

Summary
Herein, a lignin-centered convergent approach to produce either aliphatic or aromatic bio-hydrocarbons is introduced. First, poplar or spruce wood was deconstructed by a lignin-first biorefining process, a technique based on the early-stage catalytic conversion of lignin, yielding lignin oils along with cellulosic pulps. Next, the lignin oils were catalytically upgraded in the presence of a phosphidated Ni/SiO2 catalyst under H2 pressure. Notably, selectivity toward aliphatics or aromatics can simply be adjusted by changes in H2 pressure and temperature. The process renders two distinct main cuts of branched hydrocarbons (gasoline: C6-C10, and kerosene/diesel: C14-C20). As the approach is H2-intensive, we examined the utilization of pulp as an H2 source via gasification. For several biomass sources, the H2 obtainable by gasification stoichiometrically meets the H2 demand of the deep converting lignin-first biorefinery, making this concept plausible for the production of high-energy-density drop-in biofuels.
Keywords: biofuels, lignin, lignin-first biorefinery, HDO, aromatics, catalytic upstream biorefining, lignin fuels, phosphidated nickel catalysts, hydrodeoxygenation, Raney Ni, hydrogen transfer
Graphical Abstract

Highlights
-
•
Lignin-first biorefinery produces gasoline and kerosene/diesel drop-in fuels
-
•
A H2 self-sufficient deep converting lignin-first biorefinery is achievable
-
•
H2 surplus releases cellulose for the production of platform chemicals
-
•
Chemicals further monetize the deep converting lignin-first biorefinery
Context & Scale
Heterogeneous catalysis is no longer limited to the conversion of lignin wastes from cellulosic-centric industries, but has expanded to offer innovative solutions for the deconstruction of lignocellulose by reductive processes. Such solutions are referred to as lignin-first biorefining. They are highly efficient at preventing the generation of recalcitrance in the lignin streams while yielding delignified pulps. Herein, a lignin-centered convergent approach rendering two main cuts of branched hydrocarbons (gasoline: C6-C10, and kerosene/diesel: C14-C20) is introduced. As the hydrodeoxygenation of lignin streams is H2-intensive, the utilization of pulp as an H2 source via gasification is proposed. The cellulosic H2 shows potential for covering the H2 demand for the production of drop-in lignin fuels. Importantly, the energy content of the lignin fuels relative to the energy content of the lignocellulose is approximately 2- to 2.5-times higher than that of cellulosic ethanol. Therefore, it is now timely to question whether cellulosic bioethanol is the most sensible target fuel from lignocellulose.
Heterogeneous catalysis is no longer limited to the conversion of lignin wastes from cellulosic-centric industries, but has expanded to offer innovative solutions for the deconstruction of lignocellulose by reductive processes. This enables the creation of a deep converting lignin-first biorefinery, shifting paradigms. Lignin-biorefinery produces gasoline and kerosene/diesel drop-in fuels. The self-sufficiency in H2 is achieved through the gasification of the delignified holocelluloses. Notably, H2 surplus releases holocellulose for the production of platform chemicals, further monetizing the lignin-first process chains.
Introduction
Stimulated by dwindling petroleum resources, and environmental and political concerns, the production of ethanol from fermentable sugars represents the backbone of the biofuel industry.1 In this context, an approach gaining momentum is the conversion of lignocellulosic biomass (e.g. forestry biomass or crop residues, instead of sugars from sugar beet or corn) into fermentable sugars for the production of bioethanol. However, taking into account the low energy density and high petrol equivalent unit cost of the cellulosic bioethanol (ca. 0.9 € L−1, excluding taxes), this process is currently far from optimal.2 Importantly, the degraded lignin generated as process waste, a fraction corresponding up to one-third of the biomass composition and 40% of lignocellulose potential fuel value, is planned to be used as a solid fuel to sustain such a process chain.3, 4, 5 Considering these points, an important question arises: is cellulose-derived bioethanol the most sensible target fuel from lignocellulosic biomass?
Alternative general strategies for the production of fuels from lignocellulosic biomass have focused on the gasification of biomass into Syngas, and subsequent Fischer-Tropsch (FT) synthesis to afford linear hydrocarbons,6 or else upgrading of the lignocellulose pyrolysis oil, derived from lignocellulose fast pyrolysis, via hydrodeoxygenation (HDO).7, 8 Nonetheless, the former process suffers from high energy consumption as a result of the indirect conversion, and the latter process requires a high external input of hydrogen gas, whereby the majority (∼95%) of the hydrogen is generated from fossil resources, and the cost for hydrogen logistics is critical.9, 10, 11 To reduce the external hydrogen requirement, efforts have been devoted to the in-situ deoxygenation during pyrolysis or the H2 recover via reforming of by-products from biomass pyrolysis.12 For instance, Anellotech is now developing the Bio-TCat process based on the pioneering research by George Huber and coworkers.13 Also, the integrated hydropyrolysis and hydroconversion (IH2) concept is capable of providing the direct production of fuel or fuel blend with less external input of H2. In that approach, part of the hydrogen is generated from reforming the light gases generated from pyrolysis.14, 15
A successful biorefinery operation should be capable of providing both liquid fuels and commodity chemicals, on a sufficiently large scale.16, 17, 18 Regarding high-energy-density fuels, lignin seems to be more attractive than cellulose and hemicellulose because of its high carbon number (lignin monomers contain nine or more atoms of carbon) and greater carbon content (60 vs. 44 wt%), and its reduced oxygen content (32 vs. 49 wt%). Again, lignin accounts for approximately 30 wt% of dry lignocellulosic biomass and 40% of its potential fuel value.19, 20, 21 On this account, fuel production from lignin is anticipated to afford compounds characterized by higher and more varied molecular weights (C6-9+), in comparison to fuels derived from the carbohydrate fraction of biomass (primarily restricted to C2-C6).22, 23 Notably, the production cost of high-octane reformulated fuel from lignin is estimated at 0.3 € L−1. This estimate is based on an economic analysis of a process encompassing a base-catalyzed depolymerization of lignin and subsequent catalytic hydroprocessing processes.24 In addition to these considerations, one characteristic feature of emerging lignin-bioengineering approaches is the possibility to increase the lignin content of energy crops.16 Overall, these points indicate the potential benefits associated with a lignin-to-fuels strategy.
