Clostridium botulinum C3 exoenzyme is an asparagine-specific ADP-ribosyltransferase. Thus, mono(ADP-ribosyl)ation reactions occur at nitrogen or sulfur atoms in different amino acids to produce N- or S-glycosides.
Identification of the target molecule for ADP-ribosylation by pierisin-1 could provide key information to understand its biological actions in the cabbage butterfly. In the present study, we sought to determine the target molecules of mono(ADP-ribosyl)ation by pierisin-1 and the chemical structures of mono(ADP-ribosyl)ated products. The results showed, for the first time, that the acceptor for mono(ADP-ribosyl)ation catalyzed by pierisin-1 is DNA, but not a protein. Mono(ADP-ribosyl)ated products could be isolated by HPLC from DNA treated with pierisin-1 in the presence of β-NAD, and their structures determined. The possible biological significance of mono(ADP-ribosyl)ation of DNA by this insect-derived ADP-ribosylating toxin is also discussed.
Materials and Methods
Materials.
2′-Deoxyguanosine (dG), β-NAD, bovine intestinal and bacterial alkaline phosphatases, calf thymus DNA, tRNAs from bakers yeast, cholera toxin A-subunit, diphtheria toxin, and pertussis toxin were obtained from Sigma. Pronase (1000 units/mg) was from Kaken Seiyaku (Tokyo). Micrococcal nuclease and phosphodiesterase I (Crotalus adamanteus venom) and II (bovine spleen) were obtained from Worthington. β-[Adenylate-32P]NAD ([32P]NAD) was purchased from NEN. [γ-32P]ATP was from ICN, and T4 polynucleotide kinase from Takara Shuzo Co. (Kyoto). Nuclease P1 was obtained from Yamasa Shoyu (Choshi, Japan). The oligonucleotides used in this study were purchased from Sawady (Tokyo). Polyethyleneimine (PEI)-cellulose TLC sheet (Polygram CEL 300 PEI) was from Macherey-Nagel (Duren, Germany).
Analysis of Transfer of Radioactivity from [32P]NAD in the Presence of Pierisin-1 or Other ADP-Ribosylating Toxins.
Pierisin-1 was purified from the pupae of P. rapae, as described (2), and aliquots (100 μg) were treated with 20 μg of Pronase for 4 h at 37°C, to cleave the protein into an N-terminal region with the ADP-ribosyltransferase domain and a C-terminal region with the receptor-binding domain (4). The Pronase-treated pierisin-1 catalyzed a more than 10-fold higher transfer of radioactivity from [32P]NAD to cell extracts than nontreated pierisin-1. Therefore, Pronase-treated pierisin-1 was used in all of the following experiments, except in the analysis of DNA adduct formation in HeLa cells by using the 32P-postlabeling method.
Human cervical carcinoma HeLa cells (RIKEN cell bank, Tsukuba, Japan) were cultured as reported (4), homogenized, and subjected to subcellular fraction by differential centrifugation (8). These samples were used directly or after sonication to disrupt organelles. Aliquots were reacted with 2.5 μg of Pronase-treated pierisin-1, 1 μCi (1 Ci = 37 GBq) of [32P]NAD and 1 μM of nonradioactive β-NAD in 50 mM Tris⋅HCl, pH 7.5, for 30 min at 37°C in a total volume of 50 μl. The reaction mixture was then treated with trichloroacetic acid and subjected to SDS/PAGE and autoradiography analysis.
When DNA, RNA, and polypeptides were used as acceptor molecules, 10 μg of a test sample was incubated with 1.0 μg of Pronase-treated pierisin-1 and 0.1 mM β-NAD containing 0.01 μCi of [32P]NAD and 1 mM EDTA in 50 mM Tris⋅HCl, pH 7.5, for 30 min at 37°C in a total volume of 10 μl. Incorporation of radioactivity from [32P]NAD by other ADP-ribosylating toxins was analyzed as described (9–12).
Isolation and Structural Analyses of Reaction Products Formed by Incubation of Nucleobase and β-NAD in the Presence of Pierisin-1.
Ten microliters of 40 mM 2′-deoxyguanosine or 1% calf thymus DNA and 2 μl of 0.1 M β-NAD were mixed with 1 μl of Pronase-treated piersin-1 (1 mg/ml) in 50 mM Tris⋅HCl buffer containing 1 mM EDTA at pH 7.5 in a total volume 100 μl and incubated at 37°C overnight. The reaction mixture for dG was then centrifuged and the supernatant was subjected to HPLC consisting of a Shimadzu LC-10A HPLC system (Shimadzu, Kyoto) equipped with a Shimadzu SPD 10Avp photodiode array detector and a column of Tosoh TSK-Gel octadecylsilyl (ODS) 80Ts (4.6 × 250 mm; Tosoh, Tokyo) at a flow rate of 1 ml/min. The solvent system was as follows: linear gradient from 3.5% acetonitrile to 15% acetonitrile over 45 min in 0.5% triethylamin⋅ acetic acid (pH 7.0).
