

Abstract
Transient global cerebral ischemia causes delayed neuronal death in the hippocampal CA1 region. It also induces an increase in cyclooxygenase 2 (COX-2), which generates several metabolites of arachidonic acid, known as prostanoids, including prostacyclin (PGI2). To determine the role of the PGI2 receptor (IP) in post-ischemic delayed cell death, wild-type and IP knockout (IP−/−) C57Bl/6 mice were subjected to 12-min bilateral common carotid artery occlusion or sham surgery, followed by 7 days of reperfusion. In the sham-operated mice, no statistical difference in CA1 hip-pocampal neuronal density was observed between the wild-type (2836±18/mm2) and IP−/− (2793±43/mm2) mice. Interestingly, in animals subjected to ischemia, surviving neuronal density in wild-type mice decreased to 50.5±7.9% and that of IP−/− mice decreased to 23.0±4.5% of their respective sham-operated controls (P<0.05). The results establish a role for the IP receptor in protecting pyramidal hippocampal neurons after this global ischemic model and suggest that IP receptor agonists could be developed to prevent delayed pyramidal neuronal cell death.
Keywords: cerebral ischemia, G-protein-coupled receptors, hippocampus, prostaglandins, stroke
Clinical studies have shown that cardiac arrest often leads to delayed neuronal death in the vulnerable pyramidal cell layer of the hippocampal CA1 region (Zola-Morgan et al., 1986). Better understanding of mechanisms of this neuronal death is important to develop therapeutic approaches. Therapies such as free radical scavengers, calcium channel blockers, and others have been effective in animal models of stroke, but have not been effective in clinical trials thus far (Stroke Therapy Academic Industry Roundtable, 2001).
Increasing evidence indicates that neuroinflammation and its consequences play an important role in the neuronal injury caused by cerebral ischemia (Nakayama et al., 1998). Inflammation is at least partially mediated by one group of biologically active lipids known as prostanoids, which are generated by the rate-limiting enzyme, cyclooxygenase (COX). COX metabolizes arachidonic acid to the intermediate prostaglandin PGH2, which is then metabolized to produce PGE2, PGF2α, PGD2, thromboxane A2, and prostacyclin (PGI2). Although both COX-1 and COX-2 enzymes are constitutively expressed, COX-2 is highly inducible, notably in stroke (Smith et al., 1991; Sairanen et al., 1998; Iadecola et al., 1999). We and others have shown that COX-2 knockout as well as treatment of mice with COX-2 inhibitors, attenuates stroke damage. We previously reported that in transgenic mice with elevated neuronal COX-2, the use of a selective COX-2 inhibitor was not effective at reducing cerebral infarct size, suggesting that chronically high COX-2 levels might have already stimulated an inflammatory cascade that the inhibitor could not limit (Doré et al., 2003). Therefore, a better understanding of the downstream pathway modulated by COX enzymes is important. The prostanoids, which bind with various affinities to G-protein-coupled receptors known as EP, FP, DP, TP, and IP (Sharif et al., 1998), are considered to play complex roles—some protective and others detrimental—in the etiology of cellular injury (Smith et al., 1991; Sairanen et al., 1998; Iadecola et al., 1999; Ahmad et al., 2005, 2006). Hence, determination of the respective role of these prostanoid receptors is essential.
The COX-catalyzed product PGI2 is released in response to ischemia (Leffler et al., 1990, 1997; Faraci and Heistad, 1998) and elicits vascular dilation on binding to its IP receptor. Evidence suggests that it is one of the most important vascular relaxants for increasing cerebral blood flow (CBF) (Willis and Leffler, 1999). In an in vitro brain trauma model, the adherence of activated platelets to percussed human cerebral microvascular endothelial cells was blocked by the addition of PGI2; in another model, leukocyte depletion and administration of iloprost, a PGI2 analog, was shown to reduce the leukocyte accumulation (Orfeo et al., 1994; Taoka et al., 1997). Therefore, here, we subjected wild-type (WT) and IP knockout (IP−/−) mice to a transient bilateral common carotid artery occlusion model to examine the role of the IP receptor in delayed neuronal injury in the hippocampus. Because PGI2 also has been reported to be involved in regulation of multiple functions in vivo, particularly in blood pressure regulation (Scotland et al., 2005), we used stringent physiologic parameter monitoring and temperature control.
EXPERIMENTAL PROCEDURES
Animals
Experiments and procedures were conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals. Protocols were approved by the Johns Hopkins Animal Care and Use Committee and were designed to minimize distress and number of animals. WT and IP−/− C57Bl/6 male mice (22–29 g) were obtained from our breeding colony, which was originated by Dr. Shuh Narumiya (Kyoto University, Kyoto, Japan) (Murata et al., 1997), and genotyped as described previously (Kobayashi et al., 2004).
Blood vessel anatomy
To determine the characteristics of cerebral blood vessel anatomy, three animals per genotype were perfused with normal saline for approximately 5 min and then injected with 0.3 mL of black latex, as described previously (Maeda et al., 1998). The vessel branches were viewed under a microscope, and photographs were acquired with Intellicam software (Matrox Graphics Inc., Dorval, QC, Canada). The posterior communicating artery (PcomA) was evaluated qualitatively in each hemisphere with a score of 0–3 following a protocol described before (Murakami et al., 1997), in which 0=no connection between anterior and posterior circulation; 1=anastomosis in the capillary phase; 2=small truncal PcomA; and 3=truncal PcomA.
