

Abstract
Background
Both hepatitis B and C viruses were transmitted through blood transfusion before implementation of donor screening. The existence of additional, yet unknown transfusion transmittable agents causing liver disease could have important public health implications.
Methods
Analyses were based on the Scandinavian Donations and Transfusions (SCANDAT2) database. Cox regression models were used to estimate the hazard ratio (HR) of developing chronic liver disease in recipients of blood from donors who later developed any chronic liver disease compared to recipients who received blood transfusion from healthy donors. We also studied whether the risk of liver disease was increased in patients who received units from ‘high-risk’ donors, defined as donors who had a higher than expected occurrence of liver disease among their previous recipients. All analyses were stratified before and after 1992 to account for the effect of screening for hepatitis C virus.
Results
A total of 1,482,922 transfused patients were included in the analyses. Analyses showed evidence of transfusion transmission of liver diseases before, but not after the implementation of hepatitis C virus screening in 1992, with HRs for any liver disease of 1.38 (95% confidence interval, 1.30-1.46) and 0.99 (95% confidence interval, 0.91-1.07), before and after 1992, respectively. Similarly, blood components from ‘high-risk’ donors conferred increased risks before, but not after 1992.
Conclusions
Our data provide no evidence for transfusion transmission of agents causing liver disease after the implementation of screening for hepatitis B and C, and suggest that if such transmission does occur, it is rare.
Keywords: Transfusion medicine, HepatitisAbstract
Background
Globally, common causes of chronic hepatitis (inflammation of the liver) include infection with hepatitis B virus (HBV), hepatitis C virus (HCV), excess alcohol consumption, obesity, and autoimmune liver disease. The association between blood transfusion and “hepatitis” was well known for many years before a specific causative agent was isolated[1], leading to the coining of the term “serum hepatitis”. In the late 1960s, the occurrence of post-transfusion hepatitis was linked to the presence of the “Australian Antigen” (now HBV surface antigen) in the blood of donors. This in turn later led to the identification of HBV as the first cause of post-transfusion hepatitis, and to subsequent testing of donors [2, 3]. However, even after the discovery of HBV, it was clear that some other hepatotropic virus (referred to as non-A-non-B hepatitis virus) was also transmitted through transfusion of blood products, leading to cases of hepatitis. The characterization of circulating antibodies to a previously unknown virus in 1989 marked the identification of HCV, which proved to be a major cause of transfusion-related liver disease[4].
Although the discovery of HCV explained most cases of non-A-non-B hepatitis, in as many as 5.4% of cases of chronic liver disease in high-income countries, no conclusive cause can be identified[5]. This is often referred to as cryptogenic cirrhosis. It has been postulated that there are other, yet undiscovered, hepatotropic infectious agents that could explain at least some fraction of the remaining “cryptogenic” liver disease. Indeed at least two studies have found blood transfusion to be a risk factor for cryptogenic cirrhosis even after HBV and HCV have been excluded[6, 7]. However, these studies included only small numbers of patients, and it is conceivable that the residual risk of cirrhosis is unrelated to the transfusion per se.
To investigate the possibility of yet unidentified agents being transmitted through blood transfusion after the introduction of screening for both HBV and HCV, we set up a large-scale, binational cohort study of 1.5 million transfused patients.
Methods
Data sources
The study was based on data from the Scandinavian Donations and Transfusions (SCANDAT2) database which contains all electronically available data on blood donations and transfusions in Sweden and Denmark dating back as far as the late 1960’s and early 1980’s, respectively[8]. The database and its creation have been described in detail previously[9]. In summary, the database contains data from prospectively maintained administrative registers on blood donations and transfusions in the Swedish and Danish blood banks and includes information on more than 1.6 million blood donors and more than 2 million transfused patients, with detailed data on links between donors and their respective recipients. The coverage of the SCANDAT2 database has been near-complete in Sweden since 1996, and in Denmark since 1998[9]. Using the unique national registration numbers that are available for residents in both countries[10], the database has been linked to nationwide healthcare and population registers (including inpatient, cause of death and cancer registers), providing detailed data on a range of health outcomes, as well as complete and unbiased long-term follow-up[11].
Study Design
Analyses followed a similar approach as in a previous study investigating the possible transfusion transmission of neurodegenerative disease[12]. We designed the study to investigate the transfusion transmission of a yet unknown agent causing chronic liver disease using two different approaches. First, we postulated that such a transmission could manifest itself as a donor-recipient disease concordance, i.e. an increased risk of disease in recipients of blood products from donors who themselves go on to develop the disease in question[13]. Alternatively, transfusion transmission could also manifest itself as disease clustering in recipients, i.e. a jointly increased risk among all transfusion recipients from an infectious donor, irrespective of whether the donor develops and is diagnosed with the disease of interest [12, 14]. The advantage of the latter approach is that it utilizes the fact that most donors donate blood to many recipients, which should increase the statistical power to detect the transfusion transmission of agents with a low clinical penetrance where only a fraction of infected patients become ill and are diagnosed[12].
Based on these notions, we set up a retrospective cohort study of all patients in the SCANDAT2 database who were transfused with any type of blood product between 1968 and 2012 (Sweden) and 1981 and 2012 (Denmark). Analyses were restricted to patients with no prior history of chronic liver disease, as ascertained from the respective country’s patient register (for specific diagnosis codes, see Supplementary Table 1). We then identified all transfusions given to the included patients during a 180-day exposure ascertain period, starting from the date of the first transfusion as recorded in the SCANDAT2 database, and then tracked all donors who contributed these units. Patients who received an autologous transfusion were excluded as were recipients of blood from unidentifiable donors, as were patients who received transfusions in areas that were not fully covered by the Swedish Patient Register at the time of the transfusion.