In the case of the reaction mixture for DNA, DNA was recovered with 8 M ammonium acetate and ethanol and digested with a nuclease mix (micrococcal nuclease at 200 units/ml and phosphodiesterase II at 2 units/ml) in 20 mM Bis-Tris/10 mM HCl buffer at pH 6.5 at 37°C for 4 h. Then, zinc(II) chloride (final concentration 5 mM) and bacterial alkaline phosphatase (1.5 units/ml) were added to the reaction solution with further incubation at pH 8.0 at 37°C for 4 h. After centrifugation, the reaction mixture was then subjected to HPLC analyses on an ODS 80Ts column and eluted with the following system: 3.5% acetonitrile in 0.5% triethylamine-acetic acid (pH 7.0) from 0–20 min, and then a linear gradient to 15% acetonitrile in 0.5% triethylamine-acetic acid (pH 7.0) to 35 min.
The reaction products thus obtained were analyzed by electrospray ionization–mass spectrometry (ESI-MS) using a JEOL JMS-7000Q instrument equipped with an HP 1100-HPLC system, and also by 1H-NMR, 13C-NMR, 1H-1H-correlated spectroscopy (2D-COSY), and heteronuclear multiple quantum coherence (HMQC) using a JEOL 600α (600 MHz) or JEOL 500α (500 MHz). Chemical shifts were reported in parts per million (ppm) downfield from the internal tetramethylsilane (0.00 ppm). J values were given in Hz. The spectral data of the reaction product, N2-(ADP-ribos-1-yl)-2′-deoxyguanosine (α:β = 1:1), are shown below. 1H-NMR (DMSO-d6, 600 MHz): 8.41 (s, H-8 A), 8.13 (s, H-2 A), 7.95 [s, NH(β)], 7.92 (s, H-8 G), 7.21 (s, NH2 A), 7.16 [d, J = 9, NH(α)], 6.17 [t, J = 6.6, H-1′ dR(G)], 5.90 [d, J = 4.8, H-1′ R(A)], 5.75 [m, H-1" R(α)], 5.59 [d, J = 9, H-1" R (β)], 4.52 [m, H-2′ R(A)], 4.38 [m, H-3" R(α)], 4.33 [m, H-3′ dR(G)], 4.29 [m, H-3′ R(A)], 4.13 (m, H-2" R), 4.09 [m, H-3" R(β)], 4.04 (m, H-4" R), 4.00 (m, H-5‴, 5⁗ R), 3.90 [d, J = 11, H-5′, 5" R(A)], 3.86 [m, H-4′ R(A)], 3.81 [m, H-4′ dR(G)], 3.51 [dd, J = ≈12, H-5′, 5" dR(G)], 2.85 [m, H-2" dR/(G)], 2.22 [m, H-2′ dR(G)]; 13C-NMR (DMSO-d6, 125 MHz): 156.75 (G-C6), 156.59 (A-C6), 155.93 (G-C2), 152.81 (G-C4), 152.60 (A-C2), 151.69 (A-C4), 139.1 (A-C8), 135.6 (G-C8), 119.50 (A-C5), 117.10 (G-C5), 87.60 [R(A)1′], 87.07 [R1"(β)], 82.83 [dR(G)4′, dR(G)1′], 82.22 [R4"(A), R4‴], 86.07 [R1"(α)], 75.16 (R2"), 73.87 [R(A)2′], 70.89 [R3", dR(G)3′], 69.99 [R5‴, R(A)3′], 64.50 [R(A)5′], 61.79 [dR(G)5′], ≈40 [dR(G)2′]; ESI-MS (+): 910 (M + H+ + TEA), 809 (M + H+), 693.
Enzymatic Hydrolysis of ADP-Ribosylated dG.
ADP-ribosylated dG (1 mg), obtained as above, was incubated with phosphodiesterase I (0.1 units/ml) and bovine intestinal alkaline phosphatase (1.5 units/ml) in 20 mM Tris⋅HCl buffer in the presence of 10 mM magnesium chloride at pH 7.5 at 37°C for 12 h. The reaction mixture was subjected to HPLC analyses on an ODS 80Ts column and eluted isocratically with 2% acetonitrile in 0.5% triethylamine-acetic acid (pH 7.0).
Analysis of ADP-Ribosylated dG in DNA from Pierisin-1-Treated HeLa Cells by the 32P-Postlabeling Method.
