

Abstract
Most plants show remarkable developmental plasticity in the generation of diverse types of new organs upon external stimuli, allowing them to adapt to their environment. Haustorial formation in parasitic plants is an example of such developmental reprogramming, but its molecular mechanism is largely unknown. In this study, we performed field-omics using transcriptomics and metabolomics to profile the molecular switch occurring in haustorial formation of the root parasitic plant, Thesium chinense, collected from its natural habitat. RNA-sequencing with de novo assembly revealed that the transcripts of very long chain fatty acid (VLCFA) biosynthesis genes, auxin biosynthesis/signaling-related genes and lateral root developmental genes are highly abundant in the haustoria. Gene co-expression network analysis identified a network module linking VLCFAs and the auxin-responsive lateral root development pathway. GC-TOF-MS analysis consistently revealed a unique metabolome profile with many types of fatty acids in the T. chinense root system, including the accumulation of a 25-carbon long chain saturated fatty acid in the haustoria. Our field-omics data provide evidence supporting the hypothesis that the molecular developmental machinery used for lateral root formation in non-parasitic plants has been co-opted into the developmental reprogramming of haustorial formation in the linage of parasitic plants.
Keywords: Developmental reprogramming, Haustorium, Metabolome, Parasitic plants, Thesium chinense, Transcriptome
Introduction
Plants and animals, separated by about 1.5 billion years of evolutionary history, have evolved their multicellular organization independently. Animal development, determined during embryogenesis, is largely buffered against environmental perturbation. Plants, however, can adapt to their environment by reprogramming their development post-embryonically. Therefore, the developmental reprogramming in plants should be precisely controlled by different layers of regulation in biological processes, responding to their natural environment. Thanks to the recent technological advances, field-omics provides a powerful strategy for investigating how plants actually employ developmental reprogramming for adaptation to their natural environment.
One example of stimulus-responsive developmental reprogramming is the formation of haustoria in parasitic plants. Approximately 4,500 species of parasitic plants have evolved independently at least 11 times (Barkman et al. 2007). In every case, each parasitic plant develops a unique multicellular organ called the haustorium (Kuijt 1969, Yoshida et al. 2016). The haustorium is formed from mature differentiated tissues, responding to the detection of the host-derived chemicals. In the root parasite family Orobanchaceae (Chang and Lynn 1986), these chemicals include 2,6-dimethoxy-p-benzoquinone and its structural analogs. The haustorium shows a completely different morphology from that of the original root tissues and provides specific parasitic functions, such as host attachment, invasion, vasculature connection and material transfer between hosts and parasites (Yoshida et al. 2016). A number of studies focusing on the molecular mechanisms underlying haustorial formation have characterized the gene expression and accumulation of metabolites (Ichihashi et al. 2015). No studies, however, have addressed both transcriptomic and metabolomic profiling of the same samples, thus molecular switches in haustorial formation are not fully understood at the genome-wide level.
The genus Thesium is a member of the family Santalaceae, which consists of approximately 1,000 species (Stevens 2001 onwards). All Santalaceae species are parasitic plants, including both stem and root parasites. This family contains economically important members, such as the sandalwood tree because its aromatic heartwood contains sandal oil, used in perfumes and medicine (Baldovini et al. 2011). Thesium chinense is a facultative root hemi-parasite and is commonly distributed in the grassland habitats of eastern Asia (Fig. 1A). Like sandalwood, T. chinense is also used in medicine for its anti-inflammatory and analgesic properties, as it is rich in metabolites such as flavonoids (Parveen et al. 2007). In addition, the ecology of the genus Thesium has been well characterized (Suetsugu et al. 2008, Dostálek and Münzbergová 2010, Suetsugu 2015). Thesium chinense can parasitize a broad range of plant species with post-attachment host selectivity (Suetsugu et al. 2008). and shows a unique strategy of seed dispersal by ants, known as myrmecochory (Suetsugu 2015). This species lends itself to a molecular investigation of the developmental reprogramming of haustorial formation in the wild, due to its accessibility and the amount of information available concerning its ecology. Here, we characterize the transcriptome and metabolome of T. chinense haustoria collected from their native habitat. Our field-omics-based approach enabled the detection of detailed changes at the transcript and metabolite level in the developmental reprogramming of haustorial formation. These data allowed us to identify the recruitment of an auxin-responsive lateral root development pathway, tightly associated with the accumulation of very long chain fatty acids (VLCFAs), into the T. chinense haustorial formation. This study provides novel insights into our understanding of the molecular basis of developmental reprogramming in natura.