To establish a complex biorefinery resembling the deep converting oil refineries (i.e. oil refineries with a Nelson Complexity Index, NCI, typically higher than 5),25 a deep converting biorefinery must overcome the following barriers: (i) the recalcitrance of the isolated lignin fraction, which is created by the conventional techiques of biomass deconstruction,16 and; (ii) the H2 demand for HDO, which required large input of CO2-intensive hydrogen for both the depolymerization of lignin to smaller molecules and their catalytic upgrading to branched alkane products so as to improve fuel perfomance and minimize soot formation upon combustion.13, 26
These difficulties have been addressed in recent literature.16 A heightened understanding of efficient lignin depolymerization (and prevention of re-condensation) has been acquired.16, 18, 27 Regarding the prevention of lignin recalcitrance generation upon its removal from the lignocellulosic matrix, our research group has demonstrated the Early-stage Catalytic Conversion of Lignin (ECCL) to offer an atom-economic method for the efficient deconstruction of lignocellulose.16, 28, 29, 30, 31 This technique produces lignin oils of low molecular weight (Mw) and high structural uniformity in addition to holocelluloses (pulps). In this quest, other groups also provided very important contributions to the lignin-first deconstruction of lignocellulose, giving irreversible momentum to this new research field upon demonstrating the feasibility of this approach under various conditions.27, 32, 33, 34, 35, 36, 37, 38 In a broader context, heterogeneous catalysis is no longer limited to the processing of technical (recalcitrant) lignin wastes, generated cellulosic-centric industries (e.g. pulp and paper industry and cellulosic bioethanol production), but has expanded to offer innovative solutions for the pulping process itself and efficient methods for lignin-first lignocellulose deconstruction, here referred to as Catalytic Upstream Biorefining (CUB) or lignin-first biorefining, which prevent the generation of lignin recalcitrance.16, 28, 29
In our approach, the CUB process is performed in the presence of Raney Ni as the catalyst for H-transfer reactions (i.e., hydrogenation, hydrodeoxygenation, and hydrogenolysis) and 2-propanol as a component of the lignin-extracting solvent mixture and an H-donor.16, 28, 29 Built on the lignin extraction from lignocellulose in 2-propanol/water (7:3, v/v), the presence of the inexpensive Raney Ni in CUB leads to reductive processes on reactive lignin fragments (e.g. Hibbert ketones) formed by solvolysis of native lignin. As a result, the EECL passivates the reactive lignin fragments and, therefore, protects the whole lignin stream from increased recalcitrance via recondensation. Notably, CUB isolates lignin as a brownish viscous oil, instead of a red-brown polymeric solid like that from the organosolv process.16, 28, 29 Lignin oils comprise up to 50–60% phenolic species (with Mw < 250 Da, as shown in the orange color inset in Figure 1). The other products (30–40%) found in the lignin oil are dimers and trimers and, to a lesser extent, lignin oligomers and polyols. The latter class of products is derived from the hydrogenolysis of hemicellulose sugars released by solvolytic processes.
Figure 1.

A Convergent Deep-Converting Lignin-First Biorefinery
A simplified representation of the convergent process for preparation of C6-C29 aliphatic fuels or aromatic chemicals from biomass: (I) lignin-first deconstruction of lignocellulose via Early-stage Catalytic Conversion of Lignin (ECCL): holocellulose and lignin oil streams are obtained;28 (II): gasification of holocellulose generates the required H2 for hydrodeoxygenation (HDO) processes, and; (III) the convergence point: the HDO of lignin oil yields aromatics (with a surplus hydrogen) or high-purity alkane fuels, depending on the process conditions employed in the HDO step. Another version of this representation, a block flow diagram is presented in Figure S6.
Herein, we present a concept for deep converting biorefinery producing either to aliphatic or aromatic hydrocarbons, based on the catalytic upstream and downstream processing of the lignin component. We demonstrate the holocellulose fraction to provide the H2 requirement via gasification/steam reforming. This approach, therefore, significantly differs from the production of cellulosic bioethanol. The proposed convergent approach for a deep converting lignin-first biorefinery is schematically represented in Figure 1: (I) CUB process leading to tandem depolymerization, passivation and isolation of a lignin stream of low molecular weight and high structural uniformity;28 (II) gasification/steam reforming of the holocellulose fraction yielding H2,39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 and; these two streams converge at (III) the HDO of the lignin oil, yielding a blend of value-added C6-C29 aliphatic or aromatic hydrocarbons, depending on the chosen reaction parameters (pressure of H2 and temperature). The observable product species range from C6 up to C29, with a predominance of hydrocarbons within distinct C6-C10, C14-C20 and to a lesser extent C22-C26 bands. This feature is of practical importance because it enables separation by distillation facilities present in the current oil refineries, to produce gasoline and kerosene/diesel fuels.
Results
HDO of the Lignin Streams
To perform the HDO of lignin oil to aliphatics or aromatics, poplar and spruce were selected as hardwood and softwood feedstocks, respectively. Hardwood and softwood have different lignin content and corresponding different S/G (syringyl/guaiacyl) ratio which plays a crucial role toward lignin depolymerization.50 Initially, lignin oils obtained from the lignin-first deconstruction of lignocellulose (step I), before HDO treatment, were analyzed using two-dimensional Gas Chromatography-Mass Spectrometry, GC×GC–MS/FID (Figure S1). Despite the different S/G value of selected samples, as indicated by the GC×GC–MS/FID (Figure S1), the decomposed fractions reveal the sufficient cleavage of the different types linkages. 2D GC×GC images indicated that the volatile products fraction in each oil was largely composed of dihydro-p-lignols and other derivatives of monolignols, together with polyols derived from hydrogenolysis of hemicellulose sugars. Polyols correspond to 5–6% of the lignin stream (i.e. approximately 36% of the xylan content relative to the original feedstock).28 Notably, the fraction of products observable by 2D GC×GC corresponding to about 55–60%, based on estimates derived from thermogravimetric analysis (TGA) under inert conditions.28 Dilignols and oligomeric species are detectable by gel-permeation chromatography. The dimers represent another one-quarter of the composition, the remaining species are the lignin oligomers with Mw between 0.4 and 3.5 kDa, as reported by us elsewhere.28
Lignin oils were each subjected to a phosphidated Ni/SiO2 catalyst. This catalyst was produced by a simple phosphidation procedure which consists of cooking a commercially available Ni/SiO2 catalyst (64% loading from Strem Chemicals) in tri-(n-octyl)phosphine (TOP) at 300°C for 6 hr. This procedure is inspired by the seminal works of Schaak et al. for the production of unsupported metal phosphide nanoparticles from metallic nanoparticles.51, 52 The phosphidated Ni/SiO2 catalyst was prepared with a TOP:Ni ratio of 6.1. To evaluate the influence of reaction conditions on product distribution, reactions were performed under high pressure-low temperature (5 MPa H2, 573 K) and low pressure-high temperature (0.5–1 MPa H2, 623 K). Notably, colorless clear solutions were obtained after both reactions. No lignin-derived insoluble char was formed. In this case, an estimated by TGA indicates that up to 90% products are volatile at 300°C (Figure S5), the GC-injector temperature.
GC×GC–MS images of the product mixtures are displayed in Figure 2 (Figure S2 for spruce). The aliphatic/aromatic product distributions were determined for each of the four product mixtures, and are listed in Table 1. Semi-quantification of the products was performed using the response of the flame-ionization detector (FID) and considering the response factors estimated by Effective Carbon Number method (ECN).53 A typical yield of bio-oil after ECCL is 20–25% relative to the initial weight of lignocellulose. In turn, the weight yield of HDO of lignin-oil to hydrocarbons is 40–45%. These together give a cumulative weight yield of around 10% of lignin-derived hydrocarbons relative to the initial lignocellulose weight.
Figure 2.

Characteristic Bio-hydrocarbons and Product Distributions Obtained from Lignin il
(A–D) GC×GC−MS(FID) traces highlighting volatile products obtained from the HDO of poplar (A and C). Distribution of products according their C-atoms (B and D). Reaction conditions: (A and B) high pressure/5 MPa - low temperature/573 K for aliphatics-directed HDO; (C and D): low pressure/0.5 MPa - high temperature/623 K for aromatics-directed HDO. The semi-quantification of the products was performed based on ECN estimation.53
Table 1.