HeLa cells (1 × 107) were treated with intact pierisin-1 for 3 h at 37°C. After the incubation, the cell medium was exchanged to remove pierisin-1 from cell culture and incubation was continued for an additional 1 h. DNA recovered from HeLa cells was digested with micrococcal nuclease and phosphodiesterase II at 37°C for 3 h and the resulting digest was 32P-postlabeled by using a nuclease P1 enrichment method as reported (13). The 32P-labeled samples were next applied to polyethyleneimine (PEI)-cellulose TLC sheet, attached to filter paper at the top, and developed with 3 M urea/0.1 M lithium chloride/3 M acetic acid to remove [32P]phosphate and [γ-32P]ATP. Modified nucleotides, which remained at the origin, were contact-transferred to another PEI-cellulose sheet and subjected to two-dimensional (2D) TLC with the following solvent system: buffer A (3.15 M lithium formate/5.95 M urea, pH 3.5) from bottom to top and buffer B (0.7 M lithium chloride/0.35 M Tris⋅HCl/5.95 M urea, pH 8.0) from left to right. Adducts were detected with a Bio-Image Analyzer (BAS2000; Fuji) after exposure to a Fuji imaging plate. The relative adduct labeling (RAL) was determined by the method of Gupta et al. (14) and calculated as the average of three assays.
For the preparation of a standard sample of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine 3′-monophosphate, 2′-deoxyguanosine 3′-monophosphate was reacted with β-NAD in the presence of Pronase-treated pierisin-1 and the product was isolated by HPLC, in a manner similar to that for the isolation of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine.
Chemical Synthesis of N2-[2,3,5-Tris-O-(trimethylsilyl)-d-ribofuranos1-yl]-O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine.
To a stirred solution of 1,2,3,5-tetrakis-O-(trimethylsilyl)-D-ribofuranose (1 mmol) in dichloromethane (1 ml), 1 mmol of iodotrimethylsilane was added at −20°C and the reaction mixture was stirred for 30 min (15). The reaction mixture was then added drop-wise to a mixture of 0.2 mmol of O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (16) and 0.3 mmol of N,N-diisopropylethylamine in dry dichloromethane (2 ml) and stirred continuously until the spot of the starting material had disappeared on TLC analysis. The resulting mixture was then extracted with cold water and brine. The organic layer was dried over anhydrous sodium sulfate, evaporated, and column chromatographed on silica gel with hexane/ethyl acetate (3:1) to yield the desired material (42%). α:β = 1:1. The spectral finding for this compound is published as supporting information on the PNAS web site, www.pnas.org.
Chemical Synthesis of N2-(D-Ribofuranos-1-yl)-2′-deoxyguanosine.
A stirred solution of N2-(2,3,5-tris-O-(trimethylsilyl)-D-ribofuranos-1-yl)-O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (0.1 mmol) in tetrahydrofuran was hydrogenated under a balloon in the presence of Pd black (20 mg) for 5 h at room temperature. The mixture was filtered through a pad of Celite, and the solid cake was washed with a small amount of tetrahydrofuran. To the filtrate, 1.0 M tetrabutylammonium fluoride in tetrahydrofuran (500 μl) was added. After stirring overnight, it was concentrated in vacuo and the residue was purified by open column chromatography using ODS (6% methanol in 0.5% triethylamine⋅acetic acid, pH 7.0) to yield 30 mg (85%) of N2-(D-ribofuranos-1-yl)-2′-deoxyguanosine as a white solid. The spectral findings were as follows. 1H NMR (DMSO-d6, 500 MHz): δ 7.94 (s 1H), 7.39 (d, J = 9, 1H), 6.14 (t, J = 6, 1H), 5.30 (d, J = 9, 1H), 4.34 (dt, J = 5, 3, 1H), 3.80 (dt, J = 7, 3, 1H), 3.75 (brs, 1H), 3.56–3.64 (m, 2H), 3.54 (dd, J = 12, 5, 1H), 3.49 (dd, J = 12, 5, 1H), 3.38 (brd, J = 9, 1H), 2.57 (ddd, J = 13, 6, 5, 1H), 2.18 (ddd, J = 13, 6, 3, 1H); ESI-MS (−): 398 (M − H+)−.
Results
Incorporation of Radioactivity from [32P]NAD into DNA by Pierisin-1.
A mixture of [32P]NAD, Pronase-treated pierisin-1, and cell-free extracts from HeLa cells was incubated, and incorporation of radioactivity was analyzed by SDS/PAGE followed by autoradiography. Most of the radioactivity was recovered in high-molecular-weight material fractions. These radioactive fractions could be digested with DNase, but not protease. Consistent with this finding, incorporation of radioactivity was mainly observed in fractions containing nuclei and mitochondria. No incorporation of radioactivity into high-molecular-weight materials was observed without the pierisin-1. The results suggest that DNA could be the acceptor for ADP-ribosylation by pierisin-1.