Murine global ischemia by transient occlusion of the bilateral common carotid artery
Forebrain global ischemia was induced as described previously (Olsson et al., 2003), with modifications for the mouse. Briefly, under halothane anesthesia (1–5%), mice were intubated with a small-animal respirator (Harvard, type 845, Harvard Inc., Holliston, MA, USA), and both common carotid arteries were occluded with aneurysm clips for 12 min. A preliminary series of experiments revealed that a 12-min occlusion period gave us the optimal and most reproducible effects with this mouse model (data not shown). Reperfusion was begun by release of the clips. After 15 min of reperfusion, animals were disconnected from the ventilator. Mice were placed into a warm incubator (33–34 °C) for 24 h before being returned to their cages. In our experience, the core body temperature can be maintained at approximately 37 °C under these conditions (data not shown). After 24 h, the animals can maintain their core temperature at approximately 37 °C without assistance. For the sham-operated groups, mice underwent the same surgical procedures as the ischemic group, with the exception that the common carotid arteries were not occluded.
Physiologic parameter measurements
Rectal and head temperatures were controlled between 36.5 and 37.5 °C by heat blanket and lamp and were monitored by a rectal probe and a subdermal probe in the parietal bone region. Arterial blood pressure was measured by right femoral artery cannulation with PE-10 tubing; these animals also were used for measurement of neuronal damage outcomes (see below). CBF in each hemisphere was measured transcranially by laser-Doppler flowmetry via probes placed on each temple. Mean arterial blood pressure (MABP) and CBF were recorded continuously during 10 min of baseline, 12 min of global ischemia, and the first 15 min of reperfusion. Change in CBF in each hemisphere was calculated as a percentage of baseline. Blood gas measurements were assessed before baseline recording and 15 min after global ischemia. To sample temperature every 30 s, transponders (IPTT-200, IPTT-300 Bio Medic Data Systems, Inc., Seaford, DE, USA) were implanted into the abdomen of the mice 1 week before surgery. Body temperature was monitored before ischemia, 30 min and 24 h after ischemia, and then once daily until animals were killed 7 days after ischemia.
Neuronal damage following forebrain global ischemia
Seven days after ischemia, the brains were harvested, embedded in paraffin, and cut into 10-μm sections, which were stained with hematoxylin and eosin. Ischemic neuronal damage in the hippocampal CA1 region, a region highly vulnerable to global ischemia, was evaluated at the coronal levels corresponding to −1.94±0.01, −2.42±0.01, and −2.90±0.01 mm from the bregma (Kitagawa et al., 1998a; Sheng et al., 1999; Olsson et al., 2003). We used the most linear section of the CA1 region, as seen under low magnification and aligned with the parallel lines of the microscope grid. The diameter (D) of the observed field (100×) was obtained by the standard grid-lined slide provided by the microscope manufacturer. Numbers of surviving neurons in the CA1 regions were counted in each hemisphere under light microscopy (100× oil lens) and expressed as the average number/mm2 (Kitagawa et al., 1998a; Sheng et al., 1999; Olsson et al., 2003).
Statistical analysis
One-way ANOVA followed by a Newman Keul’s multiple comparisons test was used to compare control and treated groups. To compare two groups, an unpaired Student’s t-test was used. Data are presented as means±standard error of the mean, and a P value<0.05 was considered statistically significant.
RESULTS
Mouse genotype and cerebral vessel anatomy
Polymerase chain reaction confirmed the IP−/− genotype of the mice (Fig. 1A). Because vessel anatomy/plasticity of the PcomA can affect outcome in the global ischemia model, we compared the major features of vessel anatomy of the mice. We found that the vessel anatomy of the PcomA of the IP−/− and WT mice was not remarkably different. Following the scale system of Murakami et al. (1997), the mean score values of PcomA plasticity were 1.5±0.2 in IP−/− (n=12) and 1.3±0.2 in WT (n=10) mice. The features of vessel anatomy of the mice are shown in Fig. 1B and C.
Fig. 1.
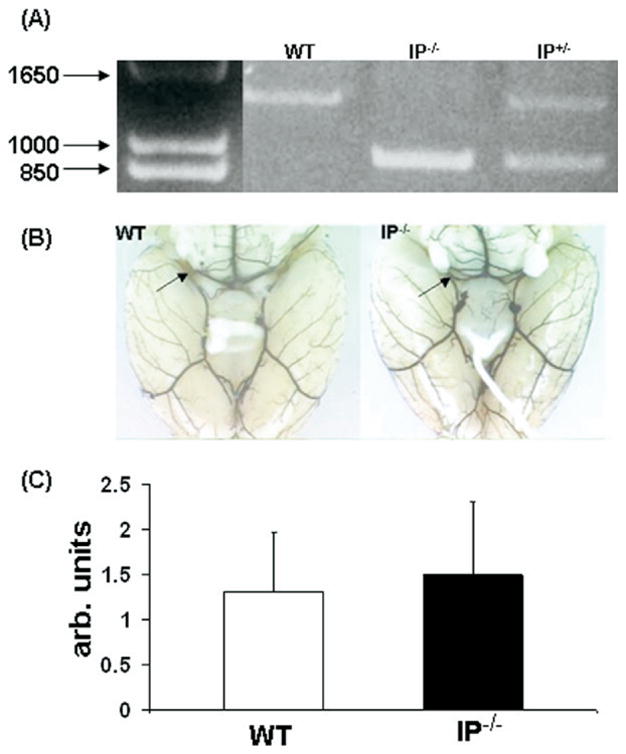
Genotype and vascular features of IP−/− and WT mice. (A) IP−/− genotype was determined by PCR. (B) Mice of each genotype were injected with black latex to assess the major features of vessel anatomy of the PcomA. (C) Status of the PcomA in WT and IP−/− mice was evaluated qualitatively by a 0–3 scoring system as described in Experimental Procedures. No significant anatomical differences in the PcomA were observed in IP−/− and WT mice. arb, Arbitrary.