Using the respective country’s patient, cancer, and cause of death registers, we identified all donors and recipients who were diagnosed with conditions associated with liver diseases. These conditions were categorized into 9 groups for analysis, starting from a first group with wide selection of liver-related conditions, and subsequently moving into the other, more selective groups (Supplementary Table 1). This approach allowed us to test for the transmission of an agent with variable clinical manifestation.
The first group included International Classification of Diseases (ICD) codes for all liver diseases, both acute and chronic, allowing for a high sensitivity for the detection of any transfusion transmittable liver disease. The second group included acute and chronic liver disease without HBV or HCV infections, to remove the influence of known viral hepatitis. The third and fourth group focused on chronic liver diseases with or without liver failure, respectively, to specifically test for the transmission of a broad spectrum of chronic liver diseases. Liver cirrhosis and its complications, portal hypertension and hepatocellular carcinoma, were included together in the fifth group but also analysed individually as three additional groups. Finally, a group included ICD codes specifically related to HBV and HCV, to assess the contribution of viral hepatitis to the transfusion associated liver disease.
Statistical analysis
Along the lines of the two approaches for studying transfusion transmission, we set up a standard survival analysis for each of the 9 above mentioned groups of liver diseases as the outcome of interest. Considering that manifestations of a yet undiagnosed liver disease may be an indication for blood transfusion, e.g. acute variceal bleeding, follow-up of recipients was delayed by 180 days from the date of the first transfusion to allow for the exclusion of recipient with yet undiagnosed liver disease. Patients were thus followed up starting 180 days after the first transfusion until date of first liver disease diagnosis, death, emigration or end of follow-up (January 31st, 2012). Patients who died or were censored within 180 days of their first transfusion were not included in the analyses.
In the first analysis, patients who received blood from a donor who developed a diagnosis of liver disease during follow-up were considered exposed, and recipients of blood from donors without such a diagnosis were considered unexposed. In the second analysis, we instead classified exposure according to a cumulative disease excess score (DES) defined as the difference between the observed and expected number of liver disease cases among all previous recipients of each donor[14]. The DES metric was computed separately for each blood donation in such a way that it incorporated information from all previous donations. For example, a donor who has made 20 donations, where we observed 10 cases of liver cirrhosis among recipients, but only expected to see 1 case would, at the time of the 21st donation, have a DES of 9 (i.e., 10–1), indicating a much greater disease occurrence among past recipients than expected. In the analysis, both the observed and expected counts were computed time-dependently, so that the DES changed with each new donation. The expected count was calculated as the sums of the predicted probabilities extracted from Poisson regression models incorporating patient age, sex, country of residence and year of donation[12].
For each of the 9 disease groups we then computed the relative risk of liver disease, in relation to exposure defined according to the two different approaches outlined above, using Cox regression models. For the analyses of donor-recipient disease concordance, we also subdivided exposure depending on the latency of liver disease observed in the donor (<5, 5-10, >10 years). For the analysis of DES score, we used the highest DES score of all contributing donors. The analyses were adjusted for the recipient age (as a natural spline with 3 knots), sex, calendar year of transfusion (as a natural spline with 3 knots), patient ABO blood group (as a categorical term), and the number of transfusions during the exposure period (as a natural spline with 5 knots). The models also accounted for regional differences by incorporating county of transfusion as a stratum term.
Recognizing that screening of all blood donations for HCV, a major cause of chronic liver disease, had been fully implemented by 1992 in both countries, all analyses were conducted separately for pre-1992 and for the 1992 and onwards periods[12]. Results from the cohort first transfused before 1992, when it is well established that HCV was readily transmitted, were used to validate the study concept and proposed analysis methodology, while results from the cohort first transfused in or after 1992—when there should have been virtually no transmission both HBV and HCV—were used to test the for the continued transmission of some other agent linked to liver disease.
In sensitivity analyses, all analyses were repeated with 90 day and 1-year exposure ascertainment periods. Furthermore, because the risk of diagnosis of chronic liver disease is related to the follow up period after transfusion, an additional sensitivity analysis was done using a maximum follow-up period in recipients of 10 or 20 years.
The statistical power to detect the transmission of a hepatotropic agent causing hepatitis after implementation of HCV screening was evaluated using a simulation approach, a variant of which we have employed previously[12]. To avoid assumptions based on a limited literature, the simulations were based on the actual dataset of patients transfused in or after 1992, with liver cirrhosis as the studied outcome. We retained the actual cirrhosis events among both donors and recipients to act as a baseline risk of cirrhosis, unrelated to an unknown transmissible agent. The actual cirrhosis events thus did not count as being of an infectious origin. Instead we randomly assigned some donors to be carriers of a transmissible agent with a variable prevalence (0.001%, 0.01%, 0.1%, or 1%). The hepatotropic agent was assumed to be 100% infectious, but to have a variable rate of spontaneous resolution of 25 or 50% which applied to both donors and recipients. The donors were randomly assigned to be diagnosed with cirrhosis 5-10 years after their first donation. If this date occurred while they were still active donors, donations after the date of diagnosis were disregarded. If the donor was randomly determined not to develop cirrhosis, or if the simulated date of cirrhosis occurred after the actual date of death of that donor, or after end of follow-up, the donor could still give rise to cirrhosis among his/her recipients, but the donor would not count as being infected in the analyses. Among recipients who received blood from a donor randomly assigned to be a carrier of the hepatotropic agent, we then randomly assigned cirrhosis events with a variable latency. Specifically, the cirrhosis events were randomly assigned to occur after 10, 15, or 20 years. The latency was assumed to be normally distributed with a standard deviation of 3 years. Again, 25%, or 50% of recipients were assigned to not develop cirrhosis, and again events occurring after the patients’ death or after end of follow-up did not count as events. Based on these assumptions, we then performed simulation analyses based on the “real” data, augmented by randomly assigned disease events among both donors and recipients. The statistical analyses and data processing were otherwise identical to the regular analyses. We ran 100 iterations for each combination of assumptions, with a variable seed.
