

Abstract
We sequenced mitochondrial genomes from five diverse diatoms (Toxarium undulatum, Psammoneis japonica, Eunotia naegelii, Cylindrotheca closterium, and Nitzschia sp.), chosen to fill important phylogenetic gaps and help us characterize broadscale patterns of mitochondrial genome evolution in diatoms. Although gene content was strongly conserved, intron content varied widely across species. The vast majority of introns were of group II type and were located in the cox1 or rnl genes. Although recurrent intron loss appears to be the principal underlying cause of the sporadic distributions of mitochondrial introns across diatoms, phylogenetic analyses showed that intron distributions superficially consistent with a recurrent-loss model were sometimes more complicated, implicating horizontal transfer as a likely mechanism of intron acquisition as well. It was not clear, however, whether diatoms were the donors or recipients of horizontally transferred introns, highlighting a general challenge in resolving the evolutionary histories of many diatom mitochondrial introns. Although some of these histories may become clearer as more genomes are sampled, high rates of intron loss suggest that the origins of many diatom mitochondrial introns are likely to remain unclear.
Keywords: group II introns, HGT, mitochondria, organelle, protists
Introduction
Mitochondrial genomes exhibit striking differences in size, sequence complexity, and architecture across eukaryotes, with animals and land plants capturing many of the extremes. Animal mitochondrial genomes are generally small (∼11–28 kb) and unichromosomal, exhibit high rates of nucleotide substitution, and harbor relatively small amounts of noncoding DNA (Moritz et al. 1987). The mitochondrial genomes of land plants are, by contrast, much larger—sometimes several megabases in size (Sloan et al. 2012)—often exist within multiple isoforms or chromosomes, and have generally low, but in some cases extremely high, rates of nucleotide substitution (Palmer and Herbon 1988; Mower et al. 2007). The mitochondrial genomes of other eukaryotes, including many algal groups, generally fall somewhere between these two extremes. The broad range of mitochondrial genome size and sequence complexity observed across eukaryotes is thought to be driven by an equally broad range of mitochondrial mutation rates (Lynch et al. 2006), though empirical support for this hypothesis has been mixed (Smith 2016a).
One uniting feature of diatom nuclear and organellar genomes is the apparent presence of varying amounts of DNA acquired from foreign sources. The diatom nuclear genome, for example, contains a mix of genes that date back to primary and secondary plastid endosymbioses (Armbrust 2004), possibly a transient green algal endosymbiont (Moustafa et al. 2009; but see Deschamps and Moreira 2012), and potentially hundreds of genes acquired by horizontal gene transfer (HGT) from archaeal and bacterial donors (Bowler 2008). Diatom plastid genomes vary substantially in size and sequence complexity, with several species possessing unique intergenic sequences that may have been acquired by horizontal transfer (Ruck et al. 2014)—a hypothesis bolstered by the sporadic presence of invasive group I and group II introns in some species, indicating that foreign DNA can, in some instances, find its way into cellular organelles (Brembu et al. 2014; Ruck et al. 2017).
Invasive introns have been known in diatom mitochondrial genomes since the discovery of a group II intron in the cox1 gene of Thalassiosira nordenskioeldii Cleve (Ehara et al. 2000). A similar intron exists at the same site in Pylaiella littoralis (L.) Kjellman (Ehara et al. 2000), a brown alga in the same lineage (Stramenopila, or “stramenopiles”) that includes diatoms. This suggests that introns are an ancestral feature of diatom mitochondrial genomes. Other mitochondrial introns have been reported in some, but not all, diatom species with sequenced genomes. For example, the cox1 gene of the araphid pennate diatom, Ulnaria acus (Kützing) Aboal, contains two group II introns that most closely resemble ones from brown algae and haptophytes (Ravin et al. 2010), the latter representing a much more distantly related group of algae. High sequence similarity between cox1 group II introns in diatoms and a distantly related stramenopile, Chattonella marina (Subrahmanyan) Hara and Chihara (Raphidophyceae), led to the hypothesis that Chattonella acquired the intron by HGT from diatoms (Kamikawa et al. 2009), though determining the proximal donor and direction of transfer is difficult for sparsely sampled data sets (Ruck et al. 2017). The full complement of mitochondrial introns in diatoms, as well as patterns of intron gain and loss across genes and species, remain poorly characterized due to the small number of sequenced diatom mitochondrial genomes and lack of a comprehensive comparative analysis of existing genomes (Ehara et al. 2000; Kamikawa et al. 2009; Ravin et al. 2010).
We sequenced mitochondrial genomes for five diatom species and added them to a data set of 11 publicly available diatom mitochondrial genomes to characterize broadscale patterns of mitochondrial genome evolution in diatoms. We describe many newly discovered group I and group II introns, which appear to preferentially insert themselves into the cox1 and rnl genes. We also characterize, as best as possible given the limited data set, the origins and ancestries of these introns. Our findings show that the relatively compact, AT-rich mitochondrial genomes of diatoms have a fairly conserved gene complement, with some species readily accepting foreign introns from mostly unknown sources.
Materials and Methods
Data Collection and Genome Sequencing
We compiled mitochondrial gene or genome data for a total of 19 diatom and 3 outgroup species (table 1). Genomes for 10 of these species were downloaded from GenBank, and the genomes of Cyclotella cryptica Reimann, Lewin and Guillard and Fragilariopsis cylindrus (Grunow) Helmcke and Krieger were downloaded from the supplementary materials of Traller et al. (2016) and Mock et al. (2017), respectively. In addition, we sequenced mitochondrial genomes for five species: Toxarium undulatum, Psammoneis japonica, Eunotia naegelii, Cylindrotheca closterium, and Nitzschia sp. (table 1). Our analyses of cox1 introns used additional data from Pseudo-nitzschia multiseries Hasle (Hasle) (Yuan et al. 2016), Asterionella formosa Hassall (Villain et al. 2017), and Thalassiosira nordenskioeldii (Ehara et al. 2000). In most cases, the genus name provided a unique identifier of the sequences in our analysis, so we use this shorthand throughout, recognizing that sampling of additional species within the genus might show genomic variation. We refer to the model diatom species, Thalassiosira pseudonana, by its taxonomically correct name, Cyclotella nana (Alverson et al. 2011). The 19 diatom species in our analyses included: five polar centrics (Mediophyceae), four of which belong to Thalassiosirales; three araphid pennate species (Bacillariophyceae); and 11 raphid pennate species (Bacillariophyceae) (table 1).
Table 1.
