

Abstract
Background
The LDL-receptor related protein 1 (LRP-1) is a scavenger receptor that regulates adaptive immunity and inflammation. LRP-1 is not known to modulate the pathogenesis of allergic asthma.
Objective
To assess whether LRP-1 expression by dendritic cells (DCs) modulates adaptive immune responses in house dust mite (HDM)-induced airways disease.
Methods
LRP-1 expression on peripheral blood DCs was quantified by flow cytometry. The role of LRP-1 in modulating HDM-induced airways disease was assessed in mice with a deletion of LRP-1 in CD11c+ cells (Lrp1fl/fl; CD11c-Cre) and by the adoptive transfer of HDM- pulsed CD11b+ DCs from Lrp1fl/fl; CD11c-Cre mice to wild-type mice.
Results
Human peripheral blood myeloid DC subsets from eosinophilic asthmatics have lower LRP-1 expression than cells from healthy, non-asthmatic subjects. Similarly, LRP-1 expression by CD11b+ lung DCs was significantly reduced in HDM-challenged wild-type mice. HDM-challenged LRP-1fl/fl; CD11c-Cre mice have a phenotype of increased eosinophilic airway inflammation, allergic sensitization, Th2 cytokine production, and mucous cell metaplasia. The adoptive transfer of HDM-pulsed LRP-1-deficient CD11b+ DCs into wild-type mice generated a similar phenotype of enhanced eosinophilic inflammation and allergic sensitization. Furthermore, CD11b+ DCs in the lungs of Lrp1fl/fl; CD11c-Cre mice have an increased ability to take up HDM antigen, whereas bone marrow-derived DCs display enhanced antigen presentation capabilities.
Conclusion
This identifies a novel role for LRP-1 as a negative regulator of DC-mediated adaptive immune responses in HDM-induced eosinophilic airway inflammation. Furthermore, the reduced LRP-1 expression by circulating myeloid DCs from eosinophilic asthmatics suggests a possible role for LRP-1 in modulating type 2-high asthma.
Keywords: LDL-receptor related protein-1 (LRP-1), dendritic cells, eosinophilic airway inflammation, house dust mite, type 2-high asthma
Graphical abstract

Introduction
The low-density lipoprotein receptor-related protein-1 (LRP-1), is a ubiquitously expressed, multifunctional, scavenger-type endocytic receptor that binds a broad spectrum of more than 40 structurally unrelated ligands, including lipoproteins (e.g., apolipoprotein E- containing chylomicrons and VLDL remnants), proteases (e.g., urokinase plasminogen activator (uPA) and tissue-type plasminogen activator (tPA)), protease-inhibitor complexes (i.e., uPA and tPA complexes with plasminogen activator inhibitor (PAI-1) and activated α-2- macroglobulin), extracellular matrix proteins (e.g., thrombospondins-1 and -2), growth factors (i.e., platelet-derived growth factor and transforming growth factor-β), bacterial toxins (e.g., Pseudomonas exotoxin A), viruses (e.g., Rhinovirus), and amyloid β (Aβ) peptides1–7. LRP-1, is one of the largest members of the low-density lipoprotein (LDL)- receptor gene family that also includes the apolipoprotein E (ApoE) receptor 2, the very low-density lipoprotein (VLDL) receptor, and megalin. LRP-1 is derived from a 600-kDa precursor protein to yield a 515 kDa α-chain and an 85 kDa β-chain that are associated in a non-covalent fashion on the cell surface. Furthermore, LRP-1 is expressed by multiple cell types, including dendritic cells, macrophages, vascular endothelial cells, neurons, Schwann cells, hepatocytes, and fibroblasts6, 8. This broad spectrum of cellular expression allows LRP-1 to participate in multiple cellular processes, such as tissue homeostasis, intracellular signaling, cell survival, trafficking, proliferation and (trans)-differentiation9–12.
Besides its canonical role in endocytosis, LRP-1 also participates in the regulation of inflammation and adaptive immune responses13–15. For example, the macrophage-specific deletion of LRP-1 decreases VLDL uptake and thereby promotes atherogenesis in hypercholesterolemic mice by increasing matrix metalloproteinase 9 activity and the production of pro-inflammatory mediators, such as TNF-α and CCL2 (MCP-1)13. Lrp1−/− mice display a phenotype of enhanced ligand-specific inflammatory responses that are regulated by NF-κB and miR-155 in macrophages7. Furthermore, the mechanism by which LRP-1 suppresses NF- κB signaling may involve the down-regulation of signaling by TNF receptor-116. Consistent with an anti-inflammatory function in macrophages, LRP-1 has been demonstrated to promote a M2 phenotype17. LRP-1 has also recently been shown to attenuate neuroinflammation and microglial immune responses by suppressing signaling via c-Jun N-terminal kinase and NF-κB pathways18.
LRP-1 modulates host defense and anti-tumor immune responses by facilitating antigen uptake and presentation. For example, an important function of LRP-1 is the binding, internalization, and clearance of apoptotic cells via the formation of a multimeric protein complex comprised of the scaffolding protein, RAN-binding protein 9 (RANBP9), and the receptor tyrosine kinase, AXL19. This multimeric protein complex facilitates efferocytosis by dendritic cells (DCs) and the subsequent cross-presentation of viral antigens expressed by apoptotic cells to CD8+ T cells, which promotes host defense against viral infections. Similarly, binding of bacterial or tumor cell-derived heat shock proteins to LRP-1, which is also known as CD91, facilitates the high efficiency uptake of exogenous antigenic peptides by antigen presenting cells for chaperone-based presentation that primes effector CD4+ and CD8+ T cells in the setting of infection or malignancy20–22. Interestingly, LRP-1 also modulates pro- inflammatory cytokine production and the initiation of inflammatory responses in LPS-induced acute lung injury by a calreticulin axis that binds surfactant proteins, SP-A and SP-D, to activate downstream p38 and NF-κB signaling pathways23.
Previously, we identified an endogenous apoE-LDL receptor pathway in the lung that regulates airway hyperreactivity and mucous cell metaplasia in house dust mite (HDM)- induced allergic asthma24. Moreover, we demonstrated that the VLDL receptor on dendritic cells (DCs) acts as a negative regulator and thereby attenuates HDM-induced airway inflammation and adaptive immune responses25. Here, we hypothesized that another LDL receptor family member, LRP-1, might similarly regulate DC function and thereby modify adaptive immune responses to HDM, which is a common and clinically relevant aeroallergen. We show that the cell-specific deletion of LRP-1 on CD11c+ cells enhances HDM-induced allergic sensitization, adaptive Th2 immune responses, and eosinophilic airway inflammation via a mechanism that involves increased antigen uptake and presentation by DCs. In addition, we demonstrate that circulating myeloid DC subsets from human subjects with eosinophilic asthma have decreased LRP-1 expression as compared to healthy, non-asthmatic subjects. We also show that LRP-1 expression by circulating myeloid DC subsets is negatively correlated with peripheral blood eosinophil counts in asthmatics, which suggests a possible role for LRP-1 in modulating adaptive immune responses in humans. Therefore, we propose that LRP-1 functions as a negative regulator of DC-mediated adaptive immune responses in HDM-induced allergic eosinophilic airway inflammation.
Methods
Quantification of LRP-1 expression on Peripheral Blood DCs
Informed consent was obtained from 38 subjects who participated in protocol 99-H-0076, which was approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute (NHLBI). All asthmatics had evidence of either a positive response to an inhaled β2-agonist or a positive methacholine bronchoprovocation challenge test, whereas all healthy, non-asthmatic subjects had a negative methacholine bronchoprovocation challenge test. The presence or absence of allergy was determined by skin testing, which was performed either at NHLBI or by their local physician, or by patient history. Allergy skin testing at NHLBI was performed using a Multi- Test II applicator, which was a generous gift from Lincoln Diagnostics (Decatur, IL) with glycerin as a negative control and histamine as a positive control (Holister-Stier, Spokane, WA). Allergy was defined by the presence of skin test reactivity to at least one of six common aeroallergens, which included house dust mite (Dermatophagoides farinae), cockroaches (Periplaneta americana and Battella germanica), cat hair, Aspergillus fumigatus, Ragweed (giant and short) and grasses (Kentucky Bluegrass, Orchard Redtop, Timothy Sweet Vernal grass, Meadow Fescue, and Perennial Ryegrass).