Physiologic parameters
To minimize possible variations in neuronal injury from changes or differences in physiologic parameters, body core temperature, MABP, and arterial blood pH and gases (PaO2 and PaCO2) were carefully monitored. We observed that MABP (which was controlled to between 60 and 80 mm Hg during the 10 min before occlusion) increased during the initial 3 min after occlusion, reaching 101±6 and 97±2 in the WT and IP−/− groups, respectively. In contrast, the MABP remained 73±3 and 67±7 mm Hg in the sham-operated WT and IP−/− groups, respectively. Upon reperfusion, the MABP of the WT and IP−/− groups that underwent forebrain global ischemia declined to 56±5 and 58±3 mm Hg, respectively, while that of the sham-operated groups remained fairly constant (70±3 and 73±7 mm Hg, respectively; Fig. 2A). No significant difference between WT and IP−/− groups was observed at any of the time points.
Fig. 2.
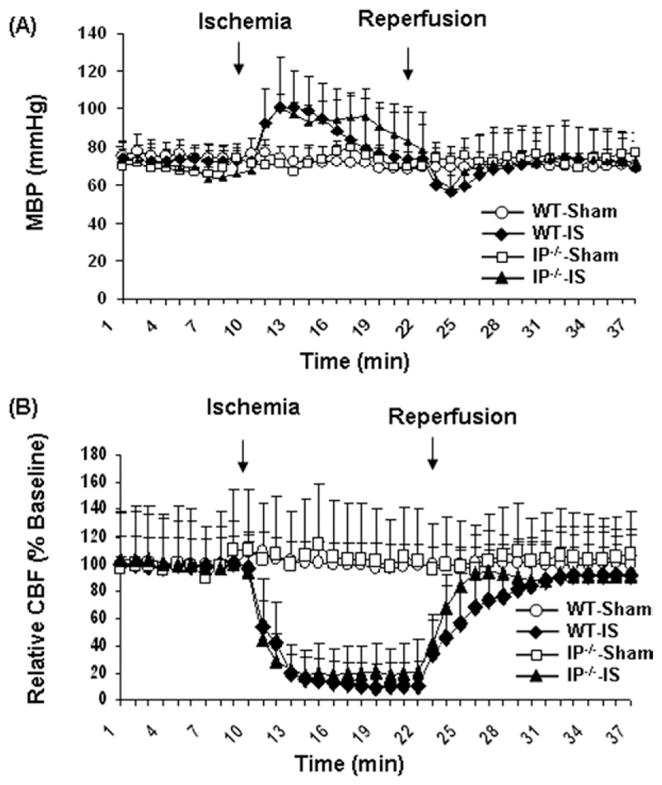
MABP and relative CBF of sham-operated mice and mice subjected to 12 min of forebrain global ischemia followed by 7 days of reperfusion. The changes in MABP (A) and CBF (B) that occurred during and immediately after ischemia were not significantly different in WT and IP−/− mice.
During ischemia, the CBF in both hemispheres of the WT and IP−/− mouse brains dropped dramatically, reaching a minimum at about 8–9 min after occlusion (Fig. 2B). There were no significant differences in the CBF decrease between left and right hemispheres or between the WT and IP−/− groups at any time during the arterial occlusion.
Blood gas analysis revealed no significant differences in pH, PaCO2, or PaO2 between either the WT or IP−/− mice that underwent ischemia (n=11 WT, 8 IP−/−) and the sham-operated mice (n=6 WT, 2 IP−/−; Table 1). At baseline, the mean core body temperature was 36.7± 0.3 °C in the sham-operated WT group, 36.0±1.3 °C in the ischemic WT group, 36.0±1.1 °C in the sham-operated IP−/− group, and 36.7±0.7 °C in the ischemic IP−/− group, and these values did not change significantly during the early period of reperfusion or throughout the 7 days of reperfusion (data not shown).
Table 1.
Measurement of arterial blood gas in mice
| Group | pH | PaCO2 | PaO2 | |||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| 10 Min pre-ischemia | 15 Min post-ischemia | 10 Min pre-ischemia | 15 Min post-ischemia | 10 Min pre-ischemia | 15 Min post-ischemia | |
| Sham WT | 7.34±0.02 | 7.31±0.03 | 38.9±1.6 | 42.2±1.5 | 132±8.9 | 123±5.3 |
| Ischemic WT | 7.34±0.01 | 7.32±0.02 | 39.1±0.6 | 39.8±1.5 | 141±4.4 | 125±6.0 |
| Sham IP−/− | 7.33±0.03 | 7.35±0 | 37.4±3.3 | 40.4±0.7 | 121±21 | 126±6 |
| Ischemic IP−/− | 7.35±0.02 | 7.41±0.02 | 39.9±1.2 | 39.4±1.9 | 134±4.9 | 119±4.5 |
Neuronal damage following forebrain global ischemia
Reperfusion periods after cerebral global ischemia vary in the literature. Most often animal survival is for 4–7 days after ischemia (Ohtaki et al., 2003; Gillingwater et al., 2004; Olsson et al., 2004; Yonekura et al., 2004). The 7-day survival time was chosen to allow sufficient time for alterations in the rate of maturation of cell death, which could be influenced by gene deletion. The number of mice that did not survive to the end of the experimental protocol was estimated at about 30–40%, but no difference was observed between the respective genotypes. The neuronal loss in the CA1 region was significantly higher in the ischemic WT and IP−/− mice than in the sham-operated mice. The density of surviving neurons in the CA1 region of the sham-operated WT mice (2836±18/mm2; n=12) was not significantly different than that of the sham-operated IP−/− mice (2793±43/mm2; n=8). In the WT mice that underwent global ischemia (n=22), the density of surviving neurons was 1431±195, or 50.5±7.9% that of the sham- operated WT mice. The density of surviving neurons dramatically decreased to 642±72/mm2 in the IP−/− mice (n=16) that underwent global ischemia, a value that was only 23.0±4.5% of the sham-operated IP−/− mice. Thus, the density of surviving neurons was significantly less in IP−/− mice than in WT after ischemia and reperfusion (P<0.05; Fig. 3). These data indicate that genetic deletion of the IP receptor increased the neuronal loss in the CA1 region of hippocampus after transient global cerebral ischemia.