Data for incorporation of radioactivity from [32P]NAD into several test substrates, including DNA and RNA, in the presence of Pronase-treated pierisin-1 are shown in Table 1. Efficient incorporation of radioactivity from [32P]NAD into calf thymus DNA and double-stranded dG⋅dC14 oligonucleotide was observed. Single-stranded dG oligonucleotide also proved to be a target, although the efficiency was around 2-fold less than that for calf thymus DNA and double-stranded dG⋅dC oligonucleotide. In contrast, 32P incorporation was not detected when double-stranded dA⋅dT oligonucleotide and single-stranded dC oligonucleotide were used for acceptors. When tRNAs from yeast were used as an acceptor instead of calf thymus DNA, the incorporation efficiency was 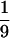 of that for DNA. Poly-L-arginine, an acceptor with the cholera toxin, did not serve as an acceptor with pierisin-1 (Table 1). Incorporation of radioactivity from [32P]NAD into DNA was not observed with other ADP-ribosylating toxins including the cholera toxin A-subunit, pertussis toxin, and diphtheria toxin under the conditions described previously (data not shown).
of that for DNA. Poly-L-arginine, an acceptor with the cholera toxin, did not serve as an acceptor with pierisin-1 (Table 1). Incorporation of radioactivity from [32P]NAD into DNA was not observed with other ADP-ribosylating toxins including the cholera toxin A-subunit, pertussis toxin, and diphtheria toxin under the conditions described previously (data not shown).
Table 1.
Incorporation of radioactivity from [32P]NAD in the presence of pierisin-1
| Substrate | Incorporation, cpm |
|---|---|
| Calf thymus DNA | 1570 |
| Double-stranded DNA (dG × 14/dC × 14) | 2010 |
| Double-stranded DNA (dA × 14/dT × 14) | 17 |
| Single-stranded DNA (dG × 14) | 820 |
| Single-stranded DNA (dC × 14) | 33 |
| Yeast tRNA | 200 |
| Poly-l-arginine | 43 |
| Non-acceptor | 20 |
Mixtures containing Pronase-treated pierisin-1 (1 μg), acceptor (10 μg), and [32P]NAD (0.01 μCi, 10 mCi/mmol) were incubated for 30 min at 37°C at pH 7.5 and the incorporated radioactivity was measured and expressed as mean values from three independent experiments.
Structural Determination of ADP-Ribosylated 2′-Deoxyguanosine.
The above results suggested that pierisin-1 catalyzed the ADP ribosylation of dG residues in DNA. To confirm this, dG was reacted with β-NAD in the presence of Pronase-treated pierisin-1 under conditions similar to those for the [32P]NAD incorporation assay described above. HPLC analysis demonstrated that a peak with a UV λ max at 257 nm, suggesting the presence of both a guanine and an adenine moiety, was detected at a retention time of 23 min (Fig. 1 a and b). Furthermore, LC-ES-MS analysis showed the compound in this peak fraction to have a molecular ion peak at m/z 809, a daughter ion peak at m/z 693 arising from the loss of a deoxyribose moiety, and an ion peak at m/z 910 corresponding to a triethylamine addition, derived from the HPLC eluent, to the parent mass at m/z 809. These mass analyses indicate formation of ADP-ribosylated dG with a molecular weight of 808. The yield of ADP-ribosylated dG from dG was around 1% under the conditions used.
Figure 1.

HPLC analysis of reaction products from dG and β-NAD in the presence of pierisin-1. (a) HPLC profile of the reaction products on an ODS column. The UV absorbance of the eluate was monitored at 254 nm, and the peak appeared by the reaction is marked with an asterisk. (b) UV absorption spectrum with a photodiode array detector of the compound in the peak fraction marked with an asterisk.
To further analyze the chemical structure, reactions of dG and β-NAD with piersin-1 were performed several times and the product was purified by HPLC for NMR spectroscopy. The 1H-NMR spectrum at 600 MHz revealed the presence of peaks for H-8 of guanine at δ 7.92 and H-8 and H-2 of adenine at δ 8.41 and δ 8.13, respectively (Fig. 2). An anomeric proton of the deoxyribose moiety of dG and also an R1′ proton of the ribose moiety of adenosine were observed at δ 6.17 and δ 5.90, respectively. Protons of NH2 at adenine N6 position appeared at δ 7.21. However, typical NH2 protons at the N2 position of dG, which were generally observed at δ 6.50, were missing. These results suggest that ADP-ribosylation of dG occurred at the amino group at the N2 position of dG.