Fig. 1.

Haustorial formation in T. chinense. (A) T. chinense in their natural habitat (near the Kizu River, Kyoto, Japan). The blue dashed line traces a T. chinense individual; blue arrowheads indicate several T. chinense individuals. (B) Gross morphology of T. chinense haustorium and host root. (C–E) Safranin O-stained lateral haustoria at various stages: host invasion (C), xylem formation (D) and host connection (E). Arrowheads indicate haustoria. (F–H) Longitudinal sections of T. chinense haustorium and host root: whole haustorium (F), basal part of the haustorium, where de novo-formed xylem connects to the parasite root xylem (G), apical part of the haustorium, where intrusive cells invaded the host xylem (H). P, parasite; H, host; Xb, xylem bridge; Px, parasite xylem; Hx, host xylem. Scale bars = 1 mm for (A–E) and 200 μm for (F–H).
Results
Haustorial formation in T. chinense
To observe the structure of T. chinense haustoria growing in the wild, we collected various stages of haustoria from the side of the Kizu River, Kyoto Prefecture, Japan for morphological and histological studies (Fig. 1B–H). Haustoria, initiated from fully mature roots as dome-shaped structures with small cells, invade the host root tissues with their tips (Fig. 1C). Xylem cells differentiate within the haustoria while it is growing. This differentiation occurs from the vascular region in the parasite root towards the direction of the host’s vascular region (Fig. 1D) and subsequently from the interface between the parasite and host towards the direction of the parasite vascular region. Eventually, the bidirectionally differentiated xylem cells meet in the middle of the haustorium to establish the interspecies xylem continuity, known as a xylem bridge (Fig. 1E). The tip of the haustoria, in which the frontline cells are known as intrusive cells, reaches the host xylem, and the physical connection between parasite and host xylem cells is observed (Fig. 1F–H). Lignin deposition at the parasite–host interface and the accumulation of starch-like compounds in haustorial cells are also detected (Fig. 1F–H). These developmental and histological characteristics are like that of the Orobanchaceae (Joel and Losner-Goshen 1994, Ishida et al. 2016), which is an evolutionarily independent lineage of T. chinense, suggesting that morphological and developmental patterns of haustorium might be highly conserved across parasitic plants. Thus, the haustorial formation of T. chinense is achieved through dynamic cellular reprogramming and the reactivation of cell proliferation and differentiation from mature root tissues.
Transcriptional regulation of haustorial formation
To understand the genome-wide transcriptional regulation behind the developmental reprogramming of haustorial formation, we prepared RNA-sequencing (RNA-seq) libraries from T. chinense haustoria and roots, as well as from the host roots (Artemisia capillaris, Eragrostis curvula and Lespedeza juncea), with all plants collected from their native habitat (Fig. 2A). A total of 256,562,563 read pairs (100 bp) was obtained after sequencing libraries on the Illumina HiSeq4000 platform. After filtering for reads with a Phred score >30 and a minimal length of 90 bases, and removal of adaptor sequences, a total of 194,933,609 read pairs was obtained. To minimize host tissue contamination, in addition to the careful collection of tissue samples and rinsing using 70% ethanol, removal of host transcripts was performed during bioinformatics analysis (Supplementary Fig. S1). First, read pairs from host roots were used for de novo assembly by the CLC Genomics Workbench to generate 1,287,640 contigs. The reads from T. chinense haustoria and roots aligning to the host contigs, which correspond to host transcript contamination (14.6% of reads), were then discarded. Using these host transcript-free reads, we performed de novo transcriptome assembly, followed by refinement (CD-HIT-EST, TransDecoder and DeconSeq), to generate the final contigs of the T. chinense root system. The final contigs have 39,756 non-redundant contigs with an N50 length of 945 bp, an average size of 713 bp and a total length of 28,354,914 bp (Table 1).
Fig. 2.