Fig. 3.
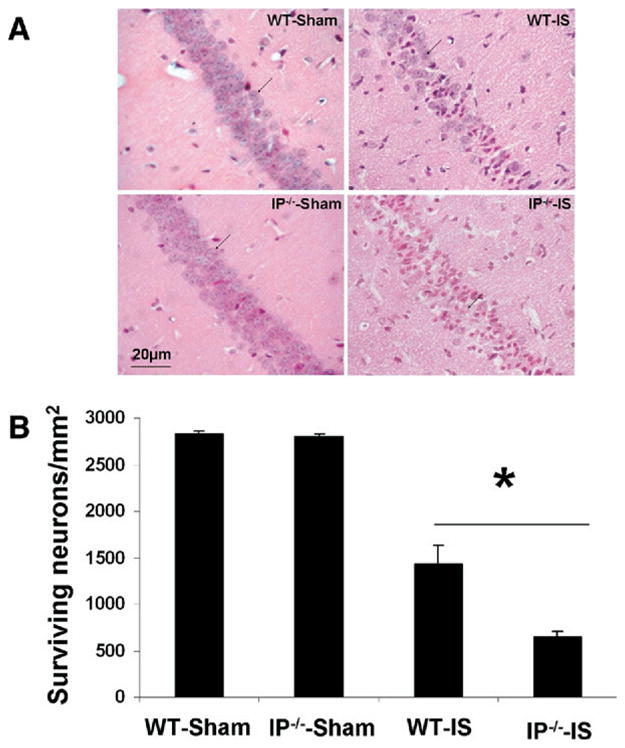
Neuronal viability in the hippocampal CA1 region of sham-operated mice and mice subjected to 12 min of global ischemia followed by 7 days of reperfusion. (A) Typical histological features of the neurons in the CA1 region (arrows point to surviving neurons). (B) Statistical results show significantly greater neuronal loss in the CA1 region of IP−/− mice than of WT mice. * P<0.05; IS: ischemia.
DISCUSSION
In this study, we showed that in mice subjected to 12 min of forebrain global cerebral ischemia followed by 7 days of reperfusion, genetic deletion of the PGI2 IP receptor results in greater delayed neuronal loss in the CA1 region of the hippocampus than that seen in WT mice. The data imply a protective role for the IP receptor in delayed pyramidal neuron cell death. Our findings in C57Bl/6 male mice are also consistent with those of other studies showing that relatively selective IP receptor agonists reduce delayed neuronal loss in gerbil and rat (Awad et al., 1983; Dohi, 1987; Karasawa et al., 2002, 2003; Takamatsu et al., 2002). However, affinity and selectivity of pharmacological analogs to the G-protein-coupled receptors are often limiting factors for optimal interpretation of the experimental results (Narumiya et al., 1999). To the best of our knowledge, this is the first study to find the IP receptor protective by using knockout animals, and therefore is free of the non-selective side effects of the PGI2 analogs or IP agonists. It is also the first study to strictly monitor several physiologic parameters and evaluate neuronal injury outcomes being conducted within one cohort of mice, an approach that may reduce discrepancies in the results (Castillo et al., 2004; Huang and McNamara, 2004).
Global cerebral ischemia causes marked damage to pyramidal neurons in the hippocampal CA1 region within days after ischemia/reperfusion in animals and humans (Zola-Morgan et al., 1986; Sheng et al., 1999; Yonekura et al., 2004). Neuronal loss in the hippocampus starts between 24 and 48 h after global ischemia and stabilizes at approximately 4 days of reperfusion, i.e. the neuronal loss does not significantly increase after 4 days (Yonekura et al., 2004). However, we believe that a 7-day reperfusion period is most reliable for two main reasons. First, in our preliminary study, we found the number of surviving neurons to be most reproducible at 7 days after reperfusion. Second, it has been shown that cerebral ischemia causes long-lasting inhibition of protein synthesis despite recovery of energy metabolism (Olsson et al., 2003). This latter point was highlighted in a study showing that gerbils that underwent sublethal ischemia needed a 7-day recirculation period before protein synthesis in hippocampal neurons returned to a normal level (Sorimachi et al., 1999, 2002). Hence, we used a 7-day post-ischemia recovery period to ensure maturation of delayed neuronal death.