Distribution of Volatile Products for Aliphatics-Directed and Aromatics-Directed HDO
| Feedstock | Aliphatics-Directed HDO |
Aromatics-Directed HDO |
||
|---|---|---|---|---|
| Aliphatics | Aromatics | Aliphatics | Aromatics | |
| Poplar | 100% | 0% | 22% | 78% |
| Spruce | 100% | 0% | 25% | 75% |
Examining the 2D GC×GC images (Figure 2, poplar and Figure S2 spruce), full conversion of starting materials into deoxygenated product species was achieved, and phenolic compounds were correspondingly absent. Interestingly, simultaneous modification of the H2 pressure and temperature of reaction brings about a dramatic change in the product mixture composition. In fact, under conditions of high H2 pressure/low temperature, the product mixtures are composed of exclusively cyclic aliphatic hydrocarbons (Figures 2A and S2A). By stark contrast, using an identical setup and catalyst, yet by simple adjustment of reaction conditions (low H2 pressure/high temperature), a product mixture incorporating predominantly aromatic hydrocarbons was instead attainable (Figures 2C and S2C). For this aromatics-directed HDO, aromatic hydrocarbons represented more than 75% of the liquid products (Table 1), corresponding to an H2 saving by ca. 30% in comparison to the aliphatics-directed HDO. The results indicate the potential ability to fine-tune the aliphatic/aromatic product distribution by the simple adjustment of reaction parameters, enabling the process to meet market demands for fuels and aromatic chemicals.
Despite the presence of volatile hydrocarbons in the entire range C6 to C29, the distribution is non-uniform – most of the compounds could be categorized into distinct C6-C10 and C14-C20 groups (Figures 2B, 2D, S2B, and S2D). The aliphatic compounds that fall within these categories are directly applicable as gasoline and kerosene/diesel fuels, respectively. The high concentration of C9 hydrocarbons is attributable to the high prevalence of species with a 4-propylphenol (nine-carbon) skeleton after step I (Figure S1). The presence of C14-C20 species may likely be explained by HDO of lignin dimers connected by C—C bonds. Notably, the formation of very distinct groups of products regarding carbon number indicates that cross-alkylation of smaller hydrocarbons, as reported for other catalytic systems,54 occurs to a very limited extent over the acidic sites of phosphidated Ni/SiO2. Low quantities of heavier hydrocarbons (C21+) were observed (≤4% total of identified volatile products, Figures 2B, 2D, S2B, and S2D), possibly arising from HDO of lignin trimers or higher oligomers. The formation of these three cuts of hydrocarbons could also be confirmed by thermogravimetric analysis (TGA) carried out under inert atmosphere (see Figure S5) indicates three main steps of weight loss (I - gasoline, 30–170°C: 50%; II – kerosene/diesel, 170–300°C: 42%; III - waxes, 280–400°C: 4%). The predominance of hydrocarbons within the two primary distinct bands (C6-C10 and C14-C20) facilitates their simple separation by distillation, as currently carried out in refineries.
To check whether non-volatile oxygenated species were still present in the product mixture, HSQC 2D NMR experiments were performed on the product mixtures obtained from poplar (Figure 3). The spectra lack characteristic signals for oxygen-containing compounds (specifically methoxyl substituents on the guaiacyl and syringyl units and other oxygenated functionalities), indicating the full HDO of the lignin oil. The elemental analysis of the HDO product shows that the sum of C and H varies from 98.5% to 99.3%. Moreover, the HSQC spectra also confirm the lack of aromatic species in the product mixtures attained under conditions of a high pressure of H2 and low temperature (aliphatics-directed HDO).
Figure 3.

Spectroscopic Proof of Full HDO to Alkanes or Alkanes Plus Aromatic Hydrocarbons
(A–C) HSQC 2D NMR spectra (in DMSO-d6) of (A) poplar-derived lignin oil before HDO (upper), and after (B) aliphatics-directed HDO, and (C) aromatics-directed HDO.
For both aliphatics- and aromatics-directed HDO of poplar lignin oil, the stability of the phosphidated Ni/SiO2 catalyst was evaluated. 2D GC×GC images for product mixtures obtained from three consecutive runs of catalyst used are shown in Figures S3 and S4 (Supplemental Information). For the aliphatics-directed HDO, the absence of signals for phenols or other oxygenated species (Figure S3, left) indicates that full HDO was achieved in each of the three runs. In the second run, trace levels of saturated oxygenated intermediates (e.g. cyclohexanol and methoxycyclohexanol) were identified, becoming more pronounced in the third run – this observation indicates slightly lesser extent of HDO achieved by the catalyst. Nevertheless, despite such traces of oxygenated compounds (i.e. contents lower than 0.2%), the phosphidated Ni/SiO2 catalyst maintains its outstanding features, as demonstrated by repeatedly high yields of target product fuel, consistently low-oxygen weight percentages in the product mixtures, and reproducible relative distributions of products. The similar high performance was also observed for the aromatics-directed HDO (Figure S3, right). However, trace levels of alkylphenols were identified in the third run. Nevertheless, percentages of aromatic hydrocarbons species remained consistently high (Table 2).
Table 2.
Identified Final Aliphatic/Aromatic Product Distributions, for First, Second and Third Runs (Catalyst Recycling Experiments) of the Phosphidated Ni/SiO2 Catalyst, for the Conversion of Poplar Lignin Oil to Aromatic Hydrocarbons
| Run | Aromatics-Directed HDO |
|
|---|---|---|
| Aliphatic | Aromatic | |
| 1st | 22% | 78% |
| 2nd | 26% | 74% |
| 3rd | 23% | 77% |
General reaction conditions: 5 (or 0.5) MPa H2, T = 573 (or 623) K for aliphatics (or for aromatics-directed) HDO. For recycling experiments, the catalyst was washed with n-hexane, dried and then reused.
Evolution of the Active Catalytic Phase
To shed light on the remarkable performance features of the phosphidated Ni/SiO2 catalyst, additional experiments were performed using guaiacol as a model substrate. This approach was adopted to evaluate the evolution of the active catalytic phase at an initial guaiacol conversion of approximated 65%. Figure 4 presents the results of the recycling experiments.
Figure 4.
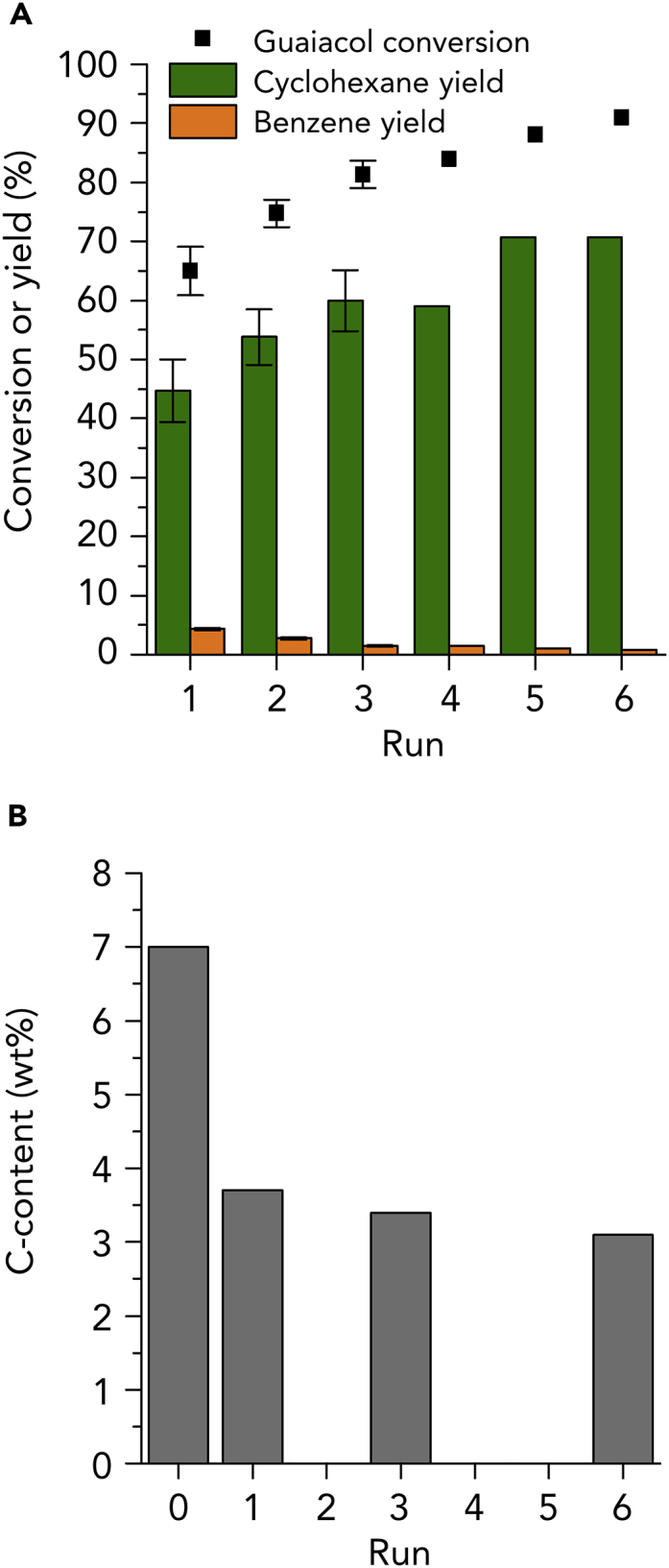
Catalyst Recycling Experiments
(A) Recycling experiments of the phosphidated Ni/SiO2 catalyst produced by liquid-phase phosphidation with TOP. Reaction conditions: phosphidated Ni/SiO2 (100 mg), guaiacol (5 mmol), n-octane (9.5 mL), 568 K, 5 MPa H2, 1 hr reaction duration. The initial three runs were performed 3 times.