Figure 2.

Chemical structure of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine (a) obtained from the reaction of dG and β-NAD catalyzed by pierisin-1, and its 1H-NMR spectrum (b) in DMSO-d6.
Homonuclear correlation spectroscopy and proton exchange experiments allowed a better definition of the chemical structure. Absorption peaks at δ 5.59 and δ 5.75, generally assigned to the absorption peaks of R1" of ADP-ribose, had an integral region corresponding to only 0.5 proton, respectively (Fig. 2). These protons correlated with absorption peaks of δ 7.16 and δ 7.95, which were D2O exchangeable, and each again had a 0.5 proton integral height. The results suggested the compound to be a mixture of α and β anomeric forms of the ribose moiety of ADP-ribose as reported to be the case with ADP-ribosylation of amino acids (17, 18). Accordingly, protons of δ 5.59 and δ 5.75 were assigned to R1"(β) and R1"(α), respectively, and absorption peaks of δ 7.16 and δ 7.95 were considered to be protons of the N2-amino group of deoxyguanosine that was covalently bound to R1" of ADP-ribose. Because of the extensive overlap of the resonances from the anomeric forms, the spectral region corresponding to the resonances arising from ribose and deoxyribose protons δ 2–δ 5, is complex. However, from 2D-COSY (correlated spectroscopy) and heteronuclear multiple quantum coherence (HMQC) experiments we were able to assign these protons, and also carbons. Based on these results, it is suggested that pierisin-1 catalyzed ADP-ribosylation at the amino group of N2 position of dG to give adduct structures of N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form (Fig. 2).
For further confirmation, we treated ADP-ribosylated dG with phosphodiesterase I and bovine intestinal alkaline phosphatase to cleave the phosphodiester bond, followed by removal of the phosphate moiety from resulting N2-(D-ribose 5′-monophosphate 1-yl)-2′-deoxyguanosine or adenosine 5′-monophosphate (Scheme S1). After separation by HPLC, the main products were collected for UV, MS, and 1H-NMR spectroscopy. As shown in Fig. 3, adenosine was detected at a retention time of 14 min. dG was also detected as a degradation product from N2-ribosylated dG. All UV spectra of four peak fractions at retention times of 11, 12, 21, and 22 min showed UV absorption patterns similar to that of dG, featuring a 1-nm bathochromic shift versus the UV λ max value of dG. Molecular ion peaks of compounds in these four peak fractions were all detected at m/z 398. 1H-NMR spectrum (see Fig. 6, which is published as supporting information on the PNAS web site) and the 2D-COSY experiments suggested that the compound in the peak fraction at a retention time of 21 min is N2-(α-D-ribofuranos-1-yl)-2′-deoxyguanosine, and that at a retention time of 11 min is its β form. Moreover, the compound in peak fraction at a retention time of 22 min is likely to be N2-(α-D-ribopyranos-1-yl)-2′-deoxyguanosine and that at 12 min is its β form.
Scheme 1.

Figure 3.

HPLC analysis of hydrolysate of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine. N2-(ADP-ribos-1-yl)-2′-deoxyguanosine was treated with phosphodiesterase I and bovine intestinal alkaline phosphatase, and separated by HPLC on an ODS column.
Chemical Synthesis of N2-(D-Ribofuranos-1-yl)-2′-deoxyguanosine.
N2-(D-Ribofuranos-1-yl)-2′-deoxyguanosine [or N2-(D-ribopyranos-1-yl)-2′-deoxyguanosine] is a novel compound. We therefore confirmed its structure by independent chemical synthesis. Because of the unstable nature of the resulting ribosyl amine in acidic or basic media, it was essential to avoid methods that use acid or alkaline hydrolysis. After testing a variety of protective groups and deprotective procedures, we finally obtained a synthetic route giving a sufficient yield, this being shown in Scheme S1. O6-Benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine was condensed with per-O-trimethylsilyl-1-iodo-D-ribofuranose. The latter was generated from per-O-trimethylsilyl-D-ribofuranose with trimethylsilyl iodide and immediately used without isolation steps. The protected N2-(D-ribofuranos-1-yl)-2′-deoxyguanosine was thus obtained in a 1:1 mixture of α and β isoforms. O6-Benzyl protection was removed by hydrogenation using palladium black in tetrahydrofuran. Subsequent deprotection of silyl protective groups was accomplished by treatment with tetrabutylammonium fluoride to yield N2-(D-ribofuranos-1-yl)-2′-deoxyguanosine as an α isoform that proved to easily epimerize in water, yielding a 1:1 mixture of α and β anomers. The physical properties of the synthetic compound of α-isoform completely matched that of presumed α-isoform obtained from enzymatically digested ADP-ribosylated dG (Scheme S1). Based on the above observations, the products formed from 2′-deoxyguanosine and β-NAD in the presence of pierisin-1 were concluded to be N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form.