Information for the Mitochondrial Gene and Genome Sequences Used In This Study
| Taxon | Genome Size (bp) | Percent Exon | Percent Intron | GC Content | No. Protein Genesb | No. rRNAs | No. tRNAs | No. Introns | Culture Strain | GenBank Accession |
|---|---|---|---|---|---|---|---|---|---|---|
| Chattonella marinaa | 44,772 | 71.2 | 1.1 | 28.4 | 34 | 3 | 25 | 2 | KA11-m-1 | NC_013837 |
| Nannochloropsis oculataa | 40,721 | 80.5 | 6.0 | 32.2 | 35 | 2 | 26 | 1 | CCMP525 | KJ410688 |
| Triparma laevisa | 39,580 | 89.0 | 0.0 | 30.4 | 34 | 2 | 26 | 0 | NIES-2565 | NC_027747 |
| Toxarium undulatum | 36,667 | 95.0 | 0.0 | 30.4 | 34 | 2 | 23 | 0 | ECT3802 | MG271847 |
| Thalassiosira nordenskioeldii | NA | NA | NA | NA | NA | NA | NA | NA | CCMP992 | AB038235 |
| Skeletonema marinoi | 38,515 | 90.4 | 0.0 | 29.7 | 34 | 2 | 24 | 0 | JK029 | NC_028615 |
| Cyclotella cryptica | 58,021 | 60.0 | 4.0 | 30.8 | 34 | 2 | 24 | 1 | CCMP332 | – |
| Cyclotella nana | 43,827 | 79.5 | 5.3 | 30.1 | 34 | 2 | 25 | 1 | CCMP1335 | NC_007405 |
| Psammoneis japonica | 73,622 | 47.3 | 36.4 | 30.8 | 34 | 2 | 27 | 11 | ECT2AJA-110 | MG148339 |
| Ulnaria acus | 46,657 | 71.2 | 12.3 | 31.8 | 32 | 2 | 24 | 3 | – | NC_013710 |
| Asterionella formosa | 61,877 | 51.7 | 4.0 | 26.6 | 34 | 2 | 25 | 1 | – | NC_032029 |
| Eunotia naegelii | 48,049 | 71.7 | 1.4 | 27.1 | 33 | 2 | 23 | 1 | FD354 | MG271846 |
| Navicula ramosissima | 48,652 | 70.9 | 21.4 | 31.1 | 34 | 2 | 22 | 6 | TA439 | KX343079 |
| Phaeodactylum tricornutum | 77,356 | 45.2 | 8.7 | 35.0 | 34 | 2 | 25 | 4 | CCAP1055/1 | HQ840789 |
| Berkeleya fennica | 35,509 | 97.4 | 0.0 | 29.7 | 34 | 2 | 24 | 1 | TA424 | NC_026126 |
| Fistulifera solaris | 39,476 | 87.7 | 0.0 | 28.1 | 34 | 2 | 24 | 0 | – | NC_027978 |
| Cylindrotheca closterium | 37,784 | 92.9 | 6.2 | 32.1 | 34 | 2 | 25 | 1 | CCMP1855 | MG271845 |
| Fragiliariopsis cylindrus | 58,295 | 60.2 | 0.0 | 31.1 | 34 | 2 | 24 | 0 | CCMP1102 | – |
| Pseudo-nitzschia multiseries | 46,283 | 65.1 | 16.3 | 31.0 | 33 | 2 | 23 | 6 | – | NC_027265 |
| Nitzschia sp. | 36,221 | 96.6 | 0.0 | 28.8 | 34 | 2 | 25 | 0 | ECT2AJA-05 | MG182051 |
| Durinskia balticac | 35,505 | 97.8 | 0.0 | 31.0 | 34 | 2 | 24 | 0 | CSIRO CS-38 | JN378735 |
| Kryptoperidinium foliaceumc | 39,686 | 87.4 | 10.9 | 32.4 | 34 | 2 | 23 | 3 | CCMP1326 | JN378734 |
Note.—Taxa for which only introns were considered (Pseudo-nitzschia multiseries, Asterionella formosa, Thalassiosira nordenskioeldii, Chattonella marina, and Nannochloropsis oculata) are not included. Culture information was not available for some taxa. Percent exonic sequence includes protein-coding, rRNA, and tRNA genes.
Outgroup taxa.
Excluding intronic ORFs.
Diatom endosymbiont of a dinoflagellate.
We extracted total DNA from Psammoneis with a Qiagen DNeasy DNA Plant Mini Kit and sequenced the mitochondrial genome by shotgun sequencing with the Pacific Biosciences RSII platform. Genomic library preparation, size selection, and sequencing were completed by the University of Delaware Sequencing and Genotyping Center using SMRTbell™ Library preparation, BluePippin size selection, and SMRT Cell sequencing. We assembled the sequencing reads using Falcon (ver. 0.4.0) (Chin et al. 2016) with length cutoff of 7,000, minimum coverage of 3, and max coverage depth and coverage difference of 100. The mitochondrial genome was identified from this assembly based on size, GC-content, BLASTN-based sequence similarity to other mitochondrial genomes, and its predicted circular-mapping topology. The assembly contained numerous single-point indels that were corrected using Quiver (ver. 2.1.0) (https://github.com/PacificBiosciences/GenomicConsensus; last accessed February 2017) and Pilon (ver. 1.2.1) (Walker et al. 2014) with default settings and by reference to an aligned read map from SAMtools (ver. 0.1.19) (Li et al. 2009).
For Toxarium, Eunotia, and Nitzschia, we extracted DNA with a Qiagen DNeasy Plant Mini Kit. We sequenced Toxarium and Eunotia DNA on the Illumina MiSeq platform housed at the Institute for Genomics and Systems Biology at Argonne National Library, with 300-bp libraries and 150-bp paired-end reads. We sequenced Nitzschia DNA with the Illumina HiSeq2000 platform housed at the Beijing Genomics Institute, with a 300-bp library and 90-bp paired-end reads. We removed adapter sequences and trimmed raw reads with Trimmomatic (ver. 0.32) and settings “ILLUMINACLIP=<TruSeq_adapters.fasta>: 2:30:10, TRAILING = 5, SLIDINGWINDOW = 6:18, HEADCROP = 9, MINLEN = 50” (Bolger et al. 2014). We assembled the trimmed reads with Ray (ver. 2.3.1) with default settings and k-mer length of 31 (Toxarium), 41 (Eunotia), or 45 (Nitzschia) (Boisvert et al. 2012). We assessed the quality of the assemblies with QUAST (ver. 2.3) (Gurevich et al. 2013) and REAPR (Hunt et al. 2013), confirmed genome-wide read coverage by mapping the trimmed reads to the assembly with Bowtie (ver. 0.12.8) (Langmead et al. 2009) and evaluated these results with SAMtools.
For Cylindrotheca, we resuspended a pellet of frozen cells in 10–15 ml of resuspension buffer (50 mM Tris [pH 8.0], 25 mM ethylenediaminetetraacetic acid, and 50 mM NaCl) and disrupted the cells by nitrogen decompression with a Parr Cell Disruption Bomb at 750–800 psi for 30 min. We lysed plastids by shaking them at 1 g for 60 min at 50°C in a solution containing 250 µl of 20% Triton X-100 and 1 ml Pronase (10 mg/ml) per 10 ml of cell slurry. We added equal weight CsCl, mixed the slurry until the CsCl was fully dissolved, then dispensed the solution into 6-ml PA Ultracrimp tubes (Sorvall) with 50 µl of ethidium bromide (EtBr) (10 mg/ml). After centrifugation at 414,728 g in a Sorvall TV-1665 rotor for 12 h, we extracted the DNA bands and removed EtBr with repeated washes in salt-saturated isopropanol. The spin was repeated with 40 µl Hoechst 33258 dye (10 mg/ml H2O), after which DNA bands were extracted and Hoechst dye removed by repeated 1:1 washes with salt-saturated isopropanol. We removed the CsCl by dialysis in TE buffer with buffer changes every 12 h for 48 h. We sequenced the DNA using the Roche 454 GS-FLX platform (titanium reagents) housed at the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois, which generated 500 bp single-end reads. We assembled the reads using the Newbler software package and used Geneious ver. 5.4 (Gene Codes Corp., Ann Arbor, MI, USA) to validate and guide finishing of the assembly.