Peripheral blood was collected in 10-ml sodium heparinized Vacutainers (Becton Dickinson, Franklin Lakes, NJ) and red blood cells (RBCs) were lysed using ACK lysing buffer. Peripheral blood cells were reacted with the following antibodies for flow cytometry: anti-human CD11c- BV785 (clone 3.9); anti-human CD1c-PE/Dazzle 594 (clone L161); anti-human CD141-PECy7 (clone M80); anti-human CD16-PerCP/Cy5.5 (clone 3G8); anti-human CD123-BV650 (clone 6H6); anti-human HLA-DR- APC Cy7 (clone W6/32); anti-human Lin 1-FITC all from BioLegend (San Diego, CA) and AF647-conjugated rabbit monoclonal antibody to LRP-1 (ab92544) from Abcam (Cambridge, UK). Cells were incubated for 15 mins with human FcγR- binding inhibitor (ThermoFisher Scientific, Waltham, MA), reacted with antibodies, and washed with PBS. The fixable viability dye was then added, followed by fixation and permeabilization. Cellular debris and doublets were excluded using physical scatter properties of RBC-lysed whole blood cells and dead cells were excluded using fixable live-dead exclusion dye. Myeloid- DCs type I (MDC1) were identified as viable/Lin-1− cells expressing CD11c+/CD1c+/CD141− /HLA-DR+, whereas myeloid-DCs type II (MDC2) were CD11c+/CD1c−/CD141+/HLA-DR+, while CD16+ DCs were identified as CD11c+/CD16+ cells and plasmacytoid DCs were CD11c− /CD123+ using Fluorescence Minus One (FMO) as a control26.
Mice
Experimental protocols were approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute, Bethesda, MD. Lrp1fl/fl (B6;129S7-Lrp1tm2Her/J) and CD11c-Cre (C57BL/6J-Tg(Itgax-cre,-EGFP)4097Ach/J) mice were purchased from The Jackson Laboratories (Bar Harbor, ME), and crossed to generate homozygous Lrp1fl/fl; CD11c (Cre) mice. The cell-specific deletion of Lrp1 from CD11c+ dendritic cells was confirmed by PCR of genomic DNA and Western blotting of cellular proteins. PCR was performed on genomic DNA from SSClo/SiglecF−/CD11c+/CD11b+/MHCIIhi mouse lung DCs and SSClo/SiglecF−/CD11c−/CD11b−/EpCAMhi/MHCIIlo lung epithelial cells (LECs), as well as SSClo/CD11c+/CD11b+/MHCIIhi splenic DCs and CD4+ T cells, which had been purified by flow cytometry, using the following PCR primers that detected the recombined Lrp1 floxed allele: primer rec1, 5’-GGT GTG ACA TAG AGT TTT AAA GAG G-3’; primer rec2, 5’-GCA AGC TCT CCT GCT CAG ACC TGG A-3’. Bone marrow-derived DCs (BMDCs) and murine lungs were lysed in RIPA buffer and 100 g of protein was separated by SDS-PAGE using 4-20% Tris- Glycine gels (Invitrogen) and transferred to nitrocellulose membranes (GE healthcare Life sciences, PA). Membranes were reacted with anti-LRP-1 (1:1000) and β-actin antibodies (Abcam). Blots were stripped using Restore Western Blot Stripping Buffer (Thermo Scientific).
HDM sensitization and challenge models
Lrp1fl/fl and Lrp1fl/fl; CD11c (Cre) mice received two intraperitoneal injections of HDM (100 μg) (Dermatophagoides pteronyssinus extract, Greer Laboratories, Lenoir, NC) with aluminum hydroxide (40 mg ml−1, Invivogen, San Diego, CA) on day 4 and day 8 followed by intranasal administration of HDM (50 μg) on days 8, 10, 12, 15 before harvest on day 17. For the adoptive transfer model, bone marrow cells from Lrp1fl/fl and Lrp1fl/fl; CD11c (Cre) mice were cultured at a density of 2 × 106 cells ml−1 in Iscove’s Modified Dulbecco’s medium (Gibco/Life Technologies, Carlsbad, CA) supplemented with 10% heat-inactivated FCS, penicillin (100 U ml−1), streptomycin (100 μg ml−1), L-glutamine (2 mM), 2-mercaptoethanol (50 μM), and recombinant mouse GM-CSF (20 ng ml−1; BioLegend, San Diego, CA). An equal volume of medium was added on day 3 and 50% of the medium was replaced on day 6. On day 8, floating non-adherent BMDCs were pulsed with HDM (100 μg ml−1) or sham-pulsed with saline and flow sorted, viable CD11c+/CD11b+/MHCIIhi BMDCs (0.05 × 106) were adoptively transferred in 20 ul of PBS via intranasal administration on day 0 to recipient naïve C57BL6 wild-type mice. All recipient mice received daily intranasal HDM challenges (50 μg) on day 9, 11, 13, 15 and endpoints were analyzed on day 15. Two independent experiments were performed for each model.
BAL and Lung Histopathology
Bronchoalveolar lavage (BAL) was performed three times with 0.5 ml PBS and red blood cells were lysed with ACK buffer for 2 min at 4° C, as previously described27. BALF cells were re-suspended in 0.3 ml RPMI-1640 containing 10% FBS and cell counts were performed using a hemocytometer, while differential cell counts were performed on Diff-Quik-stained cytospin slides (Siemens, Deerfield, Illinois). Lungs were inflated with 10% formalin to a pressure of 25 cm H2O, fixed in 10% formalin for 24 h, dehydrated through gradient ethanol, and embedded in paraffin. Sagittal sections were cut at a thickness of 5 μm and stained with hematoxylin and eosin and periodic acid Schiff (PAS). Quantification of mucous cell metaplasia was performed as previously described. All the airways present (large (conducting), medium (central), and small (distal)) within a representative lung section were inspected. The number of airways that contained PAS-positive cells was counted and mucous cell metaplasia is reported as the percentage of airways that contained PAS-positive cells, as previously described24.
Quantification of HDM-specific IgE and IgG1
Levels of HDM-specific serum IgE and IgG1 were quantified, as previously described27. 96 well plates were coated overnight with 0.01% HDM in PBS and blocked with 1% BSA in PBS. Serum samples were added to the plates for 1 hr, washed 6X with PBS with 0.05% Tween‐20 prior to incubation with biotinylated anti-mouse IgE or IgG1 (Pharmingen, San Jose, CA) for 1 hr. Plates were washed 6 additional times and streptavidin-HRP (R&D Systems, Minneapolis, MN) was added for 30 min and the amount of bound HDM-specific antibody was determined by the addition of TMB substrate.
Measurement of Airway Hyperresponsiveness
Mice were anesthetized and airway resistance to different doses of methacholine was measured using an Elan RC Fine Pointe system (Buxco, Wilmington, NC), as previously described24. Mice were mechanically ventilated following tracheal cannulation with a 19-gauge beveled metal catheter, with a constant inspiratory flow before nebulization with increasing doses of methacholine (0-10 mg/ml). Airway resistance was recorded at 10-seconds interval for 3 mins and average values are represented as cm-H2O/ml/s.
House dust mite re-stimulation of mediastinal lymph node cells
Single cell suspensions from both right and left mediastinal lymph nodes (MLN) of mice were generated. MLN cells (0.2 × 106/ml) were cultured in round-bottom 96-well plates and re-stimulated with HDM (100 μg/ml) for 72 hours at 37°C in RPMI medium with 10 % FBS. The concentrations of IL-4, IL-5 IL-13, or IFN-γ in the supernatants were measured using sandwich ELISA kits with limits of sensitivity of 15.6 pg/ml for IL-4, 31.25 pg/ml for IL-5 and IFN-γ, and 62.5 pg/ml for IL-13 (R & D Systems, Minneapolis, MN).
Lung dendritic cell migration assay
HDM was labeled with AF-647 protein labeling kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Mice received intratracheal administration of AF647-HDM (50 μg per administration of sensitization) dissolved in 80 μl of PBS using a 24-gauge sterile polyurethane catheter connected to the outlet of a micropipette, as previously described27. At 72 hours after injection, migrating DCs were quantified in digested lungs and MLNs by flow cytometry as CD11c+/MHCIIhi/AF-647-HDM+ cells.
Flow cytometry
Lungs cells were isolated by enzymatic digestion with liberase (100 μg/ml, Roche, Indianapolis, IN) and DNase I (0.2 mg/ml; DN25, Sigma, St. Louis, MO) (total volume 1-2 ml/lung) and incubated at 37°C for 25 min with agitation. Cells from digested lungs were incubated with Fc Block™ (BD Biosciences, San Jose, CA) to react with Fcγ III/II receptors before surface staining. Mouse DC subsets were identified and characterized by using anti- mouse Siglec F-PE-CF594, CD11c-APC Cy7, MHCII-PE Cy7, CD11b-PerCP Cy5.5, CD103- APC, PDCA1-AF488, Mar-1-efluor450, CD64-PE, TLR-4-APC, Dectin-1-PE, CD205- PerCPCy5.5, and CD206-BV785 antibodies, all from BioLegend (San Diego, CA); CD80- PECy5, CD86-605NC, and DC-SIGN-eFluor660 all from ThermoFisher Scientific, Waltham, MA); Dectin-2-AF647 (BioRad, Hercules, CA); and TLR 5-PE (Novus, Littleton, CO). CD4+ regulatory T cells (Treg) were identified using APC-conjugated anti-CD3, BV650-conjugated anti-CD4, and PE-Cy7-conjugated anti-CD25, all from ThermoFisher Scientific. For intracellular Foxp3 staining, cells were fixed and permeabilized with Foxp3 staining buffer (ThermoFisher Scientific), then reacted with a PE-conjugated anti-mouse Foxp3 antibody (0.5 μg per 106 cells) (Clone NRRF-30, ThermoFisher Scientific). Data were acquired on a LSRII (BD Biosciences, USA) equipped with 407, 488, 532, and 633 LASER lines using DIVA 8 software and analyzed with the Flow Jo software version 9.9.5 (Treestar, San Carlos, CA). Cellular debris was excluded using a forward light scatter/side scatter plot.