In the experimental models, it has been difficult to obtain uniform injury because of the variability of the PcomAs (Kitagawa et al., 1998b). Some investigators have used a cardiac arrest model that excludes the effect of the PcomAs on the outcomes of ischemic injury (Thel and O’Connor, 1999; Kawahara et al., 2002; Abella et al., 2004). However, in the cardiac arrest model, the neuronal injury is highly dependent on the duration of cardiac arrest; an increase in the duration of cardiac arrest is always associated with a large increase in mortality. Furthermore, it has been reported that an increase in head temperature could produce a comparable increase in neuronal injury, so distinct hyperthermic pathways might be involved (Kawahara et al., 2002). Resuscitation in the mouse model is achieved by chest compressions at a rate of approximately 400 beats per min; variations in this technique could potentially increase variability within groups and between experimenters (Thel and O’Connor, 1999; Abella et al., 2004). To minimize potential inconsistency, others have tried to establish a threshold of cerebral flow reduction to limit variations induced by blood flow reduction. It is also known that physiologic parameters, such as blood pressure and blood gas, critically influence the outcome of ischemic neuronal death (Castillo et al., 2004; Huang and McNamara, 2004). We believe that tight monitoring of physiologic parameters to within normal range is critical for the outcome of CA1 injury in the global ischemia model. To minimize the effect of physiologic changes on neuronal injury, we closely monitored the physiologic parameters of blood pressure, relative CBF, and blood gases, and evaluated CA1 injury in a single cohort of animals.
PGI2 and its receptor (IP) play multiple physiological roles in regulating intracellular cAMP levels and CBF (Whittle et al., 1978; Boie et al., 1994; Nakagawa et al., 1994; Willis and Leffler, 1999). In the brain, IP receptors are expressed in the hippocampus, cerebral cortex, thalamus, striatum, and septum (Breyer et al., 2001) and are linked to heterotrimetric G-proteins that stimulate adenylyl cyclase to produce cAMP. Intracellular cAMP concentration has been shown to increase after stimulation of the recombinant human IP receptors in the vasculature (Whittle et al., 1978; Boie et al., 1994; Nakagawa et al., 1994). In addition, IP, which is widely distributed in the microvessels, participates in various aspects of vascular function, including vascular dilation (Regan et al., 1994; Wright et al., 2001). PGI2 is also released in response to ischemia (Leffler et al., 1990, 1997; Faraci and Heistad, 1998), and evidence suggests that it is an important vascular relaxant for increasing blood flow to the CNS (Willis and Leffler, 1999). PGI2 also has anti-inflammatory properties: it inhibits platelet aggregation, leukocyte activation, and leukocyte–endothelial interactions during ischemia (Whittle et al., 1978; Dougherty et al., 1982; Anwaar et al., 1998; Catella-Lawson, 2001). However, it should be noted that the inflammatory response to global ischemia is less than that observed in focal ischemia. Evidence has shown that microglia and astroglia are activated, but whether this activation is in response to delayed neuronal damage in global ischemia still is under debate (Stoll et al., 1998).
Evidence showing that PGI2 activity decreases interstitial glucose, and increases lactate and glycerol in an experimental model of brain edema induced by lipopolysaccharide (Gardenfors et al., 2004) suggests that PGI2 may be involved in the damage following cerebral ischemia through modulation of glucose and glycerol metabolism. However, numerous studies have demonstrated that PGI2 analogs reduce memory impairment in rodents with cerebral embolism, suggesting a role for the IP receptor in modulation of neurological impairment (Awad et al., 1983; Dohi, 1987; Karasawa et al., 2002, 2003; Takamatsu et al., 2002). Researchers have observed that PGI2 analogs provide neuronal protection and improve the outcome of learning and memory defects in the cerebral ischemia models of focal as well as global ischemia (Awad et al., 1983; Dohi, 1987; Karasawa et al., 2002, 2003; Takamatsu et al., 2002). In a gerbil global ischemia model and in a rat cerebral embolism ischemia model, it was shown that PGI2 analogs reduced the neuronal loss in the hippocampal CA1 region and decreased memory impairments (Uchiyama-Tsuyuki et al., 1995; Matsuda et al., 1997; Cui et al., 1999; Satoh et al., 1999), further advocating the potential development of PGI2 analogs for treating patients with ischemic injury, particularly delayed neuronal injury.
Clinical trials of PGI2 analogs have been conducted in patients with vascular diseases. For example, PGI2 analogs iloprost and cicaprost have been used to treat patients with peripheral ischemia (Beischer et al., 1998; Ciuffetti et al., 2003) and as therapeutic agents for ischemic CNS injury (Moncada, 1982; Gryglewski et al., 1983). A clinical trial of cicaprost failed to help patients with a specific disease such as Raynaud’s syndrome (Lau et al., 1991), but evidence suggests that PGI2 is effective in treating heart failure that occurs as a result of cardiac bypass surgery (Ocal et al., 2005). The effect of PGI2 and its analogs on acute ischemic stroke outcome has not been clearly established in clinical trials because of low patient numbers, necessitating additional clinical trials to obtain conclusive results regarding the ability of PGI2 and its analogs to treat acute ischemic stroke (Bath, 2004). Considering that PGI2 analogs can also bind to other prostanoid receptors, albeit with lower affinity, adverse effects may occur as a result of unspecific binding (Narumiya and FitzGerald, 2001). Therefore, our use of IP−/− mice to investigate the role of the IP receptor in delayed neuronal injury provides new insight and rationale for using IP receptor agonists as pharmacologic interventions to be tested in preclinical and clinical trials.