Genome Annotation
We used the National Center for Biotechnology Information’s (NCBI) BLASTN software (ver. 2.7.1+) to search each newly sequenced genome against databases of known genes from previously sequenced diatom mitochondrial genomes and used NCBI’s ORFfinder software (https://www.ncbi.nlm.nih.gov/orffinder; last accessed December 2017) to confirm start and stop codon positions. We used ARAGORN (Laslett and Canback 2004) to annotate tRNA genes. Newly sequenced genomes are available from GenBank (table 1).
Intron Analyses
We characterized the phylogenetic distribution, intron class, genomic insertion site, and putative origin of mitochondrial introns for the diatom and outgroup taxa in our data set. We performed an all-vs-all BLASTN search of all mitochondrial introns with “somewhat similar” settings “-dust no -word_size 9 -reward 2 -penalty -3 -gapopen 5 -gapextend 2” to cluster introns into putatively homologous groups based on sequence similarity. Each group was assigned a name consisting of the host gene, intron type (g1 or g2), and a unique single-letter identifier (e.g., cox1_g2_A). To identify related introns outside of diatoms, we individually searched each intron sequence against GenBank’s nr database with BLASTN and kept all subject sequences with e-value < 1e-12 and qcovs > 50. We used MUSCLE with default settings (Edgar 2004) to align intron groups that included a total of at least five query and subject sequences. We trimmed the resulting five alignments using Gblocks (ver. 0.91b) with default settings (Castresana 2000) and used IQ-TREE (ver. 1.5.3) (Nguyen et al. 2015) to find the best-fitting model of nucleotide substitution and infer the intron phylogeny. Workflows and scripts for intron BLAST searches, alignments, and tree inferences are available on GitHub (https://github.com/andrewalverson/diatom_mt/tree/master/intron_phylogenies; last accessed April 2018). We used NCBI’s ORFfinder program to find intron-encoded proteins (IEPs), and we characterized functional domains within IEPs through BLASTP searches against the nr database. Intron maps were rendered from annotated FASTA files using a script that has been archived on GitHub (https://github.com/andrewalverson/diatom_mt; last accessed April 2018).
Multiple Sequence Alignment and Phylogenetic Analyses
We generated multiple sequence alignments for 36 mitochondrial genes using MUSCLE, as implemented in MEGA ver. 7.0.14 (Kumar et al. 2008), and then manually adjusted the alignments to enforce codon structure. We trimmed alignments using Gblocks (ver. 0.91b) with settings “-b3 = 8, -b4 = 3, -b5 = a” and used IQ-TREE to build a phylogenetic tree for each gene using the best-fit model of nucleotide substitution identified by IQ-TREE using the Bayesian Information Criterion. In addition to single-gene analyses, all protein-coding genes were concatenated into a single alignment with AMAS (Borowiec 2016), and the tree was inferred using the single best substitution model identified by IQ-TREE for the concatenated alignment. For each alignment, branch support was estimated with 2,000 ultrafast bootstrap pseudoreplicates (Minh et al. 2013) and 1,000 SH-like approximate likelihood-ratio tests (SH-aLRT). Gene and intron alignments and IQ-TREE command files are available as Supplementary Material (supplementary data S1, Supplementary Material online).
Results
Genome Characteristics
Three of the five newly sequenced genomes (Psammoneis, Eunotia, and Nitzschia) mapped as circular chromosomes, whereas the other two (Toxarium and Cylindrotheca) could not be circularized, leaving open the possibility that they are either linear or incompletely sequenced. Importantly, the uncircularized Toxarium and Cylindrotheca genomes had complete gene sets. Diatom mitochondrial genomes ranged in size from 35.5 to 77.4 kb (median = 39.7 kb), with Phaeodactylum (77.4 kb), Psammoneis (73.6 kb), and Asterionella (61.9 kb) possessing the largest genomes in our analysis (table 1). The G + C content was similar to diatom plastid genomes (Ruck et al. 2014), ranging from 26.6 to 35.0%. Coding sequences (protein, rRNA, and tRNA genes) comprised the vast majority of the total sequence (median = 79%, N = 21) in all but the three largest genomes, in which coding sequences comprised roughly half (45–52%) of the total sequence (table 1). Coding sequences comprised >97% of the sequence in the highly reduced genomes of Berkeleya and the diatom endosymbiont of the dinoflagellate, Durinskia. The percentage of intronic sequence was generally low (median = 4%, N = 21), but considering only the set of intron-containing genomes (N = 14), the median percentage of intronic DNA was 6%. Introns comprised more than one-third (36%) of the sequence in the large, intron-rich genome of Psammoneis. Some of these estimates should be considered approximations, however, due to possible incomplete sequencing of some of the genomes.
Diatom mitochondrial genomes contain a core set of 34 protein-coding and two rRNA (rns and rnl) genes that are largely or entirely conserved across taxa (supplementary table S1, Supplementary Material online). The atp1, rpl31, and rn5 genes, which are present in many other stramenopiles (Wei et al. 2013), are missing in diatoms (supplementary table S1, Supplementary Material online). Among the diatoms in our data set, Ulnaria is missing the rps2 and rps7 genes, and Eunotia is missing the atp8 gene. Most taxa shared a core set of 22 tRNA genes, some of which are present in multiple copies in some [e.g., trnE(uuc) in Psammoneis] or all [trnM(cau)] taxa (supplementary table S1, Supplementary Material online). The trnG(ucc) gene is present in Toxarium and the three stramenopile outgroups, suggesting repeated losses of this gene across diatoms. All sampled members of the Thalassiosirales (Cyclotella and Skeletonema) have a trnU(uca) gene, encoding selenocysteine, the selenium analog of cysteine.