OT-II co-culture
The ability of DCs to induce ex vivo antigen-specific T-cell proliferation was assessed using CFSE-labeled splenic CD4 T cells from naïve (DO11.10 TCR (C.Cg- Tg(DO11.10)10Dlo/J) transgenic mice (Jackson Laboratories, Bar Harbor, ME) that express a transgenic MHCII-restricted TCR that recognizes the OVA peptide antigen27. BMDCs from Lrp1fl/fl and Lrp1fl/fl; CD11c (Cre) mice were pulsed overnight with 1 μg ml−1 of OVA 323-339 peptide (AnaSpec, Fremont, CA, USA) or PBS as a control and viable CD11c+ cells were isolated by flow sorting using an anti-mouse CD11c-APC-Cy7 antibody (BD Biosciences). Naïve CD4+ T cells were purified from the spleens of DO11.10 TCR mice by flow sorting using an anti-mouse CD4-BV 650 antibody (ThermoFisher Scientific) and were labelled with 5 μM CFDA-SE (carboxyfluoresceine diacetate succinimidyl ester; Cayman Chemical, MI), for 15 min. 1 × 105 OVA peptide-specific CD4+ OTII cells were co-cultured with 2 × 104 CD11c+ BMDCs in 96-well plates for 4 days and T cell proliferation was quantified by flow cytometry using CFSE dye dilution. CD4+-gated cells were analyzed using proliferation profile, percent divided, and division index as calculated by the FlowJo Proliferation Platform28.
Statistics
Data were analyzed using Graph Pad Prism version 7.0b using either an unpaired t test for normally distributed data or a Mann-Whitney test for non-normally distributed data. Multiple comparisons were analyzed using a one-way ANOVA with a Sidak’s multiple comparison test or a two-way ANOVA. Correlation analyses between LRP-1 expression and peripheral blood eosinophil counts were performed by Spearman correlation. Data are presented as mean + SEM. A P value < 0.05 was considered significant.
Results
LRP-1 Expression is Decreased on Peripheral Blood Myeloid Dendritic Cells from Eosinophilic Asthmatics
Elevated peripheral blood eosinophils are a biomarker of type 2-high asthma29–32. Here, we hypothesized that LRP-1 expression by peripheral blood DCs might be modified in asthmatics with high peripheral blood eosinophils counts (> 260 cells/μl) as compared to asthmatics with low peripheral blood eosinophils counts (< 260 cells/μl) or healthy, non-asthmatics who also had low peripheral blood eosinophil counts (< 260 cells/μl) (Figure 1A)33–35. Demographic information regarding the high and low eosinophil groups are presented in Repository eTable 1. LRP-1 expression was quantified by flow cytometry on four specific peripheral blood DCs subsets; Lin−/HLA-DR+/CD11c+/CD1c+/CD141− myeloid DC type I (MDC1), Lin−/HLA-DR+/CD11c+/CD1c−/CD141+ myeloid DC type II (MDC2), Lin−/HLA-DR+/CD16+/CD11C+ (CD16+ DCs) or Lin−/HLA-DR+/CD11c+/CD123+ (plasmacytoid DCs, pDCs) (Repository eFigure 1). As shown in Figure 1B, LRP-1 expression by the MDC1, MDC2, and CD16+ myeloid DC subsets was significantly decreased in the eosinophil-high asthmatic group as compared to the healthy, non-asthmatic group, whereas there was no difference between the groups regarding LRP-1 expression by plasmacytoid DCs. Furthermore, when all eosinophil-high and eosinophil-low asthmatic subjects were combined into a single cohort, there were significant negative correlations between peripheral blood eosinophil counts and LRP-1 expression by the MDC1 (Spearman ρ = −0.46, p = 0.019), MDC2 (Spearman ρ = −0.51, p = 0.008), and CD16+ (Spearman ρ = -0.39, p = 0.047) myeloid DC subsets, whereas there was a trend for a negative correlation among plasmacytoid DCs (Spearman ρ = -0.37, p = 0.06). Collectively, these results show that myeloid DC subsets from eosinophil-high asthmatics have lower levels of LRP-1 expression than healthy, non-asthmatic subjects and that a negative correlation exists between LRP-1 expression by myeloid DC subsets and peripheral blood eosinophil counts in asthmatics.
Figure 1. Cell surface LRP-1 expression is reduced on myeloid dendritic cell subsets from eosinophilic asthmatics.

A) Asthmatics (n = 14) and healthy, non-asthmatics (n = 12) with low peripheral blood eosinophil counts (< 260 cells/l), or asthmatics with high (> 260 cells/μl) peripheral blood eosinophil counts (n = 12) (* P< 0.0001, one-way ANOVA with Sidak’s multiple comparison test). B) Mean fluorescence intensity (MFI) of LRP-1 expression was assessed by flow cytometry on viable/Lin−/HLA-DR+ peripheral blood dendritic cell subsets that express CD11c+/CD1c+/CD141− (MDC1), CD11c+/CD1c−/CD141+ (MDC2), CD11c+/CD16+ (CD16+ DCs), and CD11c−/CD123+ (pDCs). Data shown as means ± SEM (*P<0.05, one-way ANOVA with Sidak’s multiple comparison test). C) The percentage of LRP-1+ CD11c+/CD11b+/Siglec F−/MHC IIhi/Mar-1−/CD64− conventional myeloid DCs (cDCs) and CD11c+/CD11b+/Siglec F−/MHC IIhi/Mar-1+/CD64+ monocyte-derived DCs (moDCs) from saline- and house dust mite (HDM)-challenged wild-type C57BL6 mice were quantified (n = 9 mice, * P< 0.0001, saline- versus HDM-challenged, Mann-Whitney test).
Next, we used a murine model to assess whether sensitization and challenge with HDM modifies the cell surface expression of LRP-1 on CD11c+/CD11b+ lung DCs. As shown in Figure 1C, LRP-1 is expressed on both CD11b+/CD11c+/Siglec F−/MHC-IIhi/Mar-1−/CD64− conventional myeloid DCs (cDC) and CD11b+/CD11c+/Siglec F−/MHC-IIhi/Mar-1+/CD64+ monocyte-derived DCs (moDC) in the lungs of saline-challenged mice36. Furthermore, following sensitization and challenge with HDM, the percentage of both LRP-1+ cDCs and moDCs in the lung were significantly reduced.
The CD11c-specific deletion of LRP-1 Augments HDM-induced Eosinophilic Airway Inflammation
Having shown that both human and murine DCs express LRP-1, experiments were next conducted to assess the role of LRP-1 on CD11c+ cells in initiating HDM-induced allergic sensitization, Th2 immune responses, and eosinophilic airway inflammation. To address this question, we created a Lrp1fl/fl; CD11c-Cre mouse, in which LRP-1 is specifically deleted in CD11c+ cells, which include both DCs and alveolar macrophages (Repository eFigure 2). Lrp1fl/fl; CD11c-Cre mice were sensitized by intraperitoneal injection of HDM plus aluminum hydroxide, followed by multiple intranasal HDM challenges (Figure 2A). As shown in Figure 2B, the total number of BALF inflammatory cells recovered from HDM-challenged Lrp1fl/fl; CD11c-Cre mice were significantly increased as compared to control Lrp1fl/fl mice, which represented increases in eosinophils, alveolar macrophages, and lymphocytes. Similarly, lung histopathology showed an increase in the extent of peribronchial inflammatory cell infiltrates (Figure 2C) and mucous cell metaplasia (Figure 2C and 2D) in HDM-challenged Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice. In contrast, there was no significant difference in airway hyperresponsiveness between HDM-exposed Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice (Figure 3A). Re-stimulation of mediastinal lymph node (MLN) cells from Lrp1fl/fl; CD11c-Cre mice with HDM showed significant increases in the production of the Th2 cytokines, IL-4, IL-5 and IL-13, whereas there was no change in interferon-γ (Figure 3B). Flow cytometry demonstrated a significant reduction in the percentage of CD3+/CD4+/CD25+/Foxp3+ regulatory T cells (Tregs) in the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice (Figure 3C). Lastly, HDM-challenged Lrp1fl/fl; CD11c-Cre mice had increases in HDM- specific serum IgE and IgG1 levels (Figure 3D) in as compared with Lrp1fl/fl mice, which is indicative of augmented allergic sensitization. Collectively, these data demonstrate that HDM- challenged Lrp1fl/fl; CD11c-Cre mice have a phenotype of increased allergic sensitization, Th2- medated adaptive immune responses, mucous cell metaplasia, and eosinophilic airway inflammation in response to HDM that was associated with a reduction in lung Tregs.