(B) Carbon content of the fresh phosphidated Ni/SiO2 catalyst and catalyst after 1, 3 and 6 recycling runs, respectively.
No deactivation was observed during six recycling runs (Figure 4, left). Surprisingly, the conversion gradually increased throughout the recycling experiments. Experiments, in which the catalyst was recycled for three runs, were repeated three times, showing that the conversion increased from 65 ± 4% (in the first run) up to 81 ± 2% (in the third run). Moreover, the yield of cyclohexane increased from 45 ± 5% (in the first run) to 60 ± 5% (in the third run). At the same time, the benzene yield decreased from 4.0 ± 0.2% down to 1.0 ± 0.1% after three runs. Continued recycling of the phosphidated catalyst reveals a conversion gradually improving and plateauing at about the fifth/sixth run. At this point, a 71% yield of cyclohexane is achieved, while the yield of benzene remained at about 1%.
To compare the phosphidated catalyst against the pristine Ni/SiO2 catalyst, a two-run recycling experiment was performed on the precursor Ni/SiO2 catalyst. Not surprisingly, the results of the catalyst test revealed that the Ni/SiO2 catalyst lost most of its performance already at the second use, as indicated by the marked reduction in the conversion (from 98% to 27%, Table S1).
To address the factors responsible for the continuing improved performance throughout the recycling experiments, the carbon content of the as-prepared and spent catalyst samples was determined. As can be seen in Figure 4B, the as-prepared catalyst showed a carbon content of 7.0%. After the first run, the carbon content decreased from 7.0% to 3.7%. In the successive runs, the carbon content slightly decreased from 3.7 to 3.0%. These results suggest that the partial removal of TOP ligands should be responsible for the increase in conversion of guaiacol throughout the recycling experiments. Nonetheless, compared to the results obtained from experiments in the presence of pristine Ni/SiO2 catalyst, the continuously improved performance of the phosphidated Ni/SiO2 catalyst throughout the catalyst recycling experiments reveals that TOP and its degradation products bounded on the catalyst surface are effective to protect the catalyst against severe deactivation.
Finally, to assess whether structural changes occurred in the catalyst throughout the recycling experiments, XRD patterns of the fresh catalyst and spent catalysts were collected (Figure 5). The XRD patterns of catalysts after one, three and six HDO runs (Figures 5B–5D) suggest that the initial Ni2P phase (Figure 5A) is transformed into another phase. Unfortunately, a comparison of the XRD pattern of the catalyst after three runs with other nickel phosphides and nickel found in the XRD library retrieved no structure. The main reflection of the spent catalyst can be found at 47°. Two additional reflections can be found at 43° and 33°. Moreover, the reflections observed in the XRD patterns become sharper with increasing number of recycling runs (Figures 5B–5D), consistent with an increase in crystallite size (from 7-10 nm to 12–18 nm).
Figure 5.
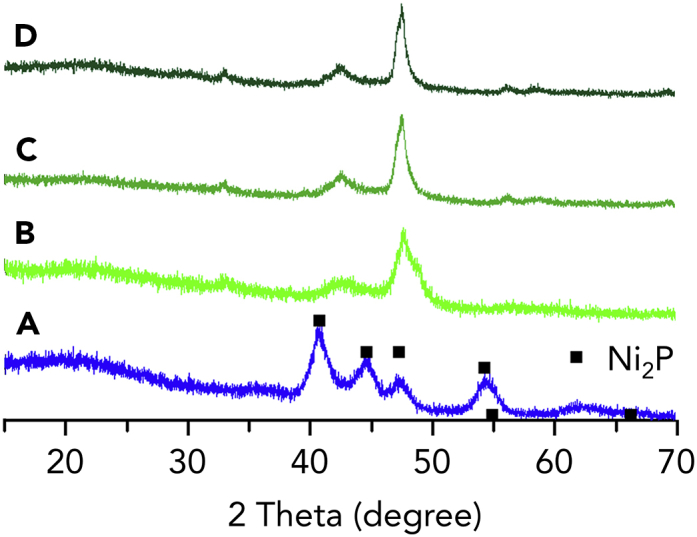
Catalyst Changes, but Performance Remarkably Improves
Comparison of the XRD patterns of the fresh phosphidated Ni/SiO2 catalyst (A) against the spent catalysts after one (B), three (C) and six (D) recycling runs.
Overall, the current results indicate that the initial Ni2P phase is not stable under the reaction conditions. However, the resulting phase shows high activity, as indicated by the catalyst tests. It seems clear that the residual TOP and its degradation products bounded on the catalyst surface remain effective in protecting the active catalytic phase against severe deactivation.
Hydrogen Balance in the Process
Currently, hydrogen is mostly obtained from the reforming of non-renewable feedstocks (estimated at 49% from natural gas, 29% from liquid hydrocarbons, either directly from naphtha or related feedstocks, or indirectly by residues conversion in refineries or as off-gases from chemical or refinery processes, 18% from coal, and 4% from electrolysis).10, 11, 55 In this manner, H2 production is associated with a large volume of CO2 emissions. In fact, CO2 equivalent emissions for hydrogen produced from natural gas corresponds to 11.88 kg CO2/H2 kg.11 Coal gasification has about twice the CO2 footprint (i.e. ca. 25-kg CO2/H2 kg) compared to centralized methane reforming.11 Therefore, a neutral hydrogen balance is critical for a neutral CO2-emission footprint in either aliphatics- or aromatics-directed HDO of lignin oils. As a “Gedankenexperiment,” we propose the utilization of hollocellulose as a feedstock for H2 production via gasification. In this manner, the (holo)cellulosic hydrogen must be sufficient for: (i) lignin depolymerization/ECCL via reductive processes (involving the passivation of reactive alkenyl/carbonylic functional groups to prevent recondensation mechanisms) or (ii) hydrogenation of acetone (formed by the H-transfer reaction) back to 2-propanol,28 and; (iii) HDO processes, as shown in Table 3. The overall hydrogen balance was calculated according to the following equation:
| Hydrogen balance = (a × x) + (b × y) - (c × z) | (Equation 1) |
where a, b and c are the hydrogen production or consumption quantities of cellulose, hemicellulose and lignin (in mol kg−1), respectively, x, y and z correspond to the cellulose, hemicellulose and lignin contents, respectively.