Mitochondrial Introns
We characterized a total of 38 mitochondrial introns from the diatom and stramenopile outgroup taxa in our data set (supplementary table S2, Supplementary Material online). Several genomes have annotated introns in the cob, cox3, rps3, rps11, and/or rns genes that were generally short (15–170 bp in length), species-specific, and lacking the characteristic signatures of canonical group I or group II introns (supplementary table S2, Supplementary Material online). Three of these sequences (in cob, cox3, and rps11) interrupt the conserved reading frame of their respective genes. We concluded that sequencing or assembly errors should be ruled out before further effort is made to understand the identities and origins of these sequences. Sequences clearly recognizable as organellar group I or group II introns were primarily restricted to the cox1 and rnl genes, with one additional group II intron in the rns gene of Phaeodactylum. The only group I introns occurred in the cox1 and rnl genes of the diatom endosymbiont of the dinoflagellate, Kryptoperidinium; both of these group I introns possessed a LAGLIDADG homing endonuclease (Stoddard 2014). Most introns contained an ORF encoding reverse transcriptase and, in many cases, zinc finger endonuclease and/or maturase domains. Psammoneis had the most intron-rich genome, with a total of 11 introns (all of them group II) in the cox1 (7 introns) and rnl (4 introns) genes (supplementary table S2, Supplementary Material online). In total, introns comprised more than one-third (36%) of the Psammoneis mitochondrial genome and were the primary source of genomic expansion in this species. Navicula had five introns that, similar to Psammoneis, were all located in the cox1 or rnl genes (supplementary table S2, Supplementary Material online). Seven taxa possessed no mitochondrial introns: Triparma, Toxarium, Skeletonema, Fragilariopsis, Nitzschia, Fistulifera, and the Durinskia endosymbiont.
In general, BLASTN searches to GenBank returned relatively few significant matches for most introns, with just five of the 18 cox1 and rnl intron groups containing more than five total sequences compiled from our study and GenBank. Phylogenetic analyses of these five intron groups revealed similarities to introns in other distantly related stramenopiles, including brown algae (Pylaiella) and raphidophytes (Chattonella) (fig. 1), though the relationships with Chattonella, in particular, may not reflect vertical inheritance and deep shared ancestry (see discussion of the cox1_g2_B intron below). One of the diatom introns (cox1_g2D) had strong matches to the fungal genera Paracoccidioides and Candida (fig. 1C). Phylogenetic trees with full taxon labels are shown in supplementary figure S1, Supplementary Material online.
Fig. 1.

—Unrooted phylogenetic trees for five intron groups (see supplementary table S2, Supplementary Material online) from the cox1 (A–D) and rnl genes (E), each of which contained at least five total sequences from our data set and GenBank’s nr database. Most of the 37 cox1 and rnl introns in our analysis have very few matches to sequenced genomes outside of diatoms. Additional details about these intron groups (e.g., cox1_g2_A) are available in supplementary table S2, Supplementary Material online, and trees with full taxon labels are shown in supplementary figure S1, Supplementary Material online. Diatoms are shown in black, and nondiatoms are shown in gray. Asterisks highlight nodes with bootstrap values >70%.
cox1 Introns
Based on sequence similarity and position in the cox1 gene, 19 of the 26 cox1 introns clustered into a total of five groups of putative homologs with two or more species (fig. 2, colored triangles), whereas the remaining seven introns were species-specific (fig. 2, open triangles). None of the introns present in more than one diatom species were shared by sister taxa. The sporadic distribution of shared introns, together with the relatively large number of species-specific introns, suggest that introns are frequently gained and/or lost from diatom mitochondrial genomes (fig. 2). Two of the cox1 intron groups, cox1_g2_A and cox1_g2_F (fig. 2, blue and yellow triangles), contained four distantly related species with levels of sequence divergence that we interpreted as consistent with vertical inheritance and recurrent loss of these introns. A similar pattern was seen for the smaller cox1_g2_E group (fig. 2, orange triangles).
Fig. 2.

—Spatial and phylogenetic distributions of introns in the cox1 gene. Groups of putative homologs share similarly colored triangles, and unfilled triangles represent species-specific introns without homologs in other species in our analysis. Each unique insertion site is marked with a dashed vertical line. All introns are of type group II except for a single group I intron (marked with a “I”) in Kryptoperidinium. The phylogeny is a reference organismal tree compiled from previous studies (Theriot et al. 2015; Parks et al. 2018). Species marked with an asterisk (*) are the hosts for endosymbiotic diatoms. Major taxonomic groups referred to in the main text are identified (“Thal” = Thalassiosirales, “Bacillario.” = Bacillariophyceae).
In all but one case, sequence similarity and insertion site in the cox1 gene supported the same set of intron groupings. The cox1_g2_B and cox1_g2_B’ groups, however, arguably represented a single homologous intron group with two distinct insertion sites (fig. 2, light and dark purple triangles at alignment positions 214 and 1,187, respectively). Phylogenetic analysis showed that introns at these two positions fell into two distinct clades (fig. 3), but the sequences could be confidently aligned, and introns at these two positions clearly shared more similarity with one another than with any other cox1 introns.
Fig. 3.
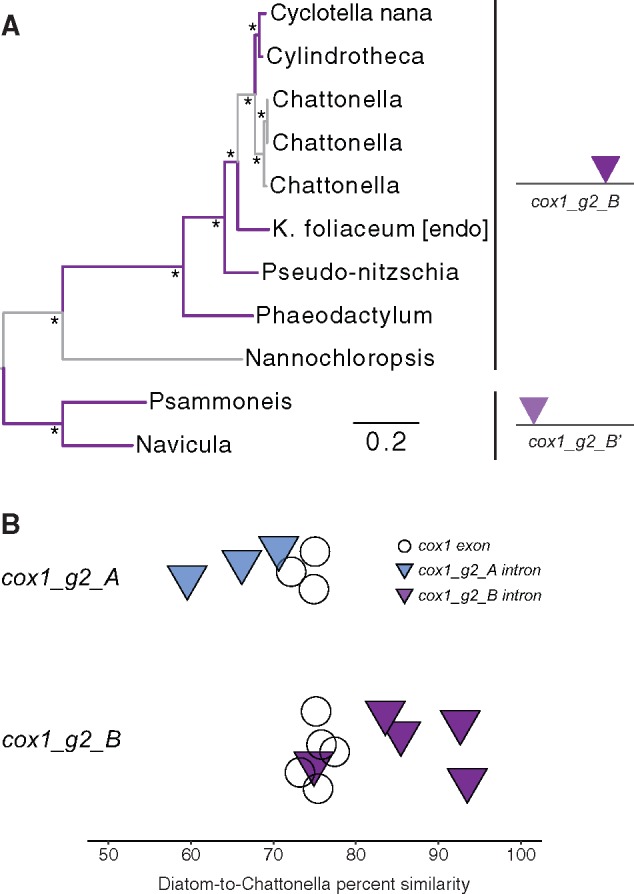
—Maximum likelihood phylogeny of the cox1_g2_B intron group. Diatoms are shown on purple branches, and nondiatom stramenopiles are shown on gray branches. The alignment included introns with two different insertion sites in the cox1 gene, shown by the positions of the purple triangles on a depiction of the cox1 coding sequences (A), similar to what is shown in figure 2. Asterisks represent 100% bootstrap support. Panel (B) shows pairwise sequence similarities between the raphidophyte, Chattonella, and each diatom in the cox1_g2_A and cox1_g2_B intron groups. For species in the cox1_g2_A group, introns uniformly show lower sequence similarity than exons. For species in the cox1_g2_B group, pairwise sequence similarities in exons were similar to what was found in cox1_g2_A, but there was a much broader range of intron similarities, with two species, Cylindrotheca and C. nana, possessing introns that were > 94% similar to the homologous copy in Chattonella; a small amount of random vertical scatter was introduced to better display the values of overlapping data points (B).