Figure 2. Eosinophilic airway inflammation and mucous cell metaplasia are increased in HDM-challenged Lrp1fl/fl; CD11c-Cre mice.

A) Lrp1fl/fl and Lrp1fl/fl; CD11c-Cre mice were sensitized with two intraperitoneal injections of 100 μg HDM (with alum 40 mg ml−1) on day 0 and day 4 and challenged on days 8, 10, 12 and 15 by intranasal administration of HDM (50 μg) before harvest and endpoint analysis on day 17. B) The number of total BALF inflammatory cells and inflammatory cell types (eosinophils (Eos), alveolar macrophages (AM), neutrophils (PMN) and lymphocytes (Lymph)) from saline- and HDM-challenged Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice (n = 7 – 16 mice per group, * P< 0.01, HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl, one-way ANOVA with Sidak’s multiple comparison test). C) Representative lung histology sections stained with hematoxylin and eosin (H&E) or Periodic acid-schiff (PAS). Scale bars 100 μm for the 200× images and 20 μm for the 1,000× images. D) Quantification of mucous cell metaplasia as assessed by the percentage of airways that contained PAS+ cells. Data are mean ± SEM (n = 5 – 7 mice per group, * P < 0.05, HDM- challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice, Mann-Whitney test). Results shown are pooled data from two independent experiments.
Figure 3. Th2 cytokine production and allergic sensitization are increased in HDM- challenged Lrp1fl/fl; CD11c-Cre mice.

A) Airway resistance (cm H2O per ml s−1) to inhaled methacholine from HDM-challenged Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice (n = 4 – 6 mice per group). (P = NS, two-way ANOVA). B) Cytokine secretion by ex vivo cultures of MLN cells that had been re-stimulated with saline or HDM (100 μg ml−1, n = 6 mice per group, * P < 0.05, HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice, one-way ANOVA with Sidak’s multiple comparison test). C) The percentage of CD4+/CD25+/FoxP3+ Tregs in the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice and Lrp1fl/fl mice (n = 5 mice per group, *P < 0.008, Mann-Whitney test). D) Serum levels of HDM-specific IgE and IgG1 (n = 9 – 10 mice per group), * P< 0.05, HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice, one-way ANOVA with Sidak’s multiple comparison test).
To confirm that the phenotype in HDM-challenged Lrp1fl/fl; CD11c-Cre mice of augmented eosinophilic inflammation in the absence of modified AHR was not specifically related to the use of aluminum hydroxide as an adjuvant during the sensitization phase, we assessed if similar results would be obtained if mice were sensitized and challenged by intranasal administration of HDM (25 μg) alone, on a daily basis, five days per week for five weeks (Repository eFigure 3A). Consistent with the results obtained using the HDM plus adjuvant model, the total number of BALF inflammatory cells recovered from Lrp1fl/fl; CD11c-Cre mice that had been sensitized and challenged with HDM alone were significantly increased as compared to control Lrp1fl/fl mice, which again represented increases in eosinophils, alveolar macrophages, and lymphocytes (Repository eFigure 3B). Again, there was no significant difference in airway hyperresponsiveness between HDM-challenged Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice (Repository eFigure 3C), while flow cytometry demonstrated that there was a significant reduction in the number of CD3+/CD4+/CD25+/Foxp3+ regulatory T cells (Tregs) in the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice (Repository eFigure 3D). BALF levels of CCL24 were also significantly increased in HDM- challenged Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice (Repository eFigure 3E). Thus, a similar phenotype of enhanced eosinophilic airway inflammation that was dissociated from airway hyperreactivity was present when Lrp1fl/fl; CD11c-Cre mice were sensitized and challenged by intranasal administration of HDM without aluminum hydroxide as an adjuvant.
The Adoptive Transfer of HDM-pulsed CD11b+ DCs from Lrp1fl/fl; CD11c-Cre mice Augments Eosinophilic Airway Inflammation in HDM-challenged recipient mice
Next, adoptive transfer experiments were performed to confirm that the augmented HDM- induced eosinophilic airway inflammatory responses in Lrp1fl/fl; CD11c-Cre mice were specifically mediated by DCs. CD11b+ BMDCs from Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice were pulsed with HDM or saline and adoptively transferred to naïve wild-type (WT) mice that subsequently received multiple intranasal HDM challenges to induce allergic airway inflammation (Figure 4A). As shown in Figure 4B, the number of total BALF inflammatory cells, which were specifically comprised of eosinophils and alveolar macrophages, were significantly increased in WT recipients of CD11b+ DCs from Lrp1fl/fl; CD11c-Cre donor mice as compared to WT recipients of CD11b+ DCs from Lrp1fl/fl donor mice. Similarly, lung histology showed increases in peribronchial inflammatory cell infiltrates (Figure 4C) and mucous cell metaplasia (Figure 4D) in WT recipients of CD11b+ DCs from Lrp1fl/fl; CD11c-Cre donor mice, as compared to WT recipients of CD11b+ DCs from Lrp1fl/fl donor mice. Serum levels of HDM- specific IgE and IgG1 (Figure 4E) and BALF levels of the C-C chemokine, CCL24 (Figure 4F), were also significantly increased in the WT recipients of CD11b+ DCs from Lrp1fl/fl; CD11c-Cre donor mice as compared to WT recipients of CD11b+ DCs from Lrp1fl/fl donor mice. In addition, the production of IL-4, but not IL-5, IL-13, or IFN-γ, was increased following HDM re-stimulation of MLN cells isolated from WT recipient mice that received the adoptive transfer of CD11b+ DCs from Lrp1fl/fl; CD11c-Cre donor mice as compared to WT recipient mice that received the adoptive transfer of CD11b+ DCs from Lrp1fl/fl donor mice (Figure 4G). Collectively, these experiments demonstrate that CD11b+ DCs from Lrp1fl/fl; CD11c-Cre donor mice significantly up-regulated eosinophilic airway inflammatory responses in WT recipient mice upon subsequent HDM challenge, as compared to WT recipient mice that received the adoptive transfer of HDM-pulsed CD11b+ BMDCs from Lrp1fl/fl donor mice. Thus, the adoptive transfer of HDM-pulsed LRP-1-deficient CD11b+ DCs into WT mice recapitulated the phenotype of enhanced allergic sensitization and eosinophilic inflammation that was observed in HDM- challenged Lrp1fl/fl; CD11c-Cre mice.
Figure 4. The adoptive transfer of CD11b+ DCs from Lrp1fl/fl; CD11c-Cre mice increases HDM-mediated eosinophilic airway inflammation.

A) BMDCs from Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl donor mice were pulsed ex vivo with saline or HDM (100 μg ml−1) for 16 hours. 2.5 × 104 CD11c+/CD11b+/MHC IIhi DCs were adoptively transferred to wild-type (WT) C57BL6 recipient mice by intranasal administration on day 0 and intranasal HDM challenges (50 μg) were administered on alternate days, from day 9 through day 15, to all recipient mice, prior to endpoint analysis on day 16. B) The number of total BALF inflammatory cells and inflammatory cell subtypes in recipient WT mice (n = 6 – 10 mice per group, *P< 0.05, HDM-pulsed Lrp1fl/fl; CD11c-Cre DCs versus HDM-pulsed Lrp1fl/fl DCs, one-way ANOVA with Sidak’s multiple comparison test). C) Representative histologic lung sections stained with H&E and PAS. Scale bars indicate 100 μm for the 200× images and 20 μm for the 1,000× images. D) Quantification of mucous cell metaplasia as assessed by the percentage of airways that contained PAS+ cells. (n = 5 mice per group, * P < 0.008, HDM-pulsed Lrp1fl/fl; CD11c-Cre DCs versus HDM- pulsed Lrp1fl/fl DCs, Mann-Whitney test). E) Serum levels HDM-specific IgE and IgG1 (n = 9 – 10 mice per group, *P < 0.01, Mann-Whitney test, HDM-pulsed Lrp1fl/fl; CD11c-Cre DCs versus HDM-pulsed Lrp1fl/fl DCs). F) BALF levels of CCL24 from WT recipient mice that received HDM-pulsed Lrp1fl/fl; CD11c-Cre DCs or HDM-pulsed Lrp1fl/fl DCs (n = 6 – 10 mice per group, *P < 0.05, unpaired t test). G) Cytokine secretion by ex vivo cultures of MLN cells that had been re-stimulated with saline or HDM (100 μg ml−1, n = 6 – 10 mice per group, * P < 0.01, HDM-pulsed Lrp1fl/fl; CD11c-Cre DCs versus HDM-pulsed Lrp1fl/fl DCs, one-way ANOVA with Sidak’s multiple comparison test). Results shown are pooled data from two independent experiments.