Table 3.
Average Molecular Formulas for Lignins and Corresponding H2 Consumption in the HDO Processes
| Type | Average Molecular Formulaa | H2 Consumption H2 (mol/lignin kg) |
|
|---|---|---|---|
| Aliphatics | Aromatics | ||
| Organosolv Lignin | C9H8.53O2.45(OCH3)1.04 | 41.0 | 25.0 |
| Pyrolysis Lignin | C8H6.3-7.3O(OCH3)0.3-0.8(OH)1-1.2 | 41.6 | 22.2 |
| Beech lignin | C9H8.83O2.37(OCH3)0.96 | 40.3 | 24.3 |
| Steam explosion lignin | C9H8.53O2.45(OCH3)1.04 | 41.0 | 25.0 |
| Dilute acid lignin | C9H8.53O2.45(OCH3)1.04 | 41.0 | 25.0 |
| Alkaline oxidation lignin | C9H8.53O2.45(OCH3)1.04 | 41.0 | 25.0 |
From Zakzeski et al.23
Scenario 1: Aliphatics-Directed HDO
For Estimation 1 (E1), as shown in Figure 6, the a and b values are 14.4 and 15.5 H2 mol/feedstock kg, respectively, which are average values calculated from the literature shown in Table S2 (cellulose, entries 1–6; hemicellulose, entries 9–10). Hydrogen consumption for the whole pathway (including CUB and HDO) was calculated (Equation 2) according to full saturation and oxygen removal of lignin, based on the organosolv lignin composition shown in Table 3 and the following hypothetical reaction:
| (Equation 2) |
Figure 6.
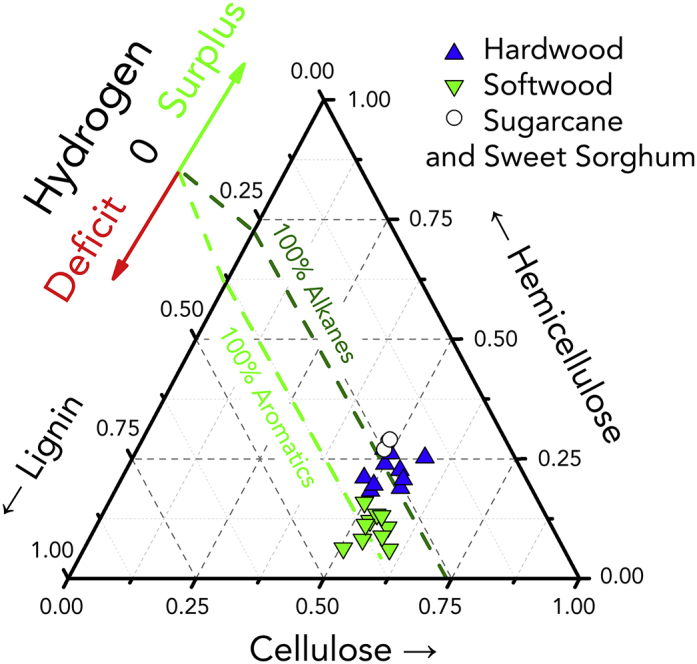
Hydrogen Balance in the Proposed Convergent Deep-Converting Lignin-First Biorefinery
Hydrogen balance for selected biomass sources (listed in Table S2): triangles (blue), hardwood (green); inverted triangles, softwood; circles, canes and sweet sorghum. The compositions of cellulose, hemicellulose and lignin are normalized on the assumption that they sum to 100 wt% of the biomass. A hydrogen balance of zero is indicated by a dark green line (for HDO leading to 100% alkane products) and a light green line (for HDO leading to 100% aromatic products). On the green side of each dash line, a surplus of the hydrogen is expected, whereas on the red side, a deficit can be found.
Calculated c values for different lignins are listed in Table 3. For calculation, the value of organosolv lignin (41.0 H2 mol/lignin kg) is taken. Therefore, the hydrogen balance is presented as:
| E1: Hydrogen balance = 14.4 x + 15.5 y – 41.0 z | (Equation 3) |
Scenario 2: Aromatic Chemicals Plus Hydrogen Production
For Estimation 2 (E2), as shown in Figure 6, the a and b values are 14.4 and 15.5 H2 mol/feedstock kg, respectively, which are average values calculated from the literature shown in Table S2 (cellulose, entries 1–6; hemicellulose, entries 9–10).
Hydrogen consumption for the whole pathway was calculated (Equation 4) according to retention of the aromatic group and removal of the oxygen content of lignin, also based on the organosolv lignin composition shown in Table 3 and the following hypothetical reaction:
| (Equation 4) |
Calculated c values for different lignins are listed in Table 3. For calculation, the value of organosolv lignin (25.0 H2 mol/lignin kg) is taken. Therefore, the hydrogen balance is presented as:
| E2: Hydrogen balance = 14.4 x + 15.5 y – 25.0 z | (Equation 5) |
As displayed in Figure 6, for either target product class (aliphatics/aromatics), the hydrogen generated from the delignified pulp is anticipated to approximately equal or, in several cases, surpass the necessary hydrogen consumption in the HDO process. At one end, surplus hydrogen may especially be found for the HDO fully directed to aromatic hydrocarbons. At the another, surplus hydrogen can be expected for the majority of hardwood feedstocks for the aliphatics-directed HDO. The comparatively low lignin content (and correspondingly higher carbohydrate content) of the hardwoods (16–30 wt%, Table S3) relative to softwood analogues (27–34 wt%, Table S3) understandably decides the hydrogen balance.
Discussion
The data indicate that the H2 generated from gasification/steam reforming will be sufficient to meet consumption to produce aliphatic hydrocarbons from hardwood lignins. However, due to the high lignin content of softwood, there will invariably exist a deficit of hydrogen. In such cases, “green” hydrogen produced by clean technologies, such as water electrolysis using renewable electricity, will be another option of H2 supply to the HDO process carried out on lignin streams. The surplus of holocellulose could well be employed as a raw material to produce platform chemicals, monetizing further the deep converting lignin-first biorefinery.
The cost of each on-site H2 supply will determine which hydrogen resource is more suitable in the case. Therefore, taking the H2 logistics into account, the deep conversion biorefinery should resemble its “old sister,” the deep conversion oil refinery. Notably, such advanced oil refineries are self-sufficient in H2. In this instance, the catalytic reforming unit usually covers the demand for H2 from the other converting units (hydrodesulfurization, hydrodemetalation, hydrodenitrogenation, hydrocracking, to mention a few), generating aromatics for improved octane-number gasoline and chemicals.56 In the case of the deep converting lignin-first biorefining, the current results demonstrate that, with the right catalyst in place, the aromatics can be obtained directly from the HDO process. In the best-case scenario, this feature will alleviate the H2 demand for the process. Therefore, the aromatic-directed HDO could hold the key for the elimination of a reforming unit for a neutral hydrogen balance in the deep converting lignin-first biorefinery. Overall, the suggested process appears feasible, whereby at least the majority of required H2 may be produced by holocellulose gasification, for all selected feedstocks, concerning either fuel or aromatics production.