All data processing and statistical analyses were done using SAS 9.4 (SAS Institute, Cary, NC). The study was approved by local ethics boards in both countries.
Results
From the SCANDAT2 database we identified a total of 426,835 patients with a valid identity who were first transfused before 1992. From these, we excluded 10,442 patients who had a diagnosis of liver disease before their first transfusion, 75,334 who died or were diagnosed with any of the liver diseases of interest within 180 days of their first transfusion, 62,880 who received blood from at least one untraceable donor, and 1,720 receiving autologous transfusion. A total of 276,459 remained for analysis in the pre-1992 period. After 1st of Jan 1992, there were a total of 1,751,969 recipients with a valid identity. From these, 61,846 were excluded due to previous liver disease, 388,957 due to death or diagnosis of any of the liver diseases of interest within 180 days of their first transfusion, 89,350 due to untraceable donor, and 5,353 due to autologous transfusion. A total of 1,206,463 patients remained for analysis in the post-1992 period.
A combined total of 1,482,922 patients were thus available for further analysis for the whole study period. Altogether, these received a total of 7,890,253 transfusions contributed by 1,318,302 blood donors, of whom, 12,511 (0.95%) developed chronic liver disease during follow-up. The basic demographics of the patients in the study are presented in Table 1. Patients transfused before 1992 were younger and had longer follow-up than those first transfused in or after 1992. The median number of transfusions received by the two groups was similar (2 vs 3). Of all recipients, 113,480 (7.6%) had follow-up longer than 20 years.
Table 1.
Characteristics of study population.
| Recipients transfused before 1992 | Recipients transfused in or after 1992 | ||
|---|---|---|---|
|
|
|
||
| No. patients, N (% of total) | 276 459 (18.6) | 1 206 463 (81.4) | |
| Female, N (%) | 161 904 (58.6) | 710 460 (58.9) | |
| Country, N (%) | |||
| Sweden | 242 242 (87.6) | 790 642 (65.5) | |
| Denmark | 34 217 (12.4) | 415 821 (34.5) | |
| Median age at first transfusion (IQR) | 61.9 (40.4-73.5) | 71.2 (56.0-80.6) | |
| Median year of first transfusion (IQR) | 1984 (1978-1988) | 2002 (1998-2007) | |
| Median no. transfusions (IQR) | 2 (2-5) | 3 (2-5) | |
| Median duration of follow-up (IQR) | 13.8 (4.3-25.0) | 4.8 (2.0-9.2) | |
| No. patients with <5 years follow-up, N (%) | 75 362 (31.3) | 624 205 (64.2) | |
| No. patients with 5-10 years follow-up, N (%) | 39 791 (19.1) | 321 653 (24.4) | |
| No. patients with 10-20 years follow-up, N (%) | 51 995 (28.1) | 256 436 (11.3) | |
| No. patients with >20 years follow-up, N (%) | 109 311 (21.5) | 4 169 (0.1) | |
| Disease of donor, N (%)* | |||
| All diagnosis | 7 767 (9.4) | 6 606 (2.2) | |
| All diagnosis-No Viral | 6 467 (8.3) | 5 918 (2.0) | |
| Cirrhosis + Liver Failure | 4 074 (5.5) | 2 825 (1.0) | |
| Cirrhosis + Chronic Hepatitis | 3 232 (4.4) | 1 787 (0.6) | |
| Cirrhosis, PHT, HCC | 2 250 (3.3) | 1 391 (0.5) | |
| Cirrhosis | 1 361 (2.0) | 661 (0.2) | |
| Viral Hepatitis | 1 973 (2.1) | 817 (0.2) | |
| Portal Hypertension | 1 049 (1.5) | 810 (0.3) | |
| Liver Cancer | 437 (0.8) | 237 (0.1) | |
| Latency of disease in donor (years), N (%)* | |||
| <5 years from donation | 1 390 (1.0) | 3 427 (0.7) | |
| 5-9 years from donation | 994 (1.3) | 1 655 (0.8) | |
| ≥10 years from donation | 5 383 (7.2) | 1 524 (0.7) | |
The sum of the number of exposed patients exceeds the total number of patients as one patient may be exposed to blood from more than one donor with a later liver disease.
Table 2 and Supplementary Table 2 presents results from the analysis comparing patients who received blood units from donors who developed a liver disease to patients who received blood units from donors who did not. Of a total of 276,459 patients transfused before 1992 in SCANDAT, 25,987 (9.4%) received transfusion from 7,767 donors who developed a liver disease during follow-up. In comparison, out of the 1,206,463 patients first transfused in or after 1992, a total of 26,641 (2.2%) were exposed to blood products from 6,606 donors who were later diagnosed with a liver disease.
Table 2.