CD11b+ Lung DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre mice have Increased Expression of CD86 and TLR5
Next, we considered whether the specific deletion of LRP-1 in CD11c+ cells would modify the number of lung DC subsets or the surface expression of co-stimulatory molecules in response to HDM challenge in mice that had been sensitized with HDM plus aluminum hydroxide. As shown in Figure 5A, there were no significant differences in the numbers of total lung CD11c+/Siglec F−/MHC IIhi DCs in HDM-challenged Lrp1fl/fl; CD11c-Cre mice as compared to HDM-challenged Lrp1fl/fl mice. In addition, there were no differences in the number of CD11b+ cDCs and moDCs or CD11b− CD103+ DCs and pDCs in the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice and Lrp1fl/fl mice (Figure 5B). Therefore, the phenotype of enhanced eosinophilic airway inflammation in HDM-challenged Lrp1fl/fl; CD11c-Cre mice was not mediated by changes in the number of DC subsets in the lung. Similarly, there were no differences in the numbers of cDCs, moDCs, CD103+ DCs and pDCs in the lungs of Lrp1fl/fl; CD11c-Cre mice and Lrp1fl/fl mice that had been sensitized and challenged by administration of intranasal HDM without adjuvant (Supplemental Repository eFigure 4).
Figure 5. Increased expression of CD86 and TLR5 by CD11c+/CD11b+ DCs from HDM- challenged Lrp1fl/fl; CD11c-Cre mice.
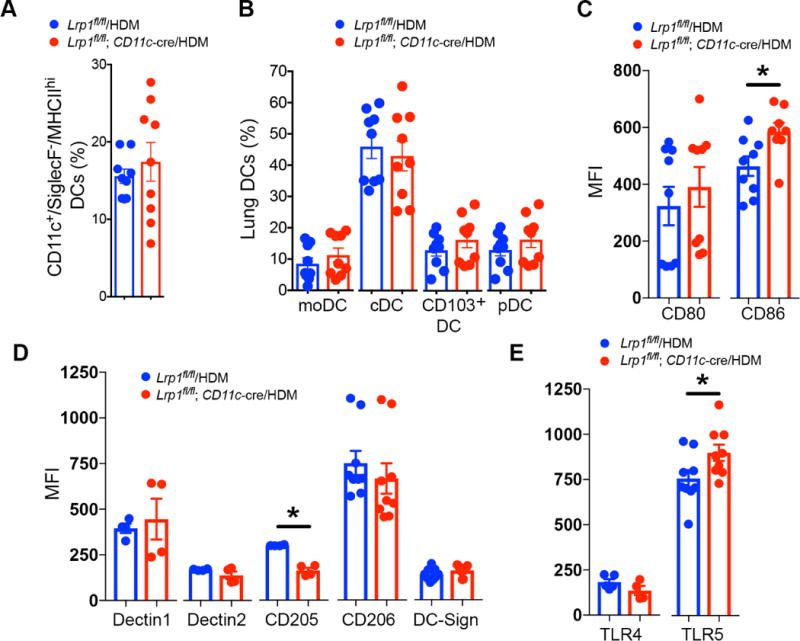
A) The percentage of CD11c+/Siglec F−/MHC IIhi total lung DCs and B) the percentage of lung DC monocyte-derived dendritic cells (moDCs), conventional myeloid dendritic cells (cDCs), CD103+ DCs, and plasmacytoid dendritic cells (pDCs) subsets as quantified by flow cytometry (P = NS, HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice, n= 9 mice per group, unpaired t test). C) Cell surface expression of CD80 and CD86 by lung CD11c+/CD11b+/SSClo/SiglecF−/MHC IIhi DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice (n = 8 – 9 mice per group, * P< 0.01, Mann-Whitney test). D) Cell surface expression of Dectin1, Dectin 2, CD205, CD206, and DC-SIGN by lung CD11c+/CD11b+/SSClo/SiglecF−/MHC IIhi DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice (n = 4 – 9 mice per group, * P = 0.029, Mann-Whitney test). E) Cell surface expression of TLR4 and TLR5 by lung CD11c+/CD11b+/SSClo/SiglecF−/MHC IIhi DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl mice (n= 4 – 9 mice per group, *P < 0.05, unpaired t test).
Since, CD11b+ cDCs and moDCs have been shown to be the primary DC subset in the lung that mediates antigen presentation and Th2 responses27, 36, 37, we characterized these DCs subsets further in Lrp1fl/fl; CD11c-Cre mice. We found that cell surface expression of the co- stimulatory molecule CD86 was increased on CD11b+ DCs from the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice, whereas there was no difference in the expression of CD80 (Figure 5C). We also considered whether cell surface expression of C-type lectin receptors (CLRs) and toll-like receptors (TLRs) by CD11b+ lung DCs might be modified in Lrp1fl/fl; CD11c-Cre mice as a possible mechanism by which allergic inflammation is augmented38–41. As shown in Figures 5D and 5E, there were no differences in the mean fluorescence intensity (MFI) of the CLRs, Dectin 1, Dectin 2, DC-SIGN, and CD206, or TLR4 by CD11b+ lung DCs from HDM- challenged Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice, whereas the expression of CD205 was decreased. This suggests that the phenotype of HDM-challenged Lrp1fl/fl; CD11c- Cre mice was not mediated at the level of CLRs. In contrast, TLR5 expression by lung CD11b+ DCs from HDM-challenged challenged Lrp1fl/fl; CD11c-Cre mice was significantly increased. These data suggest that the up-regulated expression of CD86 and TLR5 by CD11b+ myeloid lung DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre mice might in part contribute to the phenotype of enhanced eosinophilic inflammation and Th2 responses.
LRP-1-deficient DCs have Increased Antigen Uptake and Presentation
We next hypothesized that antigen uptake and presentation functions of LRP-1-deficient DCs might be enhanced as a mechanism by which HDM-induced eosinophilic airway inflammation is augmented in Lrp1fl/fl; CD11c-Cre mice. AF-647-labelled HDM was administered to the lungs of Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice and antigen uptake was assessed by flow cytometry. As shown in Figure 6A, there was a significant increase in HDM uptake by CD11b+ lung DCs from Lrp1fl/fl; CD11c-Cre mice as compared to Lrp1fl/fl mice (14.28 % ± 2.6 vs. 2.46 % ± 0.31), whereas there was no difference in the uptake of HDM antigen by lung CD11b− DCs. In addition, there was no difference in the frequency of AF-647-HDM+ CD11b+ or CD11b− DCs from Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice that migrated to MLNs following uptake of HDM antigen in the lung (Figure 6B). Collectively, these experiments show that lung DCs from Lrp1fl/fl; CD11c-Cre mice have an enhanced antigen uptake capability, whereas subsequent migration to MLNs was not modified.
Figure 6. Lrp1-deficient DCs have augmented antigen uptake and presentation capabilities.

A and B) Uptake of Alexa Fluor 647-labeled HDM (50 μg) by CD11c+/SSClo/SiglecF−/MHCIIhi CD11b+ or CD11b− DCs in the lungs (A) and MLNs (B) of Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice 72 hours after intranasal administration (n= 10 mice per group, * P < 0.05, unpaired t test). C) CD11c+ BMDCs from Lrp1fl/fl and Lrp1fl/fl; CD11c-Cre mice were pulsed with the OVA 323-339 peptide and incubated at a 1:5 ratio with CFSE- labelled splenic CD4+ DO11.10 T cells for 4 days. OVA-specific proliferation is presented as % divided OT-II cells (n = 12 – 15 per group, * P < 0.0001, one-way ANOVA with Sidak’s multiple comparison test). D) Th2 cytokines released by co-cultures of OVA 323-339 peptide-pulsed BMDCs and CFSE-labelled CD4+ DO11.10 T cells (n= 10 per group, * P < 0.05, Lrp1fl/fl; CD11c-Cre versus Lrp1fl/fl, unpaired t test). Results shown are pooled data from two independent experiments.