Compared to the bioethanol produced from a cellulose-centered biorefinery, the drop-in fuel fractions obtained by the deep converting lignin-first biorefinery seem to be more advantageous regarding both carbon and weight yields. In fact, the carbon and weight yields of conventional bioethanol production (considering the fermentation of C6-sugars only) are both usually at around 9%. This value compares unfavorably with the potential carbon and weight yields of saturated hydrocarbons obtained from lignins of hardwood feedstocks, estimated up to 32% and 20%, and softwood feedstocks as 36% and 23%, respectively (Table S3). Furthermore, considering the process thermal efficiency (PTE), that is, the energy content of the product fuel relative to the energy content of the biomass feedstock, cellulosic ethanol presents a limited PTE value of 11% (Table S3),17 the PTE values are estimated to be as a high as 45% for the hardwood Boxelder, 51% for the softwood Alligator Juniper, and 30–33% for the canes and sweet sorghum. From our experimental results, the best weight yield of the hydrocarbons was ca. 40–45%, relative to the total lignin content in the lignocellulose sources. This value range translates into an estimated PTE of approximately 20–25%, more than twice as high as that for bioethanol. Therefore, it is plausible to expect that the deep converting lignin-first biorefinery delivers high-density fuels from lignin with considerable energetic benefits over cellulosic bioethanol fuel.
The traditional approach to biomass liquefaction has involved initial gasification, to afford syngas (CO + H2), which is subsequently employed in the FT synthesis of hydrocarbons. By contrast, this contribution demonstrates the production of valuable branched C6-C10, C14-C20 and (to a lesser extent) C21+ hydrocarbons from lignin, without the requirement to rebuild high-molecular-weight hydrocarbons from C1 units. The gasification-FT approach yields hydrocarbons with an Anderson–Schulz–Flory distribution of Cn-alkanes. At a high probability of chain growth, the FT-product streams present a high content of waxes/paraffins. The dewaxing of the FT-product streams requires (expensive) advanced hydrocracking and hydroisomerisation units, to obtain high-performance gasoline, kerosene and diesel fractions. Such additional units dramatically increase the complexity index of such refineries, i.e. the CAPEX.
The features distinguishing our new approach from the traditional gasification-FT pathway are summarized in Figure 7.55
Figure 7.

Lignin Fuels versus Fischer-Tropsch Fuels
Comparison between products distributions obtained from the deep converting lignin-first biorefinery (green) and the traditional biomass gasification–Fischer-Tropsch synthesis route (gray).
Conclusions
In summary, we have proposed a strategy for refinery-like lignin conversion into aliphatic or aromatic hydrocarbons from lignocellulosic feedstocks. This deep converting biorefinery benefits from three primary advantages: (i) the integrated convergent procedure is self-sufficient in terms of H2, whereby for the majority of biomass feedstock species no external input of CO2-intensive hydrogen gas is required; (ii) the hydrocarbon products are predominantly oxygen-free C6-C10 and C14-C20 species, easily separable via distillation, with direct applicability as high-quality gasoline/diesel fuels or bio-derived platform chemicals simlar to those obtained from petrochemistry, and; (iii) the aliphatic/aromatic character of the product mixture can be wholly reversed via trivial adjustment of reaction parameters (foremost pressure of hydrogen gas and temperature), whilst maintaining an identical reaction setup and catalyst/solvent system, offering a high degree of flexibility with regard to fluctuating market values of fuels, hydrogen and aromatic commodity chemicals. Furthermore, the proposed deep converting lignin-first biorefinery seems to be conceptually scalable and readily integrated into existing refineries, whereby analogous gasification and catalytic processes (e.g. hydrodesulfurization, HDS) are already performed on an industrial scale.
Experimental Procedures
Materials
Ni on silica catalyst (64% loading, Strem chemicals), tri-(n-octyl)phosphine (Strem chemicals, 97%), and di-n-octyl ether (Sigma, 99%) were purchased and used as received. Wood feedstocks (5 mm chips for poplar and spruce) were purchased (J. Rettenmaier & Söhne) and were employed in this investigation.
Catalyst Preparation
In a typical experiment, the standard Schlenk line techniques were employed, and the phosphidation of the Ni on silica was carried out under argon. Ni/SiO2 (200 mg) was dispersed in di-n-octyl ether (3 mL) before tri-(n-octyl)phosphine (6 mL) was added (equivalent to a TOP:Ni ratio of 6.1), and then the dispersion was stirred for 30 min at room temperature. The sample was then increased to 300°C for 6 hr. 2-propanol (3 × 20 mL) was used to wash the catalyst after cooling down, and then the catalyst was separated by centrifugation. After dried at 80°C under vacuum, the catalyst was obtained as a black powder.
Lignocellulose Deconstruction via Lignin-First Approach
The procedure was previously reported by us elsewhere.28 Typically, the biomass feedstocks (300 g), Raney Ni (200 g wet) and solvent (3 L; 2-propanol:water 7:3 v/v) were added into a 5-L autoclave, then heated to 180°C at 3°C/min under mechanical stirring. The reaction was performed at 180°C for 3 hr under autogenous pressure. After the autoclave cooled down to room temperature, the liquor was placed into a glass bottle. Using an overhead mechanical stirring, the catalyst was recovered by magnetic separation by using magnets placed on the external surface of the glass bottle. Note that Raney Ni is a stable recyclable catalyst, as described by us in ref.28 The spent Raney Ni was stored in 2-propanol for further use. The liquid was separated by filtration from the rest solid (cellulosic pulp). By using rotary evaporation at 60°C under vacuum, the lignin oil (brownish viscous oil) was then obtained.
Hydrodeoxygenation of Lignin Oil
For hydrodeoxygenation (HDO) reactions of the lignin-derived bio-oil, catalyst (100 mg), bio-oil (200 mg) and n-hexane (5 mL) were measured into the reactor vessel. Catalytic conversion was performed in a home-made 45 mL stainless-steel autoclave, filled with a 0.5(poplar)/1(spruce) or 5 MPa pressure of H2 (chemical-/fuel-directed pathways, respectively) at room temperature before the reaction. Reactions were conducted at 623 or 573 K (chemical-/fuel-directed pathways, respectively) for 20 hr. Following reaction, the reactor was submerged in an ice-bath. The resultant liquid was treated with ethyl acetate (5 mL) to obtain a homogeneous solution. MgSO4 was added to adsorb water in the product, and a membrane filter (0.45 μm) was employed to remove solid particulate material before product analysis using GC×GC−MS/FID techniques. For the recycling experiments, the spent catalyst was separated from the solvent by centrifugation, was washed with n-hexane, and was separated again by centrifugation. The resultant recycled catalyst was dispersed in ethyl acetate to ensure total transfer into the autoclave – after transfer, the ethyl acetate was evaporated.
GC×GC–MS/FID Analysis
Samples were analyzed using 2D GC×GC−MS(FID) (first column: ZB-1HT Inferno 30 m, 0.25 mm ID, df 0.25 μm; second column: BPX50, 1 m, 0.15 mm ID, df 0.15 μm) in a GC-MS 2010 Plus (Shimadzu) chromatogram equipped with a ZX1 thermal modulation system (Zoex). The injector temperature was 300°C. The temperature program began at 40°C for 5 min, and subsequently was increased at a rate of 5.2°C min−1 until reaching a temperature of 300°C; the program culminated with an isothermal step at 300°C for 5 min. The modulation applied for the comprehensive GC×GC analysis was a hot jet pulse (400 ms) every 9 s/6 s (before and after HDO). The 2D chromatograms were processed with GC Image software (Zoex). The products were identified according to the matching of the MS spectra with MS libraries NIST 08, NIST 08s, and Wiley 9. Semi-quantification of the products was performed using integration of GC peaks using Effective Carbon Number concept (ECN).53 Since measured FID respond factor for various hydrocarbons varying from 0.90 to 1.07,53, 57 it is reasonable to assume the wide hydrocarbons we obtained has the respond factor of 1.