Horizontal Transfer of the cox1_g2_B Intron
Phylogenetic analysis of the cox1_g2_B group revealed strong incongruence between the intron tree and species trees inferred using both mitochondrial (supplementary fig. S2, Supplementary Material online) and nuclear genomic data (Parks et al. 2018). For example, monophyly of raphid pennate diatoms is uncontested (Theriot et al. 2015; Medlin 2016; Parks et al. 2018), but they (Cylindrotheca and Phaeodactylum) were polyphyletic in the intron tree (fig. 3A). Instead, introns from Chattonella (a raphidophyte), the raphid pennate diatom Cylindrotheca, and the polar centric diatom C. nana were very closely related (fig. 3A) and shared unusually high (>94%) sequence similarity (fig. 3B).
Assuming equal ages of cox1 introns and exons, we would expect intron sequences to exhibit levels of sequence divergence proportional to their corresponding exons. The raphidophyte Chattonella shared two introns with diatoms, cox1_g2_A and cox1_g2_B (fig. 2, blue and purple triangles), which allowed us to compare patterns of sequence divergence in these two groups as a surrogate measure of HGT. Consistent with expectations, pairwise sequence similarity in the cox1_g2_A introns between Chattonella and the three diatoms ranged from 59–71%, which was slightly lower than exon divergence, which ranged from 72–76% for these same taxa (fig. 3B). The other intron group that included Chattonella, the cox1_g2_B group, contained five distantly related diatoms (fig. 2, dark purple triangles). In this group, pairwise sequence similarity of cox1 exons between Chattonella and each diatom again ranged from 72–75%, but sequence similarity between Chattonella and diatom introns was as high as 95% in some cases (fig. 3B), reflecting the very recent shared ancestry between the Chattonella, Cylindrotheca, and C. nana introns (fig. 3A).
rnl Introns
Introns within the rnl gene were fewer in number than in cox1 and had a much more restricted phylogenetic distribution, occurring only in araphid and raphid pennate diatoms (Bacillariophyceae) and neither of the stramenopile outgroups (fig. 4). This pattern points to repeated losses of these introns in the grade of radial and polar centric diatoms and/or recent gains of these introns in the pennate clade. As for cox1, Psammoneis had the most intron-rich rnl gene, with a total of four group II introns (fig. 4). Much like the cox1 introns, sequence similarity and insertion site in the rnl gene supported the same set of intron groupings. The 10 rnl group II introns were spread across six different locations in the rnl gene, with two of the six sites shared across multiple species (fig. 4, shaded triangles). Each of the other groups contained just one intron, consistent with recent, lineage-specific acquisitions or recurrent losses of those introns. Some of the rnl introns contained as many as two ORFs encoding a reverse transcriptase and, in a few cases, a zinc-finger endonuclease domain. Overall, rnl introns tended to possess fewer ORFs and fewer functional domains than cox1 introns.
Fig. 4.

—Spatial and phylogenetic distributions of introns in the rnl gene. Groups of putative homologs share similarly shaded triangles, and unfilled triangles represent species-specific introns without homologs in other species in our analysis. Each unique insertion site is marked with a dashed vertical line. All introns are of type group II except for a single group I intron (marked with a “I”) in Kryptoperidinium. The phylogeny is a reference organismal tree compiled from previous studies (Theriot et al. 2015; Parks et al. 2018). Species marked with an asterisk (*) are the hosts for endosymbiotic diatoms.
Fission of the nad11 Gene
The nad11 gene underwent fission into two separate pieces in the common ancestor of raphid pennate diatoms (supplementary table S3, Supplementary Material online; see figs. 2 and 4 for clade designations). Each subunit has its own start and stop codons and corresponds to one of the two functional domains of the nad11 protein, the iron-sulfur binding (nad11a) and molybdopterin-binding domains (nad11b) (Imanian et al. 2012). The length of intervening sequence between the two domains ranged from 14 nt to as much as 22 kb, and in some cases exons for the two domains were located on opposite strands. The longest intervening sequences contained other genes. We did not find flanking intronic sequences around the two subunits, as would be expected if the two halves of the gene were separated by a trans-spliced intron.
Discussion
Mitochondrial Genome Evolution in Diatoms
We sequenced mitochondrial genomes for five diverse diatoms to fill taxonomic gaps in the existing set of mitochondrial genome sequences and used these data to characterize broadscale patterns of genome evolution across diatoms. The set of genomes was still small (N = 16 taxa) and biased toward raphid pennates and Thalassiosirales. The largest remaining sampling gaps include all coscinodiscophytes (radial centrics) and many mediophytes (polar centrics). Nevertheless, some generalizations are likely to hold up to further sampling. Diatom mitochondrial genomes have low G + C content (≤ 35%) and are modestly sized (median length = 39.7 kb) and gene-dense. With a few exceptions, gene content is strongly conserved across species. The missing atp8 gene from Eunotia was the most striking absence, though this gene, while generally conserved, has been lost in many different lineages (von Nickisch-Rosenegk et al. 2001; Yang et al. 2015; Tanifuji et al. 2016). Additional genome sequencing in this part of the diatom tree, or additional nuclear genomic or transcriptomic data for Eunotia, will help show whether atp8 has, in fact, been lost or transferred to the nuclear genome in some diatoms. Intact atp8 genes have evaded detection in some red algae (Salomaki and Lane 2017), but we are confident of its absence from the high-depth, QUAST- and REAPR-validated Eunotia assembly.
Among this relatively small sample of diatoms, genome size varied by >2-fold, from 35.5 to 77.4 kb. At least two different drivers of genome expansion were evident: the presence of large repeat-rich sequences, as in Phaeodactylum (Oudot-Le Secq and Green 2011), or the presence of many large introns, as in Psammoneis (supplementary table S2, Supplementary Material online). The 11 group II introns in Psammoneis account for more than one-third (26.8 kb) of its mitochondrial genome. In Psammoneis, cox1 introns alone sum to >18 kb, exceeding the 1.5 kb of cox1 exonic sequence by a factor of 12.