Next, CD11c+ BMDCs from Lrp1fl/fl; CD11c-Cre and Lrp1fl/fl mice were used to investigate whether LRP-1 expression modifies antigen-specific T cell proliferation and Th2 cytokine production. Co-culture experiments of splenic CD4+ T cells from mice expressing the major histocompatibility complex II (MHC II)-restricted DO11.10 T-cell receptor that recognizes the OVA 323-339 peptide showed that CD11c+ BMDCs from Lrp1fl/fl; CD11c-Cre mice mediate significant increases in both T cell proliferation (Figure 6C) and Th2 cytokine production (Figure 6D) as compared with CD11c+ BMDCs from Lrp1fl/fl mice following ex vivo stimulation with the OVA 323-339 peptide. Collectively, these experiments show that LRP-1-deficient CD11c+ DCs have an enhanced ability to present antigen to CD4+ T cells and thereby induce effector Th2 inflammatory responses.
Discussion
CD11b+ cDCs play key roles in initiating Th2 cell differentiation and effector cytokine production that mediate allergic airway inflammation42–44. We have previously shown that the VLDL receptor functions as a negative regulator of DC function in experimental HDM-induced asthma. However, limited data exists regarding the function of another LDL receptor family member, LRP-1, in mediating the ability of DCs to induce allergen-mediated adaptive immune responses in asthma21, 45–47. For example, LRP1 polymorphisms have been associated with food allergen sensitization in asthmatic children who participated in the Mexico City Childhood Asthma Study48. In other settings, LRP-1 expression by DCs has been shown to mediate antigen-specific immune responses. LRP-1 on DCs can facilitate the receptor-mediated uptake of a wide variety of antigens, including soluble tetanus toxin C fragment (TTC) or activated α2-macroglobulin-TTC complexes, that elicit T cell proliferation and enhance primary antigen-specific immune responses45, 49, 50. Furthermore, LRP-1 has been shown to bind and internalize immunogenic heat shock proteins (HSPs), such as HSP70, HSP90, gp96, and calreticulin22, 46, 51, which are processed and presented by MHC class I or MHC class II molecules to provide the appropriate signal for T cell priming21. LRP-1 also mediates DC responses in the setting of host defense against viral infections and in cancer19, 51, 52.
Peripheral blood eosinophil counts can be used as a biomarker to identify eosinophilic asthmatics and are also correlated with asthma severity29, 53, 54. Here, we show that eosinophil-high asthmatics have decreased LRP-1 expression on circulating MDC1, MDC2, and CD16+ myeloid DC subsets as compared to healthy, non-asthmatic subjects. Furthermore, we show LRP-1 expression by MDC1, MDC2, and CD16+ myeloid DC subsets is negatively correlated with peripheral blood eosinophil counts in asthmatics. Interestingly, LRP- 1+/Lin−/CD11c+ DCs, but not CD14+ monocytes are the primary antigen presenting cells (APCs) in circulating blood that facilitate receptor-mediated antigen uptake and presentation45. This is clinically relevant as these DC subsets also express the high-affinity IgE receptor (FcεR1), which contributes to the pathogenesis of allergic asthma55. Similarly, we show that HDM suppresses LRP-1 expression by CD11b+ cDCs and moDCs in the murine lung. This is consistent with prior reports demonstrating down-regulation of cell surface LRP-1 expression on myeloid cells in response to bacterial LPS and cigarette smoke56, 57. Collectively, these findings support the possibility that reduced LRP-1 expression by circulating myeloid DCs in eosinophilic asthmatics, as well as by CD11b+ cDCs and moDCs lung DCs from HDM- challenged mice, modify the severity of eosinophilic asthma.
We used two murine models to investigate whether LRP-1 expression by DCs modulates the pathogenesis of allergic sensitization to HDM and the subsequent induction of eosinophilic airway inflammation and Th2 adaptive immune responses in the lung. First, we created a mouse with a specific deletion of LRP-1 in CD11c+ cells, such as DCs and alveolar macrophages, to characterize the role of LRP-1 expression in HDM-induced airways disease. We show that HDM-challenged Lrp1fl/fl; CD11c-Cre mice display a phenotype of augmented allergic sensitization and enhanced Th2 immune responses to HDM that were associated with increased eosinophilic airway inflammation and mucous cell metaplasia. HDM re-stimulation of draining mediastinal lymph node cells from Lrp1fl/fl; CD11c-Cre mice showed increased production of the Th2 cytokines, IL-4, IL-5 and IL-13, which are key regulators of eosinophilic airway inflammation and mucous cell metaplasia58–60. Furthermore, there was a reduction CD4+ Tregs, which function to suppress eosinophilic airway inflammation and Th2 immune responses in the lungs of HDM-challenged Lrp1fl/fl; CD11c-Cre mice. This suggests that the decrease in lung Tregs in HDM-challenged Lrp1fl/fl; CD11c-Cre mice might foster a permissive immunologic environment that augments eosinophilic airway inflammation61, 62. In addition, CD11b+ lung DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre mice had increased expression of CD86 (B7-2), which mediates co-stimulatory signaling upon interacting with CD28 on CD4+ T cells to promote Th2 immune responses63, 64. CD11b+ lung DCs from HDM-challenged Lrp1fl/fl; CD11c-Cre mice also had increased expression of TLR5, which promotes allergic sensitization to flagellin-containing aeroallergens65. Thus, the concomitant decrease in Tregs, and increased expression of CD86 and TLR5 on CD11b+ lung DCs, may in part contribute to the phenotype of enhanced allergic sensitization and eosinophilic airway inflammation in HDM-challenged Lrp1fl/fl; CD11c-Cre mice.
Interestingly, the phenotype of augmented eosinophilic airway inflammation in HDM- challenged Lrp1fl/fl; CD11c-Cre mice that were sensitized with HDM plus adjuvant (i.e., aluminum hydroxide) was not associated with concomitant modifications in AHR. This finding was confirmed when HDM alone was used for both allergic sensitization and challenge, which shows that the dissociation between AHR and eosinophilic airway inflammation was not merely a consequence of a sensitization model that utilized aluminum hydroxide as an adjuvant. A dissociation between eosinophilic airway inflammation and AHR has been previously reported both in murine models, as well as in human studies, which suggests that distinct pathogenic pathways can regulate the development of these cardinal manifestations of asthma66–72. Consistent with this concept, our results suggest that LRP-1 regulates HDM-induced eosinophilic airway inflammation, but not AHR, in Lrp1fl/fl; CD11c-Cre mice.
CD11b+ cDCs that reside in the airway continuously sample the local environment and take up inhaled aeroallergens, such as HDM, after which they migrate to nearby draining lymph nodes to present antigens to T cells73. As such, CD11b+ cDCs are the major lung DC subset that mediates the priming and activation of CD4+ effector T cells in allergen-mediated airway inflammation27, 74, 75. Furthermore, following sensitization and challenge with HDM, migratory CD11b+ cDCs have been identified as the primary DC subset that induces Th2 immune responses in lymph nodes, while the function of CD11b+ moDCs is to produce pro- inflammatory cytokines and facilitate allergen presentation and allergen-mediated eosinophilic inflammation in the lung36. Here, we show that the adoptive transfer of HDM-pulsed CD11b+ DCs from Lrp1fl/fl; CD11c-Cre mice have an increased ability to induce allergic sensitization, as indicated by elevated serum levels of HDM-specific IgE and IgG1, as well as eosinophilic airway inflammation. In addition, BALF levels of the C-C chemokine, CCL24 (eotaxin-2), which has chemotactic activity towards eosinophils, were increased76. Collectively, these results confirm the conclusion that the phenotype of increased allergic sensitization and eosinophilic airway inflammation that we observed in HDM-challenged Lrp1fl/fl; CD11c-Cre mice was specifically mediated by CD11b+ DCs.
Uptake of HDM antigen in the lung is almost exclusively mediated by the CD11b+ DC subset, whereas CD11b− DCs can also mediate HDM uptake at high doses36, 38. Following antigen uptake in the lung, DCs are transported to draining MLNs by a CCR7-dependent process, where they induce the expansion of naïve T cells to mount a Th2 immune response77. By sub- dividing the lung and MLN DC populations, we show that CD11b+ DCs from the lungs of naïve Lrp1fl/fl; CD11c-Cre mice have an increased ability to take up HDM antigen, whereas there was no difference in the migration of HDM+ CD11b+ DCs to the draining MLNs. In addition, we show that OVA-peptide pulsed CD11c+ BMDCs Lrp1fl/fl; CD11c-Cre mice have enhanced antigen presentation capabilities as evidenced by their ability to increase the proliferation of naïve DO11.10 T cells, as well as up-regulate the production of IL-5 and IL-13 Th2 effector cytokines. Thus, increased uptake of HDM antigen, as well as enhanced antigen presentation and induction of Th2 immunity, may contribute to the phenotype of augmented eosinophilic airway inflammation in HDM-challenged Lrp1fl/fl; CD11c-Cre mice.