Our findings may also be helpful in clarifying the mechanism by which the COX-2 inhibitors can cause side effects, such as the reported increased risk of cardiovascular disease. In addition, these results are likely to be useful for the development of PGI2 mimetics that are permeable to the blood–brain barrier and more selective than those currently available. Finally, since regulation of the IP receptors can improve neuronal survival in the CA1 region of the hippocampus in global ischemia, a drug to stimulate the IP receptor might also be useful for preventing cognitive-related deficit after heart transplant surgery or cardiac arrest.
Acknowledgments
The authors would like to thank Claire Levine for her assistance in preparing this manuscript and Adam Sapirstein, Zengjing Yang, Ité Alonzo, Ellen Gordes, and every member of the Doré laboratory team for their helpful contributions. Part of this work was presented at the 2006 annual meeting of the Society for Neuroscience, Atlanta, GA. Grant support: This work was supported in part by the National Institutes of Heath: NINDS (NS046400) and the NIA (AG022971) (S.D.).
Abbreviations
- CBF
cerebral blood flow
- COX
cyclooxygenase
- IP
prostacyclin receptor
- MABP
mean arterial blood pressure
- PcomA
posterior communicating artery
- PGI2
prostacyclin
- WT
wild type
References
- Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation. 2004;109:2786–2791. doi: 10.1161/01.CIR.0000131940.19833.85. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Ahmad M, de Brum-Fernandes AJ, Doré S. Prostaglandin EP4 receptor agonist protects against acute neurotoxicity. Brain Res. 2005;1066:71–77. doi: 10.1016/j.brainres.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- Anwaar I, Gottsater A, Ohlsson K, Mattiasson I, Lindgarde F. Increasing levels of leukocyte-derived inflammatory mediators in plasma and cAMP in platelets during follow-up after acute cerebral ischemia. Cerebrovasc Dis. 1998;8:310–317. doi: 10.1159/000015873. [DOI] [PubMed] [Google Scholar]
- Awad I, Little JR, Lucas F, Skrinska V, Slugg R, Lesser RP. Treatment of acute focal cerebral ischemia with prostacyclin. Stroke. 1983;14:203–209. doi: 10.1161/01.str.14.2.203. [DOI] [PubMed] [Google Scholar]
- Bath PM. Prostacyclin and analogues for acute ischaemic stroke. Cochrane Database Syst Rev. 2004;(3):CD000177. doi: 10.1002/14651858.CD000177.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beischer W, Dembski JC, Gruss JD, Hofgartner F, Horsch A, Horsch S, Kuhlmann HW, Loose DA, Mietaschk A, Schwilden ED, Spengel F, Spitzer W, Staben P, Stallkamp B, Sturzebecher CS, Tokhi M, von Bilderling P. Low-dose iloprost infusions compared to the standard dose in patients with peripheral arterial occlusive disease Fontaine stage IV. DAWID Study Group. Vasa. 1998;27:15–19. [PubMed] [Google Scholar]
- Boie Y, Rushmore TH, Darmon-Goodwin A, Grygorczyk R, Slipetz DM, Metters KM, Abramovitz M. Cloning and expression of a cDNA for the human prostanoid IP receptor. J Biol Chem. 1994;269:12173–12178. [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Castillo J, Leira R, Garcia MM, Serena J, Blanco M, Davalos A. Blood pressure decrease during the acute phase of ischemic stroke is associated with brain injury and poor stroke outcome. Stroke. 2004;35:520–526. doi: 10.1161/01.STR.0000109769.22917.B0. [DOI] [PubMed] [Google Scholar]
- Catella-Lawson F. Vascular biology of thrombosis: platelet-vessel wall interactions and aspirin effects. Neurology. 2001;57:S5–S7. doi: 10.1212/wnl.57.suppl_2.s5. [DOI] [PubMed] [Google Scholar]
- Ciuffetti G, Sokola E, Lombardini R, Pasqualini L, Pirro M, Mannarino E. The influence of iloprost on blood rheology and tissue perfusion in patients with intermittent claudication. Kardiol Pol. 2003;59:197–204. [PubMed] [Google Scholar]
- Cui Y, Kataoka Y, Satoh T, Yamagata A, Shirakawa N, Watanabe Y, Suzuki M, Yanase H, Kataoka K. Protective effect of prostaglandin I(2) analogs on ischemic delayed neuronal damage in gerbils. Biochem Biophys Res Commun. 1999;265:301–304. doi: 10.1006/bbrc.1999.1671. [DOI] [PubMed] [Google Scholar]
- Dohi S. Effects of a intravenous or subarachnoid PGI2 analog (OP-41483) on cerebral and spinal cord blood flow in dogs. Masui. 1987;36:1790–1795. [PubMed] [Google Scholar]
- Doré S, Otsuka T, Mito T, Sugo N, Hand T, Wu L, Hurn PD, Traystman RJ, Andreasson K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol. 2003;54:155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- Dougherty JH, Jr, Levy DE, Rawlinson DG, Ruff R, Weksler BB, Plum F. Experimental cerebral ischemia produced by extracranial vascular injury: protection with indomethacin and prostacyclin. Neurology. 1982;32:970–974. doi: 10.1212/wnl.32.9.970. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Gardenfors F, Nilsson A, Ungerstedt U, Nordstrom CH. Adverse biochemical and physiological effects of prostacyclin in experimental brain oedema. Acta Anaesthesiol Scand. 2004;48:1316–1321. doi: 10.1111/j.1399-6576.2004.00522.x. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Haley JE, Ribchester RR, Horsburgh K. Neuroprotection after transient global cerebral ischemia in Wld(s) mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Nowak S, Kostka-Trabka E, Kusmiderski J, Dembinska-Kiec A, Bieron K, Basista M, Blaszczyk B. Treatment of ischaemic stroke with prostacyclin. Stroke. 1983;14:197–202. doi: 10.1161/01.str.14.2.197. [DOI] [PubMed] [Google Scholar]