Demonstration of the Formation of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine in DNA Treated with β-NAD in the Presence of Pierisin-1.
Calf thymus DNA was treated with β-NAD in the presence of Pronase-treated pierisin-1 at pH 7.5 at 37°C overnight. The recovered DNA was digested sequentially with micrococcal nuclease, phosphodiesterase II, and bacterial alkaline phosphatase. When digests were analyzed by HPLC, chromatograms showed that approximately half of the dG in the DNA converted to ADP-ribosylated dG, whose corresponding peak was newly detected at a retention time of 26 min (Fig. 4). On the other hand, the amounts of dC, dA, and T remained unchanged. The retention time and UV spectrum of the peak fraction at a retention time of 26 min were exactly the same as those for the N2-(ADP-ribos-1-yl)-2′-deoxyguanosine mentioned above. The 1H-NMR spectrum of this peak also perfectly matched that of a mixture of N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β-form. Thus, N2-(ADP-ribos-1-yl)-2′-deoxyguanosine was proven to be generated through the reaction of calf thymus DNA and β-NAD in the presence of pierisin-1.
Figure 4.

HPLC chromatogram of hydrolysate of DNA treated with β-NAD and pierisin-1. Calf thymus DNA reacted with β-NAD in the presence of pierisin-1 was recovered and digested with a nuclease mixture and alkaline phosphatase, followed by HPLC analysis.
Detection of ADP-Ribosylated dG Adducts in DNA from HeLa Cells Treated with Pierisin-1.
N2-(ADP-ribos-1-yl)-2′-deoxyguanosine in DNA could be detected in intact pierisin-1-treated HeLa cells by the 32P-postlabeling method. After incubation of HeLa cells with intact pierisin-1, DNA was extracted and the formation of ADP-ribosylated dG was investigated under nuclease P1 conditions. Two clear DNA adduct spots were detected, and the same two spots were also observed when 2′-deoxyguanosine 3′-monophosphate was reacted with β-NAD in the presence of Pronase-treated pierisin-1 (Fig. 5). These two spots would presumably be corresponding to N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine 3′,5′-diphosphate and its β form. The relative adduct labeling of ADP-ribosylated dG was 3.51 ± 0.68 per 105 nucleotides when HeLa cells were treated with pierisin-1 at a dose of 100 ng/ml. When 1 and 10 ng/ml of pierisin-1 were used, the relative adduct levels were 3.13 ± 1.38 and 7.32 ± 0.68 per 106 nucleotides, respectively.
Figure 5.
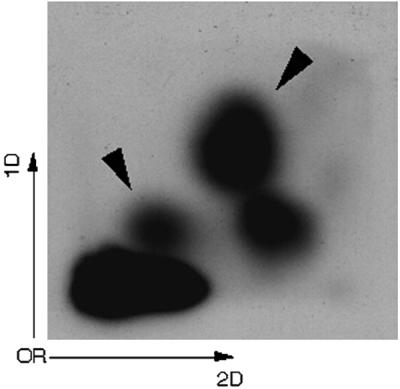
Autoradiograms of DNA adducts in HeLa cells treated with pierisin-1. HeLa cells were treated with 100 ng/ml of pierisin-1 for 3 h. The exposure time of imaging plate was 1 h. DNA adduct spots derived from pierisin-1 treatment are indicated by arrowheads.
Discussion
In the present study, we investigated identity of the acceptor for ADP-ribosylation by pierisin-1 in the presence of β-NAD, and found that dG residues in DNA were reacted with β-NAD to yield N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form. It is well documented that ADP-ribosylation by bacterial toxins, such as cholera and diphtheria toxins, occurs at specific amino acids in proteins. Thus, we extensively searched for acceptor candidates in proteins in cell-free extracts from HeLa cells, which are highly sensitive to pierisin-1-induced apoptosis. However, no candidate protein was detected. Moreover, we confirmed that the three ADP-ribosylating toxins, cholera toxin, pertussis toxin, and diphtheria toxin, showed no ability to incorporate radioactivity from [32P]NAD into DNA. Thus, pierisin-1 was here, for the first time, demonstrated to be a very unique ADP-ribosyltransferase targeting the N2 amino group of dG residues in DNA, rather than specific amino acid residues in a protein as is the case with bacteria-derived ADP-ribosylating toxins.