In conclusion, the present study demonstrates, for what we believe is the first time, a role for LRP-1 in modulating dendritic cell function in the pathogenesis of Th2 immune responses to HDM in experimental allergen-induced asthma. Our data are consistent with the conclusion that LRP-1 functions as a negative regulator of CD11b+ DCs, which thereby attenuates HDM- induced allergic sensitization and eosinophilic airway inflammation. We propose that the mechanism by which LRP-1 negatively regulates DC function involves attenuated antigen uptake and presentation capabilities, which reduces allergic sensitization and Th2 immune responses in the lung. Furthermore, we demonstrate that LRP-1 expression is reduced on circulating myeloid DC subsets from eosinophilic asthmatics, which also suggests a possible role for LRP-1 in modulating the type 2-high asthma endotype in humans. Therefore, these findings support a novel role for the LDL receptor family member, LRP-1, as a negative regulator of DC function during the pathogenesis of allergic asthma.
Supplementary Material
Key messages.
LRP-1 expression by circulating myeloid DC subsets from eosinophil-high asthmatics is reduced as compared to healthy, non-asthmatic subjects. In addition, LRP-1 expression by human peripheral blood myeloid DC subsets is negatively correlated with peripheral blood eosinophil counts in asthmatics.
LRP-1 functions as a negative regulator of dendritic cell-mediated adaptive immune responses in house dust mite-induced eosinophilic airway inflammation in mice by modulating antigen uptake and presentation.
Acknowledgments
This research was funded by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland.
Financial Support: Division of Intramural Research, National Heart, Lung, and Blood Institute, NIH, Bethesda, Maryland.
Abbreviations
- DCs
dendritic cells
- MDC
myeloid dendritic cells
- BMDC
bone marrow–derived dendritic cells
- cDCs
conventional dendritic cells
- moDCs
monocyte-derived dendritic cells
- HDM
house dust mite
- OVA
ovalbumin
- BAL
bronchoalveolar lavage
- MLN
mediastinal lymph node
- AHR
airway hyperresponsiveness
- TLR
Toll-like receptor
- Tregs
regulatory T cells
- Foxp3
Forkhead box protein 3
- AF
Alexa Fluor
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. Journal of Clinical Investigation. 2001;108:779–84. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiological Reviews. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaultier A, Hollister M, Reynolds I, Hsieh EH, Gonias SL. LRP1 regulates remodeling of the extracellular matrix by fibroblasts. Matrix Biology. 2010;29:22–30. doi: 10.1016/j.matbio.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (Lrp) Annual Review of Biochemistry. 1994;63:601–37. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 5.Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid- beta clearance in Alzheimer's disease. Frontiers in Aging Neuroscience. 2014;6:93. doi: 10.3389/fnagi.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher P, Herz J. Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem Pharmacol. 2011;81:1–5. doi: 10.1016/j.bcp.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantuano E, Brifault C, Lam MS, Azmoon P, Gilder AS, Gonias SL. LDL receptor- related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc Natl Acad Sci U S A. 2016;113:1369–74. doi: 10.1073/pnas.1515480113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonias SL, Campana WM. LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am J Pathol. 2014;184:18–27. doi: 10.1016/j.ajpath.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker TA, Williams L, Koenig K, Kothari H, Komissarov AA, Florova G, et al. Lipoprotein receptor-related protein 1 regulates collagen 1 expression, proteolysis, and migration in human pleural mesothelial cells. American Journal of Respiratory Cell and Molecular Biology. 2012;46:196–206. doi: 10.1165/rcmb.2011-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takayama Y, May P, Anderson RGW, Herz J. Low density lipoprotein receptor-related protein 1 (LRP1) controls endocytosis and c-CBL-mediated ubiquitination of the platelet- derived growth factor receptor beta (PDGFR beta) Journal of Biological Chemistry. 2005;280:18504–10. doi: 10.1074/jbc.M410265200. [DOI] [PubMed] [Google Scholar]
- 11.Song H, Li YH, Lee J, Schwartz AL, Bu GJ. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Research. 2009;69:879–86. doi: 10.1158/0008-5472.CAN-08-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boucher P, Gotthardt M, Li WP, Anderson RGW, Herz J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–32. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 13.Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circulation Research. 2007;100:670–7. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- 14.Weckbach LT, Gola A, Winkelmann M, Jakob SM, Groesser L, Borgolte J, et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta(2) integrins (CD11/CD18) Blood. 2014;123:1887–96. doi: 10.1182/blood-2013-06-510875. [DOI] [PubMed] [Google Scholar]
- 15.May P. The low-density lipoprotein receptor-related protein 1 in inflammation. Current Opinion in Lipidology. 2013;24:134–7. doi: 10.1097/MOL.0b013e32835e809c. [DOI] [PubMed] [Google Scholar]
- 16.Gaultier A, Arandjelovic S, Niessen S, Overton CD, Linton MF, Fazio S, et al. Regulation of tumor necrosis factor receptor-1 and the IKK-NF-kappa B pathway by LDL receptor-related protein explains the antiinflammatory activity of this receptor. Blood. 2008;111:5316–25. doi: 10.1182/blood-2007-12-127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.May P, Bock HH, Nofer JR. Low density receptor-related protein 1 (LRP1) promotes anti-inflammatory phenotype in murine macrophages. Cell Tissue Res. 2013;354:887–9. doi: 10.1007/s00441-013-1699-2. [DOI] [PubMed] [Google Scholar]
- 18.Yang L, Liu CC, Zheng H, Kanekiyo T, Atagi Y, Jia L, et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J Neuroinflammation. 2016;13:304. doi: 10.1186/s12974-016-0772-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian M, Hayes CD, Thome JJ, Thorp E, Matsushima GK, Herz J, et al. An AXL/LRP-1/RANBP9 complex mediates DC efferocytosis and antigen cross- presentation in vivo. J Clin Invest. 2014;124:1296–308. doi: 10.1172/JCI72051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nature Immunology. 2000;1:151–5. doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- 21.Pawaria S, Binder RJ. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nature Communications. 2011;2:521. doi: 10.1038/ncomms1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tobian AAR, Canaday DH, Boom WH, Harding CV. Bacterial heat shock proteins promote CD91-dependent class I MHC cross-presentation of chaperoned peptide to CD8+ T cells by cytosolic mechanisms in dendritic cells versus vacuolar mechanisms in macrophages. J Immunol. 2004;172:5277–86. doi: 10.4049/jimmunol.172.9.5277. [DOI] [PubMed] [Google Scholar]
- 23.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRP alpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115:13–23. doi: 10.1016/s0092-8674(03)00758-x. [DOI] [PubMed] [Google Scholar]
- 24.Yao XL, Fredriksson K, Yu ZX, Xu XL, Raghavachari N, Keeran KJ, et al. Apolipoprotein E Negatively Regulates House Dust Mite-induced Asthma via a Low-Density Lipoprotein Receptor-mediated Pathway. American Journal of Respiratory and Critical Care Medicine. 2010;182:1228–38. doi: 10.1164/rccm.201002-0308OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fredriksson K, Mishra A, Lam JK, Mushaben EM, Cuento RA, Meyer KS, et al. Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. Journal of Immunology. 2014;192:4497–509. doi: 10.4049/jimmunol.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, et al. BDCA-2, BDCA-3 and BDCA-4: Three markers for distinct subsets of dendritic cells in human peripheral blood. Journal of Immunology. 2000;165:6037–46. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 27.Mishra A, Brown AL, Yao XL, Yang ST, Park SJ, Liu CY, et al. Dendritic cells induce Th2-mediated airway inflammatory responses to house dust mite via DNA-dependent protein kinase. Nature Communications. 2015;6:6224. doi: 10.1038/ncomms7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19857–62. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. New England Journal of Medicine. 2013;368:2455–66. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 30.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. New England Journal of Medicine. 2011;365:1088–98. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 31.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. New England Journal of Medicine. 2009;360:973–84. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–9. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- 33.Hanania NA, Wenzel S, Rosen K, Hsieh HJ, Mosesova S, Choy DF, et al. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med. 2013;187:804–11. doi: 10.1164/rccm.201208-1414OC. [DOI] [PubMed] [Google Scholar]
- 34.Zhang XY, Simpson JL, Powell H, Yang IA, Upham JW, Reynolds PN, et al. Full blood count parameters for the detection of asthma inflammatory phenotypes. Clin Exp Allergy. 2014;44:1137–45. doi: 10.1111/cea.12345. [DOI] [PubMed] [Google Scholar]
- 35.Arron JR, Izuhara K. Asthma biomarkers: what constitutes a ‘gold standard’? Thorax. 2015;70:105–7. doi: 10.1136/thoraxjnl-2014-206069. [DOI] [PubMed] [Google Scholar]