- Huang Y, McNamara JO. Ischemic stroke: “acidotoxicity” is a perpetrator. Cell. 2004;118:665–666. doi: 10.1016/j.cell.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol (Berl) 1999;98:9–14. doi: 10.1007/s004010051045. [DOI] [PubMed] [Google Scholar]
- Karasawa Y, Hitomi T, Komiyama H, Isobe Y, Kobayashi T, Yoshida S, Nakaike S, Araki H. Effect of TTC-909 in a middle cerebral artery thrombosis model in stroke-prone spontaneously hypertensive rats. Eur J Pharmacol. 2002;449:127–133. doi: 10.1016/s0014-2999(02)01945-3. [DOI] [PubMed] [Google Scholar]
- Karasawa Y, Komiyama H, Yoshida S, Hino N, Katsuura Y, Nakaike S, Araki H. Effect of TTC-909 on cerebral infarction following permanent occlusion of the middle cerebral artery in stroke prone spontaneously hypertensive rats. J Pharmacol Sci. 2003;91:305–312. doi: 10.1254/jphs.91.305. [DOI] [PubMed] [Google Scholar]
- Kawahara N, Kawai K, Toyoda T, Nakatomi H, Furuya K, Kirino T. Cardiac arrest cerebral ischemia model in mice failed to cause delayed neuronal death in the hippocampus. Neurosci Lett. 2002;322:91–94. doi: 10.1016/s0304-3940(02)00101-5. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou JC, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998a;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab. 1998b;18:570–579. doi: 10.1097/00004647-199805000-00012. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CS, McLaren M, Saniabadi A, Scott N, Belch JJ. The pharmacological effects of cicaprost, an oral prostacyclin analogue, in patients with Raynaud’s syndrome secondary to systemic sclerosis—a preliminary study. Clin Exp Rheumatol. 1991;9:271–273. [PubMed] [Google Scholar]
- Leffler CW, Mirro R, Armstead WM, Busija DW, Thelin O. Prostanoid synthesis and vascular responses to exogenous arachidonic acid following cerebral ischemia in piglets. Prostaglandins. 1990;40:241–248. doi: 10.1016/0090-6980(90)90012-k. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Smith JS, Edrington JL, Zuckerman SL, Parfenova H. Mechanisms of hypoxia-induced cerebrovascular dilation in the newborn pig. Am J Physiol. 1997;272:H1323–H1332. doi: 10.1152/ajpheart.1997.272.3.H1323. [DOI] [PubMed] [Google Scholar]
- Maeda K, Hata R, Hossmann KA. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport. 1998;9:1317–1319. doi: 10.1097/00001756-199805110-00012. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Wen TC, Karasawa Y, Araki H, Otsuka H, Ishihara K, Sakanaka M. Protective effect of a prostaglandin I2 analog, TEI-7165, on ischemic neuronal damage in gerbils. Brain Res. 1997;769:321–328. doi: 10.1016/s0006-8993(97)00724-5. [DOI] [PubMed] [Google Scholar]
- Moncada S. Eighth Gaddum memorial lecture. University of London Institute of Education, December 1980. Biological importance of prostacyclin. Br J Pharmacol. 1982;76:3–31. doi: 10.1111/j.1476-5381.1982.tb09186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Epstein CJ, Chan PH. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke. 1997;28:1797–1804. doi: 10.1161/01.str.28.9.1797. [DOI] [PubMed] [Google Scholar]
- Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, Ichikawa A, Aze Y, Tanaka T, Yoshida N, Ueno A, Ohishi S, Narumiya S. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Tanaka I, Usui T, Harada M, Sasaki Y, Itoh H, Yoshimasa T, Namba T, Narumiya S, Nakao K. Molecular cloning of human prostacyclin receptor cDNA and its gene expression in the cardiovascular system. Circulation. 1994;90:1643–1647. doi: 10.1161/01.cir.90.4.1643. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Ocal A, Kiris I, Erdinc M, Peker O, Yavuz T, Ibrisim E. Efficiency of prostacyclin in the treatment of protamine-mediated right ventricular failure and acute pulmonary hypertension. Tohoku J Exp Med. 2005;207:51–58. doi: 10.1620/tjem.207.51. [DOI] [PubMed] [Google Scholar]
- Ohtaki H, Mori S, Nakamachi T, Dohi K, Yin L, Endo S, Okada Y, Shioda S. Evaluation of neuronal cell death after a new global ischemia model in infant mice. Acta Neurochir Suppl. 2003;86:97–100. doi: 10.1007/978-3-7091-0651-8_22. [DOI] [PubMed] [Google Scholar]