Relative risks chronic liver disease in relation to occurrence of the same diseases in the contributing blood donor(s), presented overall and by latency in the donors before 1992.*
| Donor diseased
|
Donor not diseased
|
|||
|---|---|---|---|---|
| Disease | Events/person-years | Hazard ratio (95% CI)† | Events/person-years | Hazard ratio(95% CI)† |
| a) Patients transfused before 1992 | ||||
| All liver disease | 1,461/377,543 | 1.38 (1.30-1.46) | 8,590/3,874,257 | 1.00 (ref) |
| <5 year latency | 180/36,075 | 1.73 (1.48-2.02) | ||
| 5-10 year latency | 191/48,855 | 1.37 (1.18-1.59) | 8,590/3,874,257 | 1.00 (ref) |
| >10 years latency | 1,090/292,613 | 1.34 (1.25-1.43) | ||
| All liver disease (excluding viral hepatitis) | 912/337,044 | 1.08 (1.00-1.16) | 7,658/3,930,336 | 1.00 (ref) |
| <5 year latency | 93/28,058 | 1.34 (1.08-1.65) | ||
| 5-10 year latency | 111/42,195 | 1.01 (0.83-1.22) | 7,658/3,930,336 | 1.00 (ref) |
| >10 years latency | 708/266,791 | 1.07 (0.98-1.16) | ||
| Liver cirrhosis | 91/83,706 | 1.34 (1.08-1.67) | 2,242/4,213,467 | 1.00 (ref) |
| <5 year latency | 9/5,961 | 2.27 (1.16-4.45) | ||
| 5-10 year latency | 11/9,333 | 1.57 (0.86-2.87) | 2,242/4,213,467 | 1.00 (ref) |
| >10 years latency | 71/68,412 | 1.25 (0.97-1.59) | ||
| Viral hepatitis | 359/82,841 | 6.26 (5.54-7.08) | 2,242/4,213,467 | 1.00 (ref) |
| <5 year latency | 66/12,761 | 7.29 (5.57-9.55) | ||
| 5-10 year latency | 53/12,112 | 7.23 (5.44-9.59) | 2,242/4,213,467 | 1.00 (ref) |
| >10 years latency | 240/57,968 | 5.86 (5.07-6.77) | ||
|
| ||||
| b) Patients transfused in or after 1992 | ||||
| All liver disease | 692/188,377 | 0.99 (0.91-1.07) | 21,672/7,059,794 | 1.00 (ref) |
| <5 year latency | 216/49,916 | 1.00 (0.87-1.16) | ||
| 5-10 year latency | 214/63,155 | 0.89 (0.77-1.02) | 21,672/7,059,794 | 1.00 (ref) |
| >10 years latency | 262/75,306 | 1.06 (0.94-1.21) | ||
| All liver disease (excluding viral hepatitis) | 570/174,502 | 0.99 (0.91-1.09) | 19,443/7,086,443 | 1.00 (ref) |
| <5 year latency | 178/44,736 | 1.03 (0.88-1.21) | ||
| 5-10 year latency | 175/58,152 | 0.90 (0.76-1.05) | 19,443/7,086,443 | 1.00 (ref) |
| >10 years latency | 217/71,614 | 1.05 (0.91-1.21) | ||
| Liver cirrhosis | 7/19,881 | 0.52 (0.23-1.18) | 3,630/7,280,689 | 1.00 (ref) |
| <5 year latency | 1/3,242 | 0.53 (0.07-3.77) | ||
| 5-10 year latency | 2/5,591 | 0.28 (0.04-1.83) | 3,630/7,280,689 | 1.00 (ref) |
| >10 years latency | 4/11,048 | 0.68 (0.25-1.83) | ||
| Viral hepatitis | 15/18,996 | 1.12 (0.62-2.02) | 3,099/7,272,465 | 1.00 (ref) |
| <5 year latency | 5/6,322 | 1.53 (0.62-3.81) | ||
| 5-10 year latency | 5/6,736 | 0.76 (0.24-2.43) | 3,099/7,272,465 | 1.00 (ref) |
| >10 years latency | 5/5,938 | 1.14 (0.42-3.15) | ||
In the donor-recipient disease concordance analysis for transfusions before 1992, transfusion of blood from a donor who later developed liver disease was associated with an increased risk for all of the 9 disease groups as compared to recipients of donors without such a diagnosis, with the exception of the liver cancer and the portal hypertension groups, where the numbers of events were small. The highest risk was seen in the recipients of blood from donors diagnosed with viral hepatitis (hazard ratio [HR], 6.26; 95% confidence interval [CI], 5.54-7.08), but associations were also seen for the all liver diseases group (HR, 1.38; 95% CI, 1.30-1.46), and for liver cirrhosis (HR, 1.34; 95% CI, 1.08-1.67) (Table 2). When analysed according to the disease latency in the donor (period from donation to the development of liver disease), a higher risk of liver disease was observed in the recipients of donors with shorter disease latency (Table 2, Supplementary Table 2). Among patients first transfused in or after 1992, we found no association between receiving blood from diseased donors and risk of liver disease, even when stratifying the donors according to their disease latency (Table 2). Estimates for the different diagnosis groups are presented in Supplementary Table 2.
Table 3 and Supplementary Table 3 present results from the analyses of the association between the disease excess score (DES) and disease risk. The highest observed DES was 15.5 for all liver disease, 12.9 for viral hepatitis B or C, 11.2 for all liver disease excluding viral hepatitis B or C, and 5.6 for patients with liver cirrhosis (Supplementary Table 4). These extreme values were all observed before 1992, with considerably lower maximum DES values observed for donors active in or after 1992 (data not shown).