Acknowledgments
R.R. acknowledges the financial support provided by the ERC Consolidator Grant (LIGNINFIRST, project number 725762). This work was also conducted within the framework of the CASCATBEL project funded by the European Commission (grant agreement no. 604307).
Author Contributions
Conceptualization, R.R. and Z.C.; Methodology, Z.C.; Investigation, Z.C., M.D., I.B.D.C.; Writing – Original Draft, Z.C., M.D., M.T.C., R.R.; Writing – Review & Editing, R.R.; Funding Acquisition, R.R.; Resources, R.R.; Supervision, R.R.
Declaration of Interests
The authors declare no conflicts of interests.
Published: April 9, 2018
Footnotes
Supplemental Information includes six figures and three tables and can be found with this article online at https://doi.org/10.1016/j.joule.2018.03.012.
Supplemental Information
References
- 1.Searchinger T., Heimlich R., Houghton R.A., Dong F., Elobeid A., Fabiosa J., Tokgoz S., Hayes D., Yu T.H. Use of U.S. croplands for biofuels increases greenhouse gases through emissions from land-use change. Science. 2008;319:1238–1240. doi: 10.1126/science.1151861. [DOI] [PubMed] [Google Scholar]
- 2.Wang L., Sharifzadeh M., Templer R., Murphy R.J. Bioethanol production from various waste papers: economic feasibility and sensitivity analysis. Appl. Energy. 2013;111:1172–1182. [Google Scholar]
- 3.Ragauskas A.J., Beckham G.T., Biddy M.J., Chandra R., Chen F., Davis M.F., Davison B.H., Dixon R.A., Gilna P., Keller M. Lignin valorization: improving lignin processing in the biorefinery. Science. 2014;344:1246843. doi: 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- 4.Balat M., Balat H., Öz C. Progress in bioethanol processing. Progr. Energy Combust. Sci. 2008;34:551–573. [Google Scholar]
- 5.Sticklen M.B. Plant genetic engineering for biofuel production: towards affordable cellulosic ethanol. Nat. Rev. Genet. 2008;9:433–443. doi: 10.1038/nrg2336. [DOI] [PubMed] [Google Scholar]
- 6.Ahrenfeldt J., Thomsen T.P., Henriksen U., Clausen L.R. Biomass gasification cogeneration – a review of state of the art technology and near future perspectives. Appl. Therm. Eng. 2013;50:1407–1417. [Google Scholar]
- 7.Tijmensen M.J.A., Faaij A.P.C., Hamelinck C.N., van Hardeveld M.R.M. Exploration of the possibilities for production of Fischer Tropsch liquids and power via biomass gasification. Biomass Bioenergy. 2002;23:129–152. [Google Scholar]
- 8.Corma A., Iborra S., Velty A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007;107:2411–2502. doi: 10.1021/cr050989d. [DOI] [PubMed] [Google Scholar]
- 9.Davda R.R., Shabaker J.W., Huber G.W., Cortright R.D., Dumesic J.A. A review of catalytic issues and process conditions for renewable hydrogen and alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported metal catalysts. Appl. Catal. B Environ. 2005;56:171–186. [Google Scholar]
- 10.Suresh B., Gubler R., Yamaguchi Y., He X. IHS Markit; 2013. Hydrogen - Chemical Economics Handbook (CEH)https://ihsmarkit.com/products/hydrogen-chemical-economics-handbook.html [Google Scholar]
- 11.Schüth F. Hydrogen: economics and its role in biorefining. In: Rinaldi R., editor. Catalytic Hydrogenation for Biomass Valorization. The Royal Society of Chemistry; 2015. pp. 1–21. [Google Scholar]
- 12.Ruddy D.A., Schaidle J.A., Ferrell Iii J.R., Wang J., Moens L., Hensley J.E. Recent advances in heterogeneous catalysts for bio-oil upgrading via "ex situ catalytic fast pyrolysis": catalyst development through the study of model compounds. Green. Chem. 2014;16:454–490. [Google Scholar]
- 13.Vispute T.P., Zhang H., Sanna A., Xiao R., Huber G.W. Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils. Science. 2010;330:1222–1227. doi: 10.1126/science.1194218. [DOI] [PubMed] [Google Scholar]
- 14.Marker, T.L., Felix, L.G., and Linck, M.B.. (2010). Hydropyrolysis of biomass for producing high quality liquid fuels, US12685352.
- 15.Marker T.L., Felix L.G., Linck M.B., Roberts M.J. Integrated hydropyrolysis and hydroconversion (IH2) for the direct production of gasoline and diesel fuels or blending components from biomass, part 1: proof of principle testing. Environ. Prog. Sustain. Energy. 2012;31:191–199. [Google Scholar]
- 16.Rinaldi R., Jastrzebski R., Clough M.T., Ralph J., Kennema M., Bruijnincx P.C., Weckhuysen B.M. Paving the way for lignin valorisation: recent advances in bioengineering, biorefining and catalysis. Angew. Chem. Int. Ed. 2016;55:8164–8215. doi: 10.1002/anie.201510351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinaldi R., Schüth F. Design of solid catalysts for the conversion of biomass. Energy Environ. Sci. 2009;2:610–626. [Google Scholar]
- 18.Schutyser W., Renders T., Van den Bosch S., Koelewijn S.-F., Beckham G., Sels B. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018;47:852–908. doi: 10.1039/c7cs00566k. [DOI] [PubMed] [Google Scholar]
- 19.Demirel Y. Energy. Springer; 2012. Energy and energy types; pp. 27–70. [Google Scholar]
- 20.Holladay J.E., White J.F., Bozell J.J., Johnson D. Pacific Northwest National Laboratory (PNNL); 2007. Top Value-added Chemicals from Biomass-Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin. [Google Scholar]
- 21.Galkin M.V., Samec J.S.M. Lignin valorization through catalytic lignocellulose fractionation: a fundamental platform for the future biorefinery. ChemSusChem. 2016;9:1544–1558. doi: 10.1002/cssc.201600237. [DOI] [PubMed] [Google Scholar]
- 22.Huber G.W., Cortright R.D., Dumesic J.A. Renewable alkanes by aqueous-phase reforming of biomass-derived oxygenates. Angew. Chem. Int. Ed. 2004;43:1549–1551. doi: 10.1002/anie.200353050. [DOI] [PubMed] [Google Scholar]
- 23.Zakzeski J., Bruijinincx P.C., Jongerius A.L., Weckhuysen B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010;110:3552–3599. doi: 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- 24.Johnson D.K., Chornet E., Zmierczak W., Shabtai J. Conversion of lignin into a hydrocarbon product for blending with gasoline. ACS Div. Fuel Chem. Preprints. 2002;47:380–381. [Google Scholar]
- 25.Kaiser M.J. A review of refinery complexity applications. Petrol. Sci. 2017;14:167–194. [Google Scholar]
- 26.Richter H., Howard J.B. Formation of polycyclic aromatic hydrocarbons and their growth to soot - a review of chemical reaction pathways. Progr. Energy Combust. Sci. 2000;26:565–608. [Google Scholar]
- 27.Renders T., Van den Bosch S., Koelewijn S.F., Schutyser W., Sels B. Lignin-first biomass fractionation: the advent of active stabilisation strategies. Energy Environ. Sci. 2017;10:1551–1557. [Google Scholar]