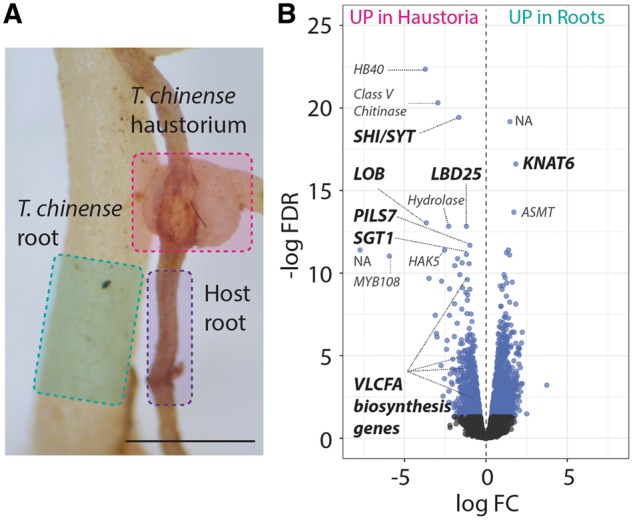
Transcriptomic change in T. chinense haustorial formation. (A) Tissues from T. chinense haustorium and root regions were analyzed and are highlighted by pink and green, respectively. To remove host contamination using bioinformatics, the host root highlighted in purple was also analyzed. The detailed bioinformatics pipeline is shown in Supplementary Fig. S1. (B) Volcano plot showing the differentially expressed transcripts between T. chinense haustoria and roots. The logarithms of the fold change (log FC) of individual genes are plotted against the negative logarithm of their FDR. Negative log FC values represent up-regulation in haustoria, and positive values represent up-regulation in roots. Blue dots represent differentially expressed genes between haustoria and roots with an FDR <0.05. The most significantly up-regulated genes, as well as VLCFA biosynthesis genes, are shown. UP in haustoria, up-regulated in haustoria; UP in roots, up-regulated in roots.
Table 1.
Summary of de novo assembly of T. chinense transcriptome
| Parameter | Number |
|---|---|
| Number of contigs | 39,756 |
| Total size of contigs | 28,354,914 |
| N50 contig length (bp) | 945 |
| Average length | 713 |
| Median length | 438 |
| Minimum length | 297 |
| Maximum length | 15,846 |
To validate the absence of host tissue contamination in the final contigs, we investigated the phylogenetic relationship of orthologs of the expressed nuclear gene encoding the photoreceptor phytochrome A (PHYA). BLAST searches using the Arabidopsis thaliana PHYA coding sequence identified one and three orthologs from the T. chinense and host contigs, respectively. These orthologs were added into a phylogenetic tree of core angiosperms (Supplementary Fig. S2; Supplementary Dataset S1). The resultant phylogenetic tree indicates that the T. chinense PHYA and host PHYA genes belong to different clades, indicating clear separation of host and parasite sequences using our informatics pipeline. Moreover, one and two host PHYA genes are divided into the distinct clades of Asterids (Artemisia capillaris) and Fabaceae (Lespedeza juncea), respectively. These are consistent with the host species identified in the wild (see the Materials and Methods) (Suetsugu et al. 2008). Our informatics pipeline thus allows the successful removal of host contamination and assembly of a high-quality T. chinense transcriptome.
For functional annotation of the final T. chinense contigs, BLAST searches against the National Center for Biotechnology Information (NCBI) non-redundant database resulted in the annotation of 30,012 transcripts (75.5% of all contigs, Supplementary Dataset S2). Also, 20,003 transcripts (50.3% of contigs) have at least one Gene Ontology (GO) term assigned (Supplementary Dataset S3). Approximately 30% of transcripts have top hits to sequences from Vitis vinifera, Theobroma cacao and Nelumbo nucifera, which belong to the Rosids or to the basal eudicots (Supplementary Fig. S3). Since T. chinense, a member of the order Santalales, is a member of the basal clade of superasterids (between the Rosids and the basal eudicots), the resulting annotation concurs with the phylogenetic relationship. This is also supported by the fact that the transcriptome of Santalum album, another member of the Santalales, also shows 54% homology hit with genes in V. vinifera (Zhang et al. 2015).
To compare the transcript abundance between the T. chinense haustoria and roots, filtered read pairs were mapped to the assembled T. chinense transcripts (45.4–52.5% mapped) followed by normalizing count reads across samples using the trimmed mean of M-values method (Supplementary Dataset S4). Differential expression analysis using a false discovery rate (FDR) <0.05 shows that many genes (2,265 transcripts corresponding to 9.1% of filtered expressed genes) are differentially expressed between haustoria and roots. A total of 801 and 1,464 transcripts display higher abundance in haustoria or roots, respectively (Fig. 2B;Supplementary Dataset S5). The reliability of the transcriptome data was validated by quantifying transcript abundances of selected differentially expressed contigs (Tc_contig_20619 and Tc_contig_32744) by reverse transcription–PCR (RT–PCR) (Supplementary Fig. S4). Consistent with the previous gene expression study of T. chinense grown in a pot (Zhang et al. 2016), genes