Table 3.
Relative risks of selected liver diseases in relation to the maximum disease excess score among all contributing blood donors, presented stratified by calendar period of transfusion.
| Maximum disease excess score among contributing blood donors* | a) Before 1992
|
b) In or after 1992
|
||
|---|---|---|---|---|
| Events/person-years | Hazard ratio (95% CI)† | Events/person-years | Hazard ratio (95% CI)† | |
| All liver disease | ||||
| < 0.0 recipients | 2,421/1,258,218 | 1.00 (ref) | 7,208/2,689,162 | 1.00 (ref) |
| 0.0, i.e. no prior donations | 2,582/1,193,869 | 0.98(0.93-1.04) | 6,079/2,107,759 | 0.98(0.95-1.02) |
| 0.1-2.5 recipients | 4,619/1,707,270 | 1.07(1.01-1.13) | 8,878/2,407,898 | 1.03(1.00-1.07) |
| 2.6-5.0 recipients | 373/87,091 | 1.40(1.24-1.58) | 199/43,287 | 0.92(0.79-1.08) |
| >5.0 recipients | 56/5,354 | 2.93(2.19-3.92) | 0/65 | 0.00 (n.e.) |
| All liver disease (excluding viral hepatitis) | ||||
| < 0.0 recipients | 2,236/1,330,080 | 1.00 (ref) | 6,730/2,782,299 | 1.00 (ref) |
| 0.0, i.e. no prior donations | 2,352/1,232,839 | 0.97(0.92-1.04) | 5,564/2,154,594 | 0.98(0.94-1.01) |
| 0.1-2.5 recipients | 3,779/1,637,517 | 1.05(0.98-1.11) | 7,572/2,286,312 | 1.03(0.99-1.07) |
| 2.6-5.0 recipients | 189/65,483 | 1.08(0.92-1.27) | 147/37,640 | 0.86(0.71-1.03) |
| >5.0 recipients | 14/1,462 | 3.17(1.80-5.60) | 0/100 | 0.00 (n.e.) |
| Liver cirrhosis | ||||
| < 0.0 recipients | 757/1,688,770 | 1.00 (ref) | 1,563/3,523,687 | 1.00 (ref) |
| 0.0, i.e. no prior donations | 724/1,435,348 | 0.91(0.81-1.01) | 1,276/2,556,476 | 0.98(0.90-1.06) |
| 0.1-2.5 recipients | 816/1,147,641 | 1.06(0.95-1.19) | 795/1,215,650 | 1.05(0.95-1.16) |
| >2.5 recipients | 36/25,272 | 1.69(1.19-2.41) | 3/4,758 | 1.03(0.33-3.24) |
| Viral hepatitis | ||||
| < 0.0 recipients | 826/1,778,734 | 1.00 (ref) | 1,346/3,725,046 | 1.00 (ref) |
| 0.0, i.e. no prior donations | 746/1,466,346 | 1.00(0.90-1.11) | 1,234/2,669,623 | 1.05(0.96-1.14) |
| 0.1-2.5 recipients | 884/1,003,571 | 1.24(1.11-1.39) | 533/894,575 | 1.00(0.89-1.13) |
| >2.5 recipients | 94/30,399 | 3.35(2.62-4.29) | 1/2,216 | 0.87(0.12-6.27) |
The diseases excess score was computed time-dependently so that for each new donation we calculated the difference between the observed and expected number of diseased patients among all previous recipients of each donor. Thus, a case excess score below zero implies that there are fewer than expected diseased patients among previous recipients and a riskiness score above zero implies that the number of events is higher than expected. Because most recipients received transfusions from more than one donor, the highest case excess score of all donors who contributed blood unit to each recipient was used in the statistical model. The donor disease excess score only included the number of diseased patients among previous recipients, i.e. not the disease status of the index patient.
Hazard ratios were adjusted for patient age, sex and ABO blood group, calendar year of transfusion, region of residence, as well as number of transfusions.
For most diagnosis groups, there was a strong and positive association between receiving blood from a donor with a high DES and disease risk in the period before 1992 (Table 3). Here, there were 5,048 (0.28%) liver-related diagnoses observed in 1,799,715 person-year of recipients of donors with a positive DES, compared to 2,421 (0.19%) events in 1,258,218 person-year of recipients of donors with negative DES. The risk of developing a liver-related diagnosis in the recipients increased with higher donor DES, with a HR increasing from 1.07 to 2.95 for recipients of donors with a DES of 0.1-2.5 or >5.0, respectively, compared to recipients of donors with negative DES.
When analysed according the predefined 9 categories of liver diseases, the strongest associations were again seen for viral hepatitis B or C (HR, 3.35; 95% CI, 2.62-4.29), all liver diseases with and without viral hepatitis (HR, 2.93; 95% CI, 2.19-3.92 and HR, 3.17; 95% CI, 1.80-5.60, respectively), and for liver cirrhosis (HR, 1.69; 95% CI, 1.19-2.41), comparing recipients of blood from donors with very high DES to those with negative DES. Similar association was seen for all the other groups (Supplementary Table 3). The analysis for the period after the beginning of 1992 showed no association with recipient disease risk even when considering the highest DES groups (Table 3 and Supplementary table 3).
Findings were similar in sensitivity analyses when we used 90-day and 1-year exposure ascertainment periods. Similarly, results were comparable when follow up period in recipients was restricted to 10 or 20 years (Supplementary Table 5).