Regarding the formation of N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine and its β form from dG and β-NAD in the presence of pierisin-1, the following two possible reaction mechanisms can be considered. It has been reported that many NAD-dependent enzymes, such as NAD glycohydrolases and ADP-ribosylcyclases, form a putative oxocarbenium ADP-ribose cation. Therefore, it is possible that pierisin-1 also catalyzes the formation of a putative oxocarbenium ADP-ribose ion as the direct product of nicotinamide elimination from NAD, and subsequent nucleophilic attack of the amino group at N2 of dG results in the formation of α- and β-isomeric conformers of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine in a fashion similar to an SN1 (first-order nucleophilic substitution) reaction mechanism (19–22). On the other hand, as a second possibility it is conceivable that pierisin-1 catalyzes ADP-ribosylation through an SN2-type reaction process (17, 23). Namely, direct back attack of exocyclic amino moiety of dG could occur at the anomeric carbon center of β-NAD, then nicotinamide moiety is eliminated to form predominantly N2-(α-ADP-ribos-1-yl)-2′-deoxyguanosine. This α-isoform would be easily epimerized to β-form as observed with facile epimerization of synthetic α-N2-(D-ribofuranos-1-yl)-2′-deoxyguanosine under aqueous conditions. Consistent with the data for enzymatic activity of pierisin-1, the yield of ADP-ribosylated dG from ADP-ribose and dG without Pronase-treated pierisin-1 was less than 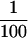 of that from β-NAD and dG in its presence.
of that from β-NAD and dG in its presence.
Pierisin-1 ADP-ribosylates DNA efficiently and this might be expected to trigger apoptotic cell death. 32P-Postlabeling analysis indicated that the relative adduct level for ADP-ribosylated dG was around 3 per 106 nucleotides when HeLa cells were treated with pierisin-1 at a dose of 1 ng/ml, about 10-fold higher than the IC50 value in HeLa cells. This suggests that ADP-ribosylated dG adducts are formed at least at levels of 1 in 107 nucleotides by treatment with a dose equivalent to the IC50 value. At present, it remains to be clarified whether ADP-ribosylation of dG in some specific regions of DNA or rather nonspecific total levels are responsible for apoptosis induction in HeLa cells by pierisin-1.
32P-Incorporation from [32P]NAD into tRNAs from yeast was here found to be around 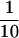 of that into calf thymus DNA. On the other hand, radioactivity from [32P]NAD was found to be more efficiently incorporated into double-stranded dG⋅dC oligonucleotide than single-stranded dG oligonucleotide. Thus, the difference in 32P incorporation from [32P]NAD between DNA and tRNA in the presence of pierisin-1 might be related to variation in ADP-ribosylation efficiency of dG and guanosine, as well as structural conformation. At present, we cannot fully exclude the possibility that RNA is a real target acceptor(s) involved in the cytotoxicity of pierisin-1.
of that into calf thymus DNA. On the other hand, radioactivity from [32P]NAD was found to be more efficiently incorporated into double-stranded dG⋅dC oligonucleotide than single-stranded dG oligonucleotide. Thus, the difference in 32P incorporation from [32P]NAD between DNA and tRNA in the presence of pierisin-1 might be related to variation in ADP-ribosylation efficiency of dG and guanosine, as well as structural conformation. At present, we cannot fully exclude the possibility that RNA is a real target acceptor(s) involved in the cytotoxicity of pierisin-1.
As far as is known, the presence of cytotoxic substance like pierisin-1 is restricted to Pieris butterflies such as P. rapae, Pieris brassicae, and Pieris napi (1, 24). The toxin accumulates in the fifth instar larvae and early phase of pupae in P. rapae (4). What is the biological significance of pierisin-1 in the Pieris butterfly? Pierisin-1 might play an important role in metamorphosis. It is also possible that it serves as a protective agent against invading organisms including the parasitic wasp. The data obtained in the present study are rather surprising because the integrity of DNA is of ubiquitous importance for living cells. Thus, pierisin-1 may be toxic to the vast majority of living organisms. Further studies are clearly now necessary to investigate the real function of pierisin-1 in P. rapae as well as the mechanisms underlying apoptosis of cancer cells caused by the unique ADP-ribosylating toxin, pierisin-1.
Supplementary Material
Acknowledgments
We are grateful to Drs. N. Kawahara (National Institute of Health Sciences), S. Ishikawa, and M. Mochizuki (Kyoritsu College of Pharmacy) for kindly performing NMR and mass spectroscopy measurements. We are also thankful to Drs. M. Maeda (National Cancer Center Research Institute), N. Minakawa, and A. Matsuda (Hokkaido University) for useful discussion and advice regarding organic synthesis. T. Kanazawa was the recipient of a Research Resident Fellowship from the Foundation of Cancer Research during the performance of this work. This study was supported by a Grant-in-Aid for Cancer Research from the Ministry of Health, Labour and Welfare, Japan.