The Patchy Distributions and Obscure Ancestries of Mitochondrial Introns in Diatoms
Mobile group I and group II introns are common in the mitochondrial genomes of plants (Cho et al. 1998; Bonen 2008), fungi (Paquin et al. 1997), some animal taxa (Hellberg 2006), and many microbial eukaryotes—including green and red algae (Smith et al. 2010; Yang et al. 2015), diatoms (Ehara et al. 2000), and other stramenopiles (Kamikawa et al. 2009; O’Brien et al. 2014). The vast majority of diatom mitochondrial introns characterized in this study were group II, and their distribution within diatom genomes was highly unbalanced, with 37 of the 38 introns located in just two genes, cox1 and rnl. This closely mirrors the pattern in red algae, where nearly all mitochondrial introns reside in the cox1 or rnl genes as well (Yang et al. 2015). This is also consistent with a more general pattern across eukaryotes—that organellar introns mostly occur in conserved housekeeping genes with important cellular functions, such as RNA genes and components of the electron transport chain (Robart and Zimmerly 2005; Mullineux et al. 2010). The cox1 and rnl introns were spread across many different insertion sites (figs. 2 and 4), and introns that we characterized as putatively homologous based on sequence similarity were positionally homologous as well. Introns that co-occurred within a genome shared little sequence similarity outside of the conserved 5′ and 3′ end sequences that are diagnostic of group II introns, pointing to an expectedly low rate of retrotransposition into ectopic sites (Nikoh and Fukatsu 2001; Lambowitz and Zimmerly 2004; Simon et al. 2005). Retrotransposition of group II introns can occur, however (Mueller et al. 1993; Lambowitz and Zimmerly 2004; Simon et al. 2005), and we uncovered one possible example of this in our data set. Introns from the cox1_g2_B (site 1208) and cox1_g2_B’ (site 239) groups shared much greater sequence similarity with one another than any other two groups, suggesting that their similarity might trace back to an ancient (at least as far back as the common ancestor of pennate diatoms) transposition event. Additional data—including the possible discovery of taxa with both introns (no such taxa were found in our data set; fig. 2)—could allow us to better pinpoint the timing of such an event and allow for more accurate reconstruction of the ancestral target sequences at these two sites. Ultimately, such analyses get to the heart of larger, fundamental questions about the origins and spread of these introns in diatoms.
Organellar introns often exhibit highly disjunct phylogenetic distributions—a pattern exemplified by diatom mitochondrial genomes and one that highlights a longstanding and clearly challenging question about the underlying cause of such patterns (Saldanha et al. 1993; Gray et al. 1998). Although patchy distributions are often considered a hallmark of HGT (Andersson et al. 2006; Andersson 2009), it is notoriously difficult to distinguish recent gains via HGT from widespread losses of an ancestrally present feature (Crisp et al. 2015; Hotopp 2017; Salzberg 2017). Many HGT hypotheses have been overturned as databases have grown, or analyses expanded, to include a broader set of taxa related to the putative recipient—instead revealing a pattern of ancestral presence and repeated loss (Salzberg et al. 2001; Stanhope et al. 2001). It appears that both processes have shaped the distributions of organellar introns in microbial eukaryotes, including diatoms. One of the primary obstacles to discriminating between these two competing hypotheses is the relative scarcity of organellar genome sequences for most lineages of microbial eukaryotes. Patchy, exemplar-based genome sampling invariably produces patterns that can only be teased apart with denser taxon sampling (Goddard and Burt 1999; Sanchez-Puerta et al. 2008; Yang et al. 2015). Nevertheless, the emerging pattern of intron evolution in diatom organellar genomes appears to follow the pattern in microbial eukaryotes as a whole, in which intron distributions reflect a combination of (mostly) recurrent loss and, in some cases, horizontal transfer.
GenBank searches showed that most mitochondrial introns in diatoms were most similar to ones in other stramenopiles (supplementary table S2, Supplementary Material online), suggesting that the ancestral stramenopile possessed an intron-rich mitochondrial genome that continues to experience independent intron losses across both diatoms and the whole of stramenopiles (Oudot-Le Secq et al. 2006; O’Brien et al. 2014). Thus, loss may be the prevailing driver of the sporadic distribution of mitochondrial introns in diatoms. The ancestry of most introns was much less clear, however, including introns that appeared, superficially, to reflect a simple pattern of recurrent loss.
Previous phylogenetic analyses of mitochondrial introns from a nondiatom stramenopile, Chattonella, and two diatoms, C. nana and the Kryptoperidinium endosymbiont (a raphid pennate), showed strong evidence for horizontal transfer of the cox1_g2_B intron between Chattonella and diatoms, with the inference that Chattonella received the intron from a diatom (Kamikawa et al. 2009). Although our analyses further support a transfer event between diatoms and Chattonella, additional diatom sampling suggested an even more complex scenario (fig. 3). As shown previously (Kamikawa et al. 2009), the Chattonella intron is closely related to, and shares strikingly high sequence similarity with, C. nana (fig. 3A). We found that the same intron was also present in the raphid pennate diatom, Cylindrotheca, and that it, too, shared exceptionally high sequence similarity and recent common ancestry with Chattonella (fig. 3B). The close relationship of the Cylindrotheca intron to homologs in a polar centric diatom (C. nana) and Chattonella—to the exclusion of the other raphid pennates (Kryptoperidinium and Phaeodactylum) in our data set—requires at least two separate events to reconcile this distribution. One hypothesis is a horizontal transfer from diatoms to Chattonella (Kamikawa et al. 2009) followed by a second diatom-to-diatom transfer to account for the close relationship (and high similarity) of the Cylindrotheca and C. nana introns. An alternative and equally parsimonious scenario would involve two separate intron transfers from Chattonella to diatoms, opposite the direction suggested by an earlier data set (Kamikawa et al. 2009). Many questions remain, however, particularly relating to the exceptionally high sequence similarity between the C. nana and Cylindrotheca introns (fig. 3), which suggests that the horizontal exchanges of these introns occurred very recently. It is noteworthy that while C. nana possessed this intron, another Cyclotella species, C. cryptica, did not. The very high similarity of the cox1_g2_B introns in C. nana and Cylindrotheca also suggests that their ancestry is distinct from cox1_g2_B introns in other diatoms, which exhibited levels of sequence divergence that were more on par with expectations (fig. 3B). Nuclear phylogenomic data have shown strong support for allopolyploid-driven whole-genome duplication in diatoms across phylogenetic scales similar to the diatom-to-diatom HGT event that may have occurred with cox1_g2_B intron (Parks et al. 2018). Species relationships based on a concatenated alignment of mitochondrial genes (supplementary fig. S2, Supplementary Material online) were congruent with large nuclear phylogenies (Parks et al. 2018), and we did not find strong or consistent evidence for incongruence between individual mitochondrial gene trees and the expected species tree (supplementary figs. S2 and S3, Supplementary Material online). This suggests that mitochondrial exchanges that may occur between diatoms are limited to mobile introns. Whereas organellar HGT tends to occur between relatively closely related species in other groups (Goddard and Burt 1999; Sanchez-Puerta et al. 2008), the events that may have occurred here would have taken place across much broader phylogenetic scales—either between distantly related diatoms or, greater still, between distantly related stramenopiles (fig. 3A). Finally, as discussed earlier, the cox1_g2_B intron may have experienced a rare retrotransposition event in the past as well (fig. 2, light and dark purple triangles), further highlighting the rich history of this single intron.