Results from the power simulations are presented in Figure 1. The ability to detect transmission increased with shorter latency, increasing prevalence among donors, and with a higher rate of cirrhosis occurrence among infected. In general, the statistical power with these sets of assumptions was higher for the first than for the second analytical approach. For common agents, with a prevalence of 1%, power was generally good with both approaches, even with a 20-year latency. However, for less common agents, the ability to detect transmission was limited to agents with mean latencies of 15 or 10 years.
Figure 1.
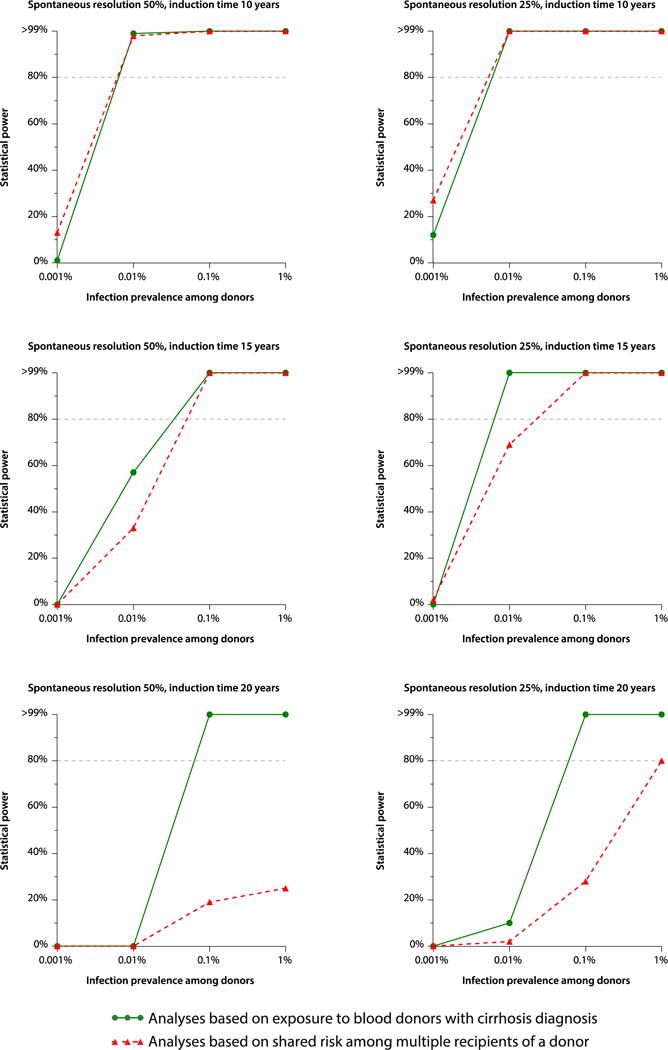
Statistical power to detect transfusion transmission in relation to prevalence of infectious agent, with either 25% or 50% of spontaneous viral resolution.
Discussion
Here we present results from a large bi-national cohort study of transfusion transmittable causes of liver disease. Using two independent analytical approaches we provide clear evidence of transfusion transmission of an agent causing a range of different forms of chronic liver disease. Importantly, our analyses only show evidence of such transmission before 1992, and not thereafter, when screening for both HBV and HCV of all blood donations was fully implemented. As expected, the risk of transfusion transmission of liver disease before 1992 was mainly driven by viral hepatitis, but even though risk estimates were less striking, results were consistent even when analyses were restricted to cirrhosis or other end-stage liver disease.
The possibility of a transmissible non-B, non-C hepatitis as a cause of chronic hepatitis has previously been suggested as an explanation of a fraction of cryptogenic liver disease—especially cases associated with blood transfusion[6, 7, 15–17]. This study, however, indicates that such associations are not driven by agents transmissible through blood transfusion, but may rather result from other risk factors more commonly found among transfused patients than in the population in general. We speculate that the residual risk of “cryptogenic cirrhosis” among transfused patients could, for example, be caused by a higher prevalence of alcohol consumption, or risk factors of non-alcoholic fatty liver disease among transfused patients.
The main strength of this study is the large number of patients included, the high-quality data of the transfusion registers, and the long and complete follow up of all individuals included. In addition, two different approaches were used to investigate the possibility of transfusion of a cause of liver disease, including an approach that would account also for infectious donors even without them developing the disease. Also, the fact that we were testing for the transmission of a broad spectrum of liver diseases in hierarchically structured groups enables us to capture different phenotypes of the same disease in the donor and recipient, or in the different recipients from an infected donor. Still, even though we base our analyses on the complete computerized blood bank history in two countries, statistical power and length of follow-up in the later follow-up period clearly limits our ability to detect transmission of very rare agents or agents with incubation times exceeding 15 years. That said, larger studies are unlikely to be conducted in the near future. Furthermore, among other strengths is the use of high-quality, population-based outcome registers, where the specificity of liver-related diagnosis such as cirrhosis and hepatocellular carcinoma is high[18].