Abbreviations
- ODS
octadecylsilyl
- dG
2′-deoxyguanosine
References
- 1.Koyama K, Wakabayashi K, Masutani M, Koiwai K, Watanabe M, Yamazaki S, Kono T, Miki K, Sugimura T. Jpn J Cancer Res. 1996;87:1259–1262. doi: 10.1111/j.1349-7006.1996.tb03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe M, Kono T, Koyama K, Sugimura T, Wakabayashi K. Jpn J Cancer Res. 1998;89:556–561. doi: 10.1111/j.1349-7006.1998.tb03297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kono T, Watanabe M, Koyama K, Kishimoto T, Fukushima S, Sugimura T, Wakabayashi K. Cancer Lett. 1999;137:75–81. doi: 10.1016/s0304-3835(98)00346-2. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe M, Kono T, Matsushima-Hibiya Y, Kanazawa T, Nishisaka N, Kishimoto T, Koyama K, Sugimura T, Wakabayashi K. Proc Natl Acad Sci USA. 1999;96:10608–10613. doi: 10.1073/pnas.96.19.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanazawa T, Watanabe M, Matsushima-Hibiya Y, Kono T, Tanaka N, Koyama K, Sugimura T, Wakabayashi K. Proc Natl Acad Sci USA. 2001;98:2226–2231. doi: 10.1073/pnas.051628898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ueda K, Hayashi O. Annu Rev Biochem. 1985;54:73–100. doi: 10.1146/annurev.bi.54.070185.000445. [DOI] [PubMed] [Google Scholar]
- 7.Krueger K M, Barbieri J T. Clin Microbiol Rev. 1995;8:34–47. doi: 10.1128/cmr.8.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spector D L, Goldman R D, Leinwand L A. Cells: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1998. [Google Scholar]
- 9.Mekalanos J J, Collier R J, Romig W R. J Biol Chem. 1979;254:5849–5854. [PubMed] [Google Scholar]
- 10.Moss J, Stanley S J, Burns D L, Hsia J A, Yost D A, Myers G A, Hewlett E L. J Biol Chem. 1983;258:11879–11882. [PubMed] [Google Scholar]
- 11.Collier R J, Kandel J. J Biol Chem. 1971;246:1496–1503. [PubMed] [Google Scholar]
- 12.Drazin R, Kandel J, Collier R J. J Biol Chem. 1971;246:1504–1510. [PubMed] [Google Scholar]
- 13.Reddy M V, Randerath K. Carcinogenesis. 1986;7:1543–1551. doi: 10.1093/carcin/7.9.1543. [DOI] [PubMed] [Google Scholar]
- 14.Gupta R C. Cancer Res. 1985;45:5656–5662. [PubMed] [Google Scholar]
- 15.Sujino K, Uchiyama T, Hindsgaul O, Seto N O L, Wakarchuk W W, Paclic M M. J Am Chem Soc. 2000;122:1261–1269. [Google Scholar]
- 16.Harwood E A, Hopkins P B, Sigurdsson S T. J Org Chem. 2000;65:2959–2964. doi: 10.1021/jo991501+. [DOI] [PubMed] [Google Scholar]
- 17.Oppenheimer N J. J Biol Chem. 1978;253:4907–4910. [PubMed] [Google Scholar]
- 18.McDonald L J, Wainschel L A, Oppenheimer N J, Moss J. Biochemistry. 1992;31:11881–11887. doi: 10.1021/bi00162a029. [DOI] [PubMed] [Google Scholar]
- 19.Scheuring J, Schramm V L. Biochemistry. 1997;36:8215–8223. doi: 10.1021/bi970379a. [DOI] [PubMed] [Google Scholar]
- 20.Ziegler M. Eur J Biochem. 2000;267:1550–1564. doi: 10.1046/j.1432-1327.2000.01187.x. [DOI] [PubMed] [Google Scholar]
- 21.Cakir-Kiefer C, Muller-Steffner H, Schuber F. Biochem J. 2000;349:203–210. doi: 10.1042/0264-6021:3490203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanner K G, Landry J, Sternglanz R, Denu J M. Proc Natl Acad Sci USA. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. . (First Published December 5, 2000; 10.1073/pnas.250422697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soman G, Narayan J, Martin B L, Graves D J. Biochemistry. 1986;25:4113–4119. doi: 10.1021/bi00362a019. [DOI] [PubMed] [Google Scholar]
- 24.Matsushima-Hibiya Y, Watanabe M, Kono T, Kanazawa T, Koyama K, Sugimura T, Wakabayashi K. Eur J Biochem. 2000;267:5742–5750. doi: 10.1046/j.1432-1327.2000.01640.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}