The ancestry and origins of many other introns, particularly the “one-off” introns present in a single diatom species, were often considerably more opaque. Many of these introns had no matches in GenBank, whereas others matched, at varying degrees, to fungi, haptophytes, or green algae (fig. 1 and supplementary table S2, Supplementary Material online). In some cases, it was not clear if these matches represented anything beyond deep shared ancestry of all group II introns, but it is noteworthy that: 1) many diatom mitochondrial introns have no matches in GenBank, 2) some matches to other lineages are quite strong, and 3) some groups show up repeatedly as the top matches to diatoms (supplementary table S2, Supplementary Material online). For example, many diatom mitochondrial introns are most similar to ones in green algae, which is also the case for the small number of known introns in diatom plastid genomes (Brembu et al. 2014; Ruck et al. 2017). The similarity between the cox1_g2_D introns of diatoms and fungi was also striking (fig. 1C). More data will hopefully show whether these relationships simply reflect database bias, more evidence of (very) deep shared ancestry and repeated intron loss, or (very) long-distance horizontal transfer. Any such transfers would likely involve an intermediary of some kind—shared viruses or bacteria, for example. There is an increasing appreciation for the importance of diatom–bacterial interactions (Amin et al. 2012), ranging from simple metabolite exchange (Durham et al. 2017) to bacterial inhibition of diatom cell division (van Tol et al. 2017). Plasmids often serve as vehicles for foreign DNA, and remnants of plasmid DNA have been found in the organelle genomes of many algal groups (Lee et al. 2016), including diatoms (Ruck et al. 2014). Moreover, plasmids can be delivered efficiently into diatoms in vitro through bacterial conjugation, providing a valuable tool for genetic transformation (Karas et al. 2015) and hinting at what might occur in the wild. Shared algal viruses, which can carry large amounts of DNA, might also mediate HGT among diatoms or across algal groups (Delaroque et al. 1999; Van Etten et al. 2002; Short 2012).
Discerning recent HGT events from recurrent losses of an ancestral gene is a notoriously challenging problem for large eukaryotic genomes, but these challenges are somewhat less pronounced for organellar genomes, which contain many fewer—and much better characterized—genes and introns, making it easier to identify and interrogate aberrant sequences. A relatively large body of evidence shows that organellar introns are indeed passed around, to varying degrees, by HGT. Not coincidentally, some of the clearest cases of HGT feature dense phylogenetic sampling (Goddard and Burt 1999; Sanchez-Puerta et al. 2008). Likewise, additional organelle genomes for diatoms, and microbial eukaryotes in general (Smith 2016b), will help further tease apart rates of intron loss and HGT as alternative explanations for the observed distributions of mitochondrial introns in diatoms.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank David Chafin, Jeff Pummill, and Pawel Wolinski for providing computational support through the Arkansas High Performance Computing Center (AHPCC), and the Chicago Botanic Garden for hosting and support of the Treubia and Fabronia computing clusters. This work was supported by the National Science Foundation (NSF) [grant numbers DEB-1353131 to A.J.A. and DEB-1353152 to N.J.W.] and multiple awards from the Arkansas Biosciences Institute to A.J.A. This research used computational resources available through the AHPCC, which were funded through multiple NSF grants and/or the Arkansas Economic Development Commission, and resources available at the Chicago Botanic Garden, which were funded by NSF (DEB-1239992 and DEB-1342873 to N.J.W.).
Literature Cited
- Alverson AJ, Beszteri B, Julius ML, Theriot EC.. 2011. The model marine diatom Thalassiosira pseudonana likely descended from a freshwater ancestor in the genus Cyclotella. BMC Evol Biol. 11:125.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SA, Parker MS, Armbrust EV.. 2012. Interactions between diatoms and bacteria. Microbiol Mol Biol Rev. 76(3):667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson JO. 2009. Gene transfer and diversification of microbial eukaryotes. Annu Rev Microbiol. 63:177–193. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Hirt RP, Foster PG, Roger AJ.. 2006. Evolution of four gene families with patchy phylogenetic distributions: influx of genes into protist genomes. BMC Evol Biol. 6:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrust EV. 2004. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306(5693):79–86. [DOI] [PubMed] [Google Scholar]
- Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J.. 2012. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 13(12):R122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen L. 2008. Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion 8(1):26–34. [DOI] [PubMed] [Google Scholar]
- Borowiec ML. 2016. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler C. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456(7219):239–244. [DOI] [PubMed] [Google Scholar]
- Brembu T, et al. . 2014. The chloroplast genome of the diatom Seminavis robusta: new features introduced through multiple mechanisms of horizontal gene transfer. Mar Genomics 16:17–27. [DOI] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Chin C-S, et al. . 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13(12):1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Qiu YL, Kuhlman P, Palmer JD.. 1998. Explosive invasion of plant mitochondria by a group I intron. Proc Natl Acad Sci U S A. 95(24):14244–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp A, Boschetti C, Perry M, Tunnacliffe A, Micklem G.. 2015. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 16(1):50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaroque N, Maier I, Knippers R, Müller DG.. 1999. Persistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J Gen Virol. 80(6):1367–1370. [DOI] [PubMed] [Google Scholar]
- Deschamps P, Moreira D.. 2012. Reevaluating the green contribution to diatom genomes. Genome Biol Evol. 4(7):683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham BP, et al. . 2017. Recognition cascade and metabolite transfer in a marine bacteria–phytoplankton model system. Environ Microbiol. 19(9):3500–3513. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara M, Watanabe KI, Ohama T.. 2000. Distribution of cognates of group II introns detected in mitochondrial cox1 genes of a diatom and a haptophyte. Gene 256(1–2):157–167. [DOI] [PubMed] [Google Scholar]
- Goddard MR, Burt A.. 1999. Recurrent invasion and extinction of a selfish gene. Proc Natl Acad Sci U S A. 96(24):13880–13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, et al. . 1998. Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res .26(4):865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G.. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29(8):1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellberg ME. 2006. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 6:24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotopp JCD. 2017. Grafting or pruning in the animal tree: lateral gene transfer and gene loss? bioRxiv 229468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, et al. . 2013. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 14(5):R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanian B, Pombert J-F, Dorrell RG, Burki F, Keeling PJ.. 2012. Tertiary endosymbiosis in two dinotoms has generated little change in the mitochondrial genomes of their dinoflagellate hosts and diatom endosymbionts. PLoS One 7(8):e43763.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikawa R, et al. . 2009. Mitochondrial group II introns in the raphidophycean flagellate Chattonella spp. suggest a diatom-to-Chattonella lateral group II intron transfer. Protist 160(3):364–375. [DOI] [PubMed] [Google Scholar]