Although the infection status of the donors was unknown at the time of the transfusion, there are still a number of confounding factors that are needed to be addressed in our analysis. The likelihood of being exposed to a possible transfusion-transmitted agent is directly related to the number of received transfusions since recipients of larger number of units have a higher probability of receiving blood from an infectious donor, making the number of transfusions an important confounding factor. Other possible confounders include risk factors for liver disease at a population level (such as calendar year and area of residence). Importantly, the allocation of blood units typically follows very predictable rules, based on a first in-first out principle, where the oldest available unit of a suitable blood type is allocated first. This, coupled with the fact that neither the future disease status of a contributing blood donor, nor the donors’ retrospectively computed DES can be known at the time of transfusion, ensures that allocation of infected units will be essentially random. Therefore, with proper adjustment for the number of transfused units, the calendar period, area of residence and blood type, residual confounding should be negligible. Since the indication for a transfusion might be related to early symptom of a yet undiagnosed liver disease in the recipient we therefore delayed the recipients’ entry into the analysis by 180 days to allow time for such a possible condition to manifest itself and be diagnosed.
As expected, the implementation of hepatitis B and C screening of all donations, as well as the exclusion of donors with high risk behaviours during the response for the HIV epidemic during the 1980’s, resulted in significantly lower occurrence of liver disease in the donor population (and of potentially exposed recipients) after 1992. This, together with the relatively shorter follow-up duration of recipients after 1992 begs the question of whether the study was sufficiently powered to detect an unknown transfusion-transmitted infection in that period. However, the power simulation analysis showed that even when considering a rare infectious agent with a prevalence of 0.01% among donors and an incubation time of 15 years, we still had a more than 80% power to detect such an infection based on the donor-recipient concordance analysis (Figure 1). Consequently, our analysis is sufficiently powered to detect transmission of hepatotropic agents with a prevalence higher than 1: 10,000 and an incubation period shorter than 15 years.
Hepatitis E virus (HEV) is usually transmitted by the faecal oral route1 Viral transmission by blood transfusion has also been reported[19]. HEV generally runs a benign course of acute hepatitis. However, in some patients who are immunocompromised, such as those with haematological malignancies or bone marrow transplantation, HEV infection can cause chronic hepatitis and liver failure, Screening for HEV in patients with acute hepatitis without risk factors for HEV, such as travel to endemic areas, is not routinely done. The results of the analyses presented here, including the absence of a signal of a transfusion transmittable agent after 1992, is not generalizable to HEV due to the very low prevalence of HEV in Scandinavian countries, the infrequent testing for that virus, and the risk of misclassification.
One important limitation of the study is the fact that SCANDAT2 is based on administrative transfusion databases that were not intended for research purposes. However, the donors and their respective recipients are very well identified in the database. Furthermore, data in the inpatient and cancer registries, which are used here for identification of the occurrence of the outcomes of interest, are regularly maintained and updated by authorities in both countries, and have been shown to have a high validity[11, 20–22]. Regardless, the use of these registries is likely to have resulted in some degree of underreporting, and therefore the possibility of misclassification of the outcomes of interest that do not warrant hospital admissions. However, the degree of such misclassification or underreporting is likely not related to the future disease status of the donor, nor the donor’s retrospectively computed DES. Therefore, we do not believe that such underreporting or misclassification would have significantly influenced our results. Other limitations are the substantial differences in the practice of transfusion medicine internationally, the changes in that practice over the period of the study, and the low prevalence of some of the hepatotropic virus in the Scandinavian population (such as hepatitis E virus, which was not included in the studied diagnoses). Caution is therefore warranted when generalizing our findings to other countries and populations, where prevalence of transfusion transmittable hepatotropic agents, such as hepatitis E, might be higher than in our Scandinavian database.
In 1996, the Danish National Board of Health recommended a HCV lookback investigation, to identify recipients of blood components from donors found to be positive for HCV since the implementation of anti-HCV screening in Denmark in 1991[23]. This would theoretically increase the prevalence and donor-recipient risk association in the Danish subset in SCANDAT. However, analyses restricted to the Swedish population only showed comparable results.
In conclusion, our results show clear evidence of a transfusion transmittable cause of chronic liver disease in the pre-1992 era, but do not support the presence of a yet undiscovered agent transmittable through blood transfusion after 1992. While we cannot exclude the possibility of transmission of very rare agents or agents with very long incubation times, these findings indicate that additional hepatotropic agents causing liver disease are unlikely to be prevalent among Scandinavian blood donors. In addition, the failure to detect any disease transmission after implementation of HCV screening confirms the success of existing blood safety efforts.
Supplementary Material
Acknowledgments
This work was supported by contracts HHSN268201100002I and HHSN268201100003I from the National Heart Lung and Blood Institute of the U.S. National Institutes of Health. The assembly of the SCANDAT2 database was made possible through grants from the Swedish research council (2011-30405, 2007-7469), the Swedish Heart-Lung Foundation (20090710), the Swedish Society for Medical Research (Edgren), the Strategic research program in Epidemiology at Karolinska Institutet (Edgren), and the Danish Council for Independent Research (2009B026).