- 36.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38:322–35. doi: 10.1016/j.immuni.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 37.Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, et al. CD11b(+) Myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. Journal of Immunology. 2009;182:623–35. doi: 10.4049/jimmunol.182.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito T, Hirose K, Norimoto A, Tamachi T, Yokota M, Saku A, et al. Dectin-1 plays an important role in house dust mite-induced allergic airway inflammation through the activation of CD11b(+) dendritic cells. Journal of Immunology. 2017;198:61–70. doi: 10.4049/jimmunol.1502393. [DOI] [PubMed] [Google Scholar]
- 39.Clarke DL, Davis NHE, Campion CL, Foster ML, Heasman SC, Lewis AR, et al. Dectin- 2 sensing of house dust mite is critical for the initiation of airway inflammation. Mucosal Immunology. 2014;7:558–67. doi: 10.1038/mi.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nature Reviews Immunology. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 41.Geijtenbeek TBH, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nature Reviews Immunology. 2016;16:433–48. doi: 10.1038/nri.2016.55. [DOI] [PubMed] [Google Scholar]
- 42.van Rijt LS, Jung S, KleinJan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c(+) dendritic cells during allergen challenge abrogates the characteristic features of asthma. Journal of Experimental Medicine. 2005;201:981–91. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stumbles PA, Thomas JA, Pimm CL, Lee PT, Venaille TJ, Proksch S, et al. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. Journal of Experimental Medicine. 1998;188:2019–31. doi: 10.1084/jem.188.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, Kool M, et al. Inflammatory dendritic cells-not basophils-are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. Journal of Experimental Medicine. 2010;207:2097–111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart JP, Gunn MD, Pizzo SV. A CD91-positive subset of CD11c(+) blood dendritic cells: Characterization of the APC that functions to enhance adaptive immune responses against CD91-targeted antigens. Journal of Immunology. 2004;172:70–8. doi: 10.4049/jimmunol.172.1.70. [DOI] [PubMed] [Google Scholar]
- 46.De Filippo A, Binder RJ, Camisaschi C, Beretta V, Arienti F, Villa A, et al. Human Plasmacytoid Dendritic Cells Interact with gp96 via CD91 and Regulate Inflammatory Responses. Journal of Immunology. 2008;181:6525–35. doi: 10.4049/jimmunol.181.9.6525. [DOI] [PubMed] [Google Scholar]
- 47.Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the toll-like receptor 2/4 pathway. Journal of Biological Chemistry. 2002;277:20847–53. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 48.Hancock DB, Romieu I, Chiu GY, Sienra-Monge JJ, Li H, Estela Del Rio-Navarro B, et al. STAT6 and LRP1 polymorphisms are associated with food allergen sensitization in Mexican children. J Allergy Clin Immunol. 2012;129:1673–6. doi: 10.1016/j.jaci.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu CT, Oury TD, Enghild JJ, Pizzo SV. Adjuvant-free in-vivo targeting. Antigen delivery by alpha 2-macroglobulin enhances antibody formation. Journal of Immunology. 1994;152:1538–45. [PubMed] [Google Scholar]
- 50.Cianciolo GJ, Enghild JJ, Pizzo SV. Covalent complexes of antigen and alpha(2)- macroglobulin: evidence for dramatically-increased immunogenicity. Vaccine. 2001;20:554–62. doi: 10.1016/s0264-410x(01)00361-9. [DOI] [PubMed] [Google Scholar]
- 51.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–13. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 52.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–46. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 53.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. New England Journal of Medicine. 1990;323:1033–9. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 54.Casciano J, Krishnan JA, Small MB, Buck PO, Gopalan G, Li C, et al. Burden of asthma with elevated blood eosinophil levels. BMC Pulm Med. 2016;16:100. doi: 10.1186/s12890-016-0263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Foster B, Metcalfe DD, Prussin C. Human dendritic cell 1 and dendritic cell 2 subsets express FcεRI: Correlation with serum IgE and allergic asthma. Journal of Allergy and Clinical Immunology. 2003;112:1132–8. doi: 10.1016/j.jaci.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 56.LaMarre J, Wolf BB, Kittler EL, Quesenberry PJ, Gonias SL. Regulation of macrophage alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein by lipopolysaccharide and interferon-gamma. J Clin Invest. 1993;91:1219–24. doi: 10.1172/JCI116283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability. American Journal of Respiratory Cell and Molecular Biology. 2007;37:748–55. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 58.Pope SM, Brandt EB, Mishra A, Hogan SP, Zimmermann N, Matthaei KI, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5-and eotaxin-dependent mechanism. Journal of Allergy and Clinical Immunology. 2001;108:594–601. doi: 10.1067/mai.2001.118600. [DOI] [PubMed] [Google Scholar]
- 59.Mould AW, Matthaei KI, Young IG, Foster PS. Relationship between interleukin-5 and eotaxin in regulating blood and tissue eosinophilia in mice. Journal of Clinical Investigation. 1997;99:1064–71. doi: 10.1172/JCI119234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. The Journal of Clinical Investigation. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaffar Z, Sivakuru T, Roberts K. CD4(+)CD25(+) T cells regulate airway eosinophilic inflammation by modulating the Th2 cell phenotype. Journal of Immunology. 2004;172:3842–9. doi: 10.4049/jimmunol.172.6.3842. [DOI] [PubMed] [Google Scholar]
- 62.Hadeiba H, Locksley RM. Lung CD25 CD4 regulatory T cells suppress type 2 immune responses but not bronchial hyperreactivity. The Journal of Immunology. 2003;170:5502–10. doi: 10.4049/jimmunol.170.11.5502. [DOI] [PubMed] [Google Scholar]
- 63.Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, et al. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–32. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 64.Masten BJ, Yates JL, Koga AMP, Lipscomb MF. Characterization of accessory molecules in murine lung dendritic cell function: Roles for CD80, CD86, CD54, and CD40L. American Journal of Respiratory Cell and Molecular Biology. 1997;16:335–42. doi: 10.1165/ajrcmb.16.3.9070619. [DOI] [PubMed] [Google Scholar]
- 65.Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll- like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nat Med. 2012;18:1705–10. doi: 10.1038/nm.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poynter ME, Cloots R, van Woerkom T, Butnor KJ, Vacek P, Taatjes DJ, et al. NF- kappa B activation in airways modulates allergic inflammation but not hyperresponsiveness. Journal of Immunology. 2004;173:7003–9. doi: 10.4049/jimmunol.173.11.7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao X, Fredriksson K, Yu ZX, Xu X, Raghavachari N, Keeran KJ, et al. Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. American Journal of Respiratory and Critical Care Medicine. 2010;182:1228–38. doi: 10.1164/rccm.201002-0308OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao X, Gao M, Dai C, Meyer KS, Chen J, Keeran KJ, et al. Peptidoglycan Recognition Protein 1 Promotes House Dust Mite-Induced Airway Inflammation in Mice. American Journal of Respiratory Cell and Molecular Biology. 2013:902–11. doi: 10.1165/rcmb.2013-0001OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. American journal of respiratory and critical care medicine. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 70.Ishii S, Nagase T, Shindou H, Takizawa H, Ouchi Y, Shimizu T. Platelet-activating factor receptor develops airway hyperresponsiveness independently of airway inflammation in a murine asthma model. J Immunol. 2004;172:7095–102. doi: 10.4049/jimmunol.172.11.7095. [DOI] [PubMed] [Google Scholar]
- 71.Makela MJ, Kanehiro A, Borish L, Dakhama A, Loader J, Joetham A, et al. IL-10 is necessary for the expression of airway hyperresponsiveness but not pulmonary inflammation after allergic sensitization. Proc Natl Acad Sci U S A. 2000;97:6007–12. doi: 10.1073/pnas.100118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–84. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lambrecht BN, Hammad H. Dendritic cell and epithelial cell interactions at the origin of murine asthma. Ann Am Thorac Soc. 2014;11(Suppl 5):S236–43. doi: 10.1513/AnnalsATS.201405-218AW. [DOI] [PubMed] [Google Scholar]
- 74.Julia V, Hessel EM, Malherbe L, Glaichenhaus N, O’Garra A, Coffman RL. A restricted subset of dendritic cells captures airborne antigens and remains able to activate specific T cells long after antigen exposure. Immunity. 2002;16:271–83. doi: 10.1016/s1074-7613(02)00276-5. [DOI] [PubMed] [Google Scholar]
- 75.Mesnil C, Sabatel CM, Marichal T, Toussaint M, Cataldo D, Drion PV, et al. Resident CD11b(+)Ly6C(−) Lung Dendritic Cells Are Responsible for Allergic Airway Sensitization to House Dust Mite in Mice. Plos One. 2012;7:e53242. doi: 10.1371/journal.pone.0053242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, Zimmermann N, et al. Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. The Journal of biological chemistry. 2005;280:13952–61. doi: 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- 77.Hammad H, Lambrecht BN, Pochard P, Gosset P, Marquillies P, Tonnel AB, et al. Monocyte-derived dendritic cells induce a house dust mite-specific Th2 allergic inflammation in the lung of humanized SCID mice: Involvement of CCR7. Journal of Immunology. 2002;169:1524–34. doi: 10.4049/jimmunol.169.3.1524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.