- Olsson T, Nygren J, Hakansson K, Lundblad C, Grubb A, Smith ML, Wieloch T. Gene deletion of cystatin C aggravates brain damage following focal ischemia but mitigates the neuronal injury after global ischemia in the mouse. Neuroscience. 2004;128:65–71. doi: 10.1016/j.neuroscience.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Olsson T, Wieloch T, Smith ML. Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade. Brain Res. 2003;982:260–269. doi: 10.1016/s0006-8993(03)03014-2. [DOI] [PubMed] [Google Scholar]
- Orfeo T, Doherty JM, Adey G, Penar PL, Shatos MA. Sublethal percussion trauma in vitro causes a persisting derangement in the nonthrombogenic properties of brain endothelial cells. J Trauma. 1994;37:347–357. doi: 10.1097/00005373-199409000-00003. [DOI] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46:213–220. [PubMed] [Google Scholar]
- Stroke Therapy Academic Industry Roundtable. Recommendations for clinical trial evaluation of acute stroke therapies. Stroke. 2001;32:1598–1606. doi: 10.1161/01.str.32.7.1598. [DOI] [PubMed] [Google Scholar]
- Sairanen T, Ristimaki A, Karjalainen-Lindsberg ML, Paetau A, Kaste M, Lindsberg PJ. Cyclooxygenase-2 is induced globally in infarcted human brain. Ann Neurol. 1998;43:738–747. doi: 10.1002/ana.410430608. [DOI] [PubMed] [Google Scholar]
- Satoh T, Ishikawa Y, Kataoka Y, Cui Y, Yanase H, Kato K, Watanabe Y, Nakadate K, Matsumura K, Hatanaka H, Kataoka K, Noyori R, Suzuki M. CNS-specific prostacyclin ligands as neuronal survival-promoting factors in the brain. Eur J Neurosci. 1999;11:3115–3124. doi: 10.1046/j.1460-9568.1999.00791.x. [DOI] [PubMed] [Google Scholar]
- Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111:796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Xu SX, Williams GW, Crider JY, Griffin BW, Davis TL. Pharmacology of [3H]prostaglandin E1/[3H]prostaglandin E2 and [3H]prostaglandin F2alpha binding to EP3 and FP prostaglandin receptor binding sites in bovine corpus luteum: characterization and correlation with functional data. J Pharmacol Exp Ther. 1998;286:1094–1102. [PubMed] [Google Scholar]
- Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Characterization of a recovery global cerebral ischemia model in the mouse. J Neurosci Methods. 1999;88:103–109. doi: 10.1016/s0165-0270(99)00018-7. [DOI] [PubMed] [Google Scholar]
- Smith WL, Marnett LJ, DeWitt DL. Prostaglandin and thromboxane biosynthesis. Pharmacol Ther. 1991;49:153–179. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- Sorimachi T, Abe H, Takeuchi S, Tanaka R. Neuronal damage in gerbils caused by intermittent forebrain ischemia. J Neurosurg. 1999;91:835–842. doi: 10.3171/jns.1999.91.5.0835. [DOI] [PubMed] [Google Scholar]
- Sorimachi T, Abe H, Takeuchi S, Tanaka R. Ischemic depolarization monitoring: evaluation of protein synthesis in the hippocampal CA1 after brief unilateral ischemia in a gerbil model. J Neurosurg. 2002;97:104–111. doi: 10.3171/jns.2002.97.1.0104. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Takamatsu H, Tsukada H, Watanabe Y, Cui Y, Kataoka Y, Hosoya T, Suzuki M. Specific ligand for a central type prostacyclin receptor attenuates neuronal damage in a rat model of focal cerebral ischemia. Brain Res. 2002;925:176–182. doi: 10.1016/s0006-8993(01)03280-2. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Uchiba M, Murakami K, Harada N, Johno M, Naruo M, Okabe H, Takatsuki K. Reduction of spinal cord injury by administration of iloprost, a stable prostacyclin analog. J Neurosurg. 1997;86:1007–1011. doi: 10.3171/jns.1997.86.6.1007. [DOI] [PubMed] [Google Scholar]
- Thel MC, O’Connor CM. Cardiopulmonary resuscitation: historical perspective to recent investigations. Am Heart J. 1999;137:39–48. doi: 10.1016/s0002-8703(99)70458-8. [DOI] [PubMed] [Google Scholar]
- Uchiyama-Tsuyuki Y, Kawashima K, Araki H, Otomo S. Prostacyclin analogue TTC-909 reduces memory impairment in rats with cerebral embolism. Pharmacol Biochem Behav. 1995;52:555–559. doi: 10.1016/0091-3057(95)00139-n. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Moncada S, Vane JR. Comparison of the effects of prostacyclin (PGI2), prostaglandin E1 and D2 on platelet aggregation in different species. Prostaglandins. 1978;16:373–388. doi: 10.1016/0090-6980(78)90216-2. [DOI] [PubMed] [Google Scholar]
- Willis AP, Leffler CW. NO and prostanoids: age dependence of hypercapnia and histamine-induced dilations of pig pial arterioles. Am J Physiol. 1999;277:H299–H307. doi: 10.1152/ajpheart.1999.277.1.H299. [DOI] [PubMed] [Google Scholar]
- Wright DH, Abran D, Bhattacharya M, Hou X, Bernier SG, Bouayad A, Fouron JC, Vazquez-Tello A, Beauchamp MH, Clyman RI, Peri K, Varma DR, Chemtob S. Prostanoid receptors: ontogeny and implications in vascular physiology. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1343–R1360. doi: 10.1152/ajpregu.2001.281.5.R1343. [DOI] [PubMed] [Google Scholar]
- Yonekura I, Kawahara N, Nakatomi H, Furuya K, Kirino T. A model of global cerebral ischemia in C57 BL/6 mice. J Cereb Blood Flow Metab. 2004;24:151–158. doi: 10.1097/01.WCB.0000096063.84070.C1. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]