- Karas BJ, et al. . 2015. Designer diatom episomes delivered by bacterial conjugation. Nat Commun. 6:6925.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Nei M, Dudley J, Tamura K.. 2008. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9(4):299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S.. 2004. Mobile group II introns. Annu Rev Genet. 38:1–35. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL.. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10(3):R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, et al. . 2016. Reconstructing the complex evolutionary history of mobile plasmids in red algal genomes. Sci Rep. 6:23744.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, et al. . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaack S.. 2006. Mutation pressure and the evolution of organelle genomic architecture. Science 311(5768):1727–1730. [DOI] [PubMed] [Google Scholar]
- Medlin LK. 2016. Evolution of the diatoms: major steps in their evolution and a review of the supporting molecular and morphological evidence. Phycologia 55(1):79–103. [Google Scholar]
- Minh BQ, Nguyen MAT, von Haeseler A.. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 30(5):1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock T, et al. . 2017. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 541(7638):536–540. [DOI] [PubMed] [Google Scholar]
- Moritz C, Dowling TE, Brown WM.. 1987. Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Syst. 18(1):269–292. [Google Scholar]
- Moustafa A, et al. . 2009. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324(5935):1724–1726. [DOI] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD.. 2007. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 7:135.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MW, Allmaier M, Eskes R, Schweyen RJ.. 1993. Transposition of group II intron aI1 in yeast and invasion of mitochondrial genes at new locations. Nature 366(6451):174–176. [DOI] [PubMed] [Google Scholar]
- Mullineux S-T, Costa M, Bassi GS, Michel F, Hausner G.. 2010. A group II intron encodes a functional LAGLIDADG homing endonuclease and self-splices under moderate temperature and ionic conditions. RNA 16(9):1818–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh N, Fukatsu T.. 2001. Evolutionary dynamics of multiple group I introns in nuclear ribosomal RNA genes of endoparasitic fungi of the genus Cordyceps. Mol Biol Evol. 18(9):1631–1642. [DOI] [PubMed] [Google Scholar]
- O’Brien MA, Misner I, Lane CE.. 2014. Mitochondrial genome sequences and comparative genomics of Achlya hypogyna and Thraustotheca clavata. J Eukaryot Microbiol. 61(2):146–154. [DOI] [PubMed] [Google Scholar]
- Oudot-Le Secq M-P, Green BR.. 2011. Complex repeat structures and novel features in the mitochondrial genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Gene 476(1–2):20–26. [DOI] [PubMed] [Google Scholar]
- Oudot-Le Secq M-P, Loiseaux-de Goër S, Stam WT, Olsen JL.. 2006. Complete mitochondrial genomes of the three brown algae (Heterokonta: phaeophyceae) Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis. Curr Genet. 49(1):47–58. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Herbon LA.. 1988. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 28(1–2):87–97. [DOI] [PubMed] [Google Scholar]
- Paquin B, et al. . 1997. The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr Genet. 31(5):380–395. [DOI] [PubMed] [Google Scholar]
- Parks MB, Wickett NJ, Alverson AJ.. 2018. Signal, uncertainty, and conflict in phylogenomic data for a diverse lineage of microbial eukaryotes (Diatoms, Bacillariophyta). Mol Biol Evol. 35(1):80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin NV, et al. . 2010. Complete sequence of the mitochondrial genome of a diatom alga Synedra acus and comparative analysis of diatom mitochondrial genomes. Curr Genet. 56(3):215–223. [DOI] [PubMed] [Google Scholar]
- Robart AR, Zimmerly S.. 2005. Group II intron retroelements: function and diversity. Cytogenet Genome Res. 110(1–4):589–597. [DOI] [PubMed] [Google Scholar]
- Ruck EC, Linard SR, Nakov T, Theriot EC, Alverson AJ.. 2017. Hoarding and horizontal transfer led to an expanded gene and intron repertoire in the plastid genome of the diatom, Toxarium undulatum (Bacillariophyta). Curr Genet. 63(3):499–507. [DOI] [PubMed] [Google Scholar]
- Ruck EC, Nakov T, Jansen RK, Theriot EC, Alverson AJ.. 2014. Serial gene losses and foreign DNA underlie size and sequence variation in the plastid genomes of diatoms. Genome Biol Evol. 6(3):644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha R, Mohr G, Belfort M, Lambowitz AM.. 1993. Group I and group II introns. FASEB J. 7(1):15–24. [DOI] [PubMed] [Google Scholar]
- Salomaki ED, Lane CE.. 2017. Red algal mitochondrial genomes are more complete than previously reported. Genome Biol Evol. 9(1):48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. 2017. Horizontal gene transfer is not a hallmark of the human genome. Genome Biol. 18(1):85.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, White O, Peterson J, Eisen JA.. 2001. Microbial genes in the human genome: lateral transfer or gene loss? Science 292(5523):1903–1906. [DOI] [PubMed] [Google Scholar]
- Sanchez-Puerta MV, Cho Y, Mower JP, Alverson AJ, Palmer JD.. 2008. Frequent, phylogenetically local horizontal transfer of the cox1 group I Intron in flowering plant mitochondria. Mol Biol Evol. 25(8):1762–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SM. 2012. The ecology of viruses that infect eukaryotic algae. Environ Microbiol. 14(9):2253–2271. [DOI] [PubMed] [Google Scholar]
- Simon D, Moline J, Helms G, Friedl T, Bhattacharya D.. 2005. Divergent histories of rDNA group I introns in the lichen family Physciaceae. J Mol Evol. 60(4):434–446. [DOI] [PubMed] [Google Scholar]
- Sloan DB, et al. . 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10(1):e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, et al. . 2010. The Dunaliella salina organelle genomes: large sequences, inflated with intronic and intergenic DNA. BMC Plant Biol. 10:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR. 2016a. The mutational hazard hypothesis of organelle genome evolution: 10 years on. Mol Ecol. 25(16):3769–3775. [DOI] [PubMed] [Google Scholar]
- Smith DR. 2016b. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics 15(5):373–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope MJ, et al. . 2001. Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature 411(6840):940–944. [DOI] [PubMed] [Google Scholar]
- Stoddard BL. 2014. Homing endonucleases from mobile group I introns: discovery to genome engineering. Mob DNA 5(1):7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanifuji G, Archibald JM, Hashimoto T.. 2016. Comparative genomics of mitochondria in chlorarachniophyte algae: endosymbiotic gene transfer and organellar genome dynamics. Sci Rep. 6:21016.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot EC, Ashworth MP, Nakov T, Ruck E, Jansen RK.. 2015. Dissecting signal and noise in diatom chloroplast protein encoding genes with phylogenetic information profiling. Mol Phylogenet Evol. 89:28–36. [DOI] [PubMed] [Google Scholar]
- Traller JC, et al. . 2016. Genome and methylome of the oleaginous diatom Cyclotella cryptica reveal genetic flexibility toward a high lipid phenotype. Biotechnol Biofuels 9(1):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten JL, Graves MV, Müller DG, Boland W, Delaroque N.. 2002. Phycodnaviridae – large DNA algal viruses. Arch Virol. 147(8):1479–1516. [DOI] [PubMed] [Google Scholar]
- van Tol HM, Amin SA, Armbrust EV.. 2017. Ubiquitous marine bacterium inhibits diatom cell division. ISME J. 11(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villain A, et al. . 2017. Complete mitochondrial genome sequence of the freshwater diatom Asterionella formosa. Mitochondrial DNA B 2(1):97–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Nickisch-Rosenegk M, Brown WM, Boore JL.. 2001. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that Platyhelminths are Eutrochozoans. Mol Biol Evol. 18(5):721–730. [DOI] [PubMed] [Google Scholar]
- Walker BJ, et al. . 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, et al. . 2013. Nannochloropsis plastid and mitochondrial phylogenomes reveal organelle diversification mechanism and intragenus phylotyping strategy in microalgae. BMC Genomics 14:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang EC, et al. . 2015. Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol Evol. 7(8):2394–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X-L, Cao M, Bi G-Q.. 2016. The complete mitochondrial genome of Pseudo-nitzschia multiseries (Baciuariophyta). Mitochondrial DNA A DNA Mapp Seq Anal. 27(4):2777–2778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.