- 28.Ferrini P., Rinaldi R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. Int. Ed. 2014;53:8634–8639. doi: 10.1002/anie.201403747. [DOI] [PubMed] [Google Scholar]
- 29.Ferrini P., Rezende C.A., Rinaldi R. Catalytic upstream biorefining through hydrogen transfer reactions: understanding the process from the pulp perspective. ChemSusChem. 2016;9:3171–3180. doi: 10.1002/cssc.201601121. [DOI] [PubMed] [Google Scholar]
- 30.Calvaruso G., Burak J.A., Clough M.T., Kennema M., Meemken F., Rinaldi R. On the reactivity of dihydro-p-coumaryl alcohol towards reductive processes catalyzed by raney nickel. ChemCatChem. 2017;9:2627–2632. [Google Scholar]
- 31.Chesi C., de Castro I.B.D., Clough M.T., Ferrini P., Rinaldi R. The influence of hemicellulose sugars on product distribution of early-stage conversion of lignin oligomers catalysed by raney nickel. ChemCatChem. 2016;8:2079–2088. [Google Scholar]
- 32.Anderson E.M., Katahira R., Reed M., Resch M.G., Karp E.M., Beckham G.T., Román-Leshkov Y. Reductive catalytic fractionation of corn stover lignin. ACS Sustain. Chem. Eng. 2016;4:6940–6950. [Google Scholar]
- 33.Galkin M.V., Samec J.S. Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis. ChemSusChem. 2014;7:2154–2158. doi: 10.1002/cssc.201402017. [DOI] [PubMed] [Google Scholar]
- 34.Klein I., Saha B., Abu-Omar M.M. Lignin depolymerization over Ni/C catalyst in methanol, a continuation: effect of substrate and catalyst loading. Catal. Sci. Technol. 2015;5:3242–3245. [Google Scholar]
- 35.Van den Bosch S., Schutyser W., Vanholme R., Driessen T., Koelewijn S.F., Renders T., De Meester B., Huijgen W., Dehaen W., Courtin C. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015;8:1748–1763. [Google Scholar]
- 36.Li C., Zhao X., Wang A., Huber G.W., Zhang T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015;115:11559–11624. doi: 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- 37.Li S.H., Liu S., Colmenares J.C., Xu Y.J. A sustainable approach for lignin valorization by heterogeneous photocatalysis. Green. Chem. 2016;18:594–607. [Google Scholar]
- 38.Schutyser W., Renders T., Van den Bossche G., Van den Bosch S., Koelewijn S.F., Ennaert T., Sels B.F. Catalysis in lignocellulosic biorefineries: the case of lignin conversion. In: Van de Voorde M., Sels B.F., editors. Nanotechnology in Catalysis. Wiley-VCH; 2017. pp. 537–584. [Google Scholar]
- 39.Asadullah M., Ito S.I., Kunimori K., Yamada M., Tomishige K. Biomass gasification to hydrogen and syngas at low temperature: novel catalytic system using fluidized-bed reactor. J. Catal. 2002;208:255–259. [Google Scholar]
- 40.Corella J., Aznar M.P., Caballero M.A., Molina G., Toledo J.M. 140 g H2/kg biomass d.a.f. by a CO-shift reactor downstream from a FB biomass gasifier and a catalytic steam reformer. Int. J. Hydrogen Energy. 2008;33:1820–1826. [Google Scholar]
- 41.Couhert C., Commandre J.M., Salvador S. Is it possible to predict gas yields of any biomass after rapid pyrolysis at high temperature from its composition in cellulose, hemicellulose and lignin? Fuel. 2009;88:408–417. [Google Scholar]
- 42.Inaba M., Murata K., Saito M., Takahara I. Hydrogen production by gasification of cellulose over Ni catalysts supported on zeolites. Energy Fuels. 2006;20:432–438. [Google Scholar]
- 43.Kimura T., Miyazawa T., Nishikawa J., Kado S., Okumura K., Miyao T., Naito S., Kunimori K., Tomishige K. Development of Ni catalysts for tar removal by steam gasification of biomass. Appl. Catal. B Environ. 2006;68:160–170. [Google Scholar]
- 44.Levin D.B., Islam R., Cicek N., Sparling R. Hydrogen production by Clostridium thermocellum 27405 from cellulosic biomass substrates. Int. J. Hydrogen Energy. 2006;31:1496–1503. [Google Scholar]
- 45.Li S., Lu Y., Guo L., Zhang X. Hydrogen production by biomass gasification in supercritical water with bimetallic Ni-M/γAl2O3 catalysts (M = Cu, Co and Sn) Int. J. Hydrogen Energy. 2011;36:14391–14400. [Google Scholar]
- 46.Taylor A.D., DiLeo G.J., Sun K. Hydrogen production and performance of nickel based catalysts synthesized using supercritical fluids for the gasification of biomass. Appl. Catal. B Environ. 2009;93:126–133. [Google Scholar]
- 47.Wu C., Wang L., Williams P.T., Shi J., Huang J. Hydrogen production from biomass gasification with Ni/MCM-41 catalysts: influence of Ni content. Appl. Catal. B Environ. 2011;108–109:6–13. [Google Scholar]
- 48.Wu C., Wang Z., Huang J., Williams P.T. Pyrolysis/gasification of cellulose, hemicellulose and lignin for hydrogen production in the presence of various nickel-based catalysts. Fuel. 2013;106:697–706. [Google Scholar]
- 49.Zhao M., Florin N.H., Harris A.T. The influence of supported Ni catalysts on the product gas distribution and H2 yield during cellulose pyrolysis. Appl. Catal. B Environ. 2009;92:185–193. [Google Scholar]
- 50.Kline L.M., Hayes D.G., Womac A.R., Labbe N. Simplified determination of lignin content in hard and soft woods via UV-spectrophotometric analysis of biomass dissolved in ionic liquids. BioResources. 2010;5:1366–1383. [Google Scholar]
- 51.Henkes A.E., Vasquez Y., Schaak R.E. Converting metals into phosphides: a general strategy for the synthesis of metal phosphide nanocrystals. J. Am. Chem. Soc. 2007;129:1896–1897. doi: 10.1021/ja068502l. [DOI] [PubMed] [Google Scholar]
- 52.Henkes A.E., Schaak R.E. Trioctylphosphine: a general phosphorus source for the low-temperature conversion of metals into metal phosphides. Chem. Mater. 2007;19:4234–4242. [Google Scholar]
- 53.Scanlon J.T., Willis D.E. Calculation of flame ionization detector relative response factors using the effective carbon number concept. J. Chromatogr. Sci. 1985;23:333–340. [Google Scholar]
- 54.Ding L.N., Wang A.Q., Zheng M.Y., Zhang T. Selective transformation of cellulose into sorbitol by using a bifunctional nickel phosphide catalyst. ChemSusChem. 2010;3:818–821. doi: 10.1002/cssc.201000092. [DOI] [PubMed] [Google Scholar]
- 55.Spath P.L., Mann M.K. National Renewable Energy Lab; 2000. Life Cycle Assessment of Hydrogen Production via Natural Gas Steam Reforming. [Google Scholar]
- 56.Marcilly C. Vol. 2. Technip Ophrys Editions; 2006. (Acido-basic Catalysis: Application to Refining and Petrochemistry). [Google Scholar]
- 57.Slemr J., Slemr F., D'Souza H., Partridge R. Study of the relative response factors of various gas chromatograph–flame ionisation detector systems for measurement of C2–C9 hydrocarbons in air. J. Chromatogr. A. 2004;1061:75–84. doi: 10.1016/j.chroma.2004.10.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.