Footnotes
DR. AMMAR MAJEED (Orcid ID: 0000-0002-7024-8787)
Conflict of interest
No author reports any relevant conflicts of interest
References
- 1.Beeson PB. Jaundice occurring one to four months after transfu- sion of blood or plasma. Report of seven cases. JAMA. 1943;121 [Google Scholar]
- 2.Stevens CE, Aach RD, Hollinger FB, et al. Hepatitis B virus antibody in blood donors and the occurrence of non-A, non-B hepatitis in transfusion recipients. An analysis of the Transfusion-Transmitted Viruses Study. Annals of internal medicine. 1984:101. doi: 10.7326/0003-4819-101-6-733. [DOI] [PubMed] [Google Scholar]
- 3.Koziol DE, Holland PV, Alling DW, et al. Antibody to hepatitis B core antigen as a paradoxical marker for non-A, non-B hepatitis agents in donated blood. Annals of internal medicine. 1986;104:488–95. doi: 10.7326/0003-4819-104-4-488. [DOI] [PubMed] [Google Scholar]
- 4.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science (New York, NY) 1989;244:359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 5.Czaja AJ. Cryptogenic Chronic Hepatitis and Its Changing Guise in Adults. Digestive Diseases and Sciences. 2011;56:3421–38. doi: 10.1007/s10620-011-1769-9. [DOI] [PubMed] [Google Scholar]
- 6.Kodali VP, Gordon SC, Silverman AL, McCray DG. Cryptogenic liver disease in the United States: further evidence for non-A, non-B, and non-C hepatitis. The American journal of gastroenterology. 1994;89:1836–9. [PubMed] [Google Scholar]
- 7.Caldwell SH, Oelsner DH, Iezzoni JC, Hespenheide EE, Battle EH, Driscoll CJ. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology. 1999;29 doi: 10.1002/hep.510290347. [DOI] [PubMed] [Google Scholar]
- 8.Edgren G, Hjalgrim H, Tran TN, et al. A population-based binational register for monitoring long-term outcome and possible disease concordance among blood donors and recipients. Vox Sanguinis. 2006;91 doi: 10.1111/j.1423-0410.2006.00827.x. [DOI] [PubMed] [Google Scholar]
- 9.Edgren G, Rostgaard K, Vasan SK, et al. The new Scandinavian Donations and Transfusions database (SCANDAT2): a blood safety resource with added versatility. Transfusion. 2015;55:1600–6. doi: 10.1111/trf.12986. [DOI] [PubMed] [Google Scholar]
- 10.Ludvigsson JF, Otterblad-Olausson P, Pettersson BU, Ekbom A. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. European Journal of Epidemiology. 2009;24:659–67. doi: 10.1007/s10654-009-9350-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludvigsson JF, Andersson E, Ekbom A, et al. External review and validation of the Swedish national inpatient register. BMC public health. 2011;11:450. doi: 10.1186/1471-2458-11-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgren G, Hjalgrim H, Rostgaard K, et al. Transmission of Neurodegenerative Disorders Through Blood Transfusion: A Cohort Study. Annals of internal medicine. 2016;165:316–24. doi: 10.7326/M15-2421. [DOI] [PubMed] [Google Scholar]
- 13.Edgren G, Hjalgrim H, Reilly M, et al. Risk of cancer after blood transfusion from donors with subclinical cancer: a retrospective cohort study. The Lancet. 2007;369 doi: 10.1016/S0140-6736(07)60779-X. [DOI] [PubMed] [Google Scholar]
- 14.Edgren G, Rostgaard K, Hjalgrim H. Methodological challenges in observational transfusion research: lessons learned from the Scandinavian Donations and Transfusions (SCANDAT) database. ISBT Science Series. 2017;12:191–5. [Google Scholar]
- 15.Wejstål R, Norkrans G, Widell A. Chronic non-A, non-B, non-C hepatitis: is hepatitis G/GBV-C involved? Scandinavian journal of gastroenterology. 1997;32:1046–51. doi: 10.3109/00365529709011223. [DOI] [PubMed] [Google Scholar]
- 16.Lai MW, Chang MH, Hsu HY. Non-A, non-B, non-C hepatitis: its significance in pediatric patients and the role of GB virus-C. The Journal of pediatrics. 1997;131:536–40. doi: 10.1016/s0022-3476(97)70057-x. [DOI] [PubMed] [Google Scholar]
- 17.Alter HJ. Transfusion transmitted hepatitis C and non-A, non-B, non-C. Vox sanguinis. 1994;67(Suppl 3):19–24. doi: 10.1111/j.1423-0410.1994.tb04539.x. [DOI] [PubMed] [Google Scholar]
- 18.Kaczynski J, Wallerstedt S. Registration of liver cancer data–a study on the reliability of the Swedish Cancer Registry. Acta oncologica (Stockholm, Sweden) 1989;28:716–7. doi: 10.3109/02841868909092300. [DOI] [PubMed] [Google Scholar]
- 19.Hewitt PE, Ijaz S, Brailsford SR, et al. Hepatitis E virus in blood components: a prevalence and transmission study in southeast England. Lancet. 2014;384:1766–73. doi: 10.1016/S0140-6736(14)61034-5. [DOI] [PubMed] [Google Scholar]
- 20.Ekström AM, Signorello LB, Hansson LE, Bergström R, Lindgren A, Nyrén O. Evaluating gastric cancer misclassification: a potential explanation for the rise in cardia cancer incidence. Journal of the National Cancer Institute. 1999;91:786–90. doi: 10.1093/jnci/91.9.786. [DOI] [PubMed] [Google Scholar]
- 21.Storm HH, Michelsen EV, Clemmensen IH, Pihl J. The Danish Cancer Registry–history, content, quality and use. Danish medical bulletin. 1997;44:535–9. [PubMed] [Google Scholar]
- 22.Schmidt M, Schmidt SA, Sandegaard JL, Ehrenstein V, Pedersen L, Sørensen HT. The Danish National Patient Registry: a review of content, data quality, and research potential. Clinical epidemiology. 2015;7:449–90. doi: 10.2147/CLEP.S91125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen PB, Groenbaek K, Krarup HB. Transfusion-acquired hepatitis C: the Danish lookback experience. The Danish HCV [hepatitis C virus] Lookback Group. Transfusion. 1999;39:188–93. doi: 10.1046/j.1537-2995.1999.39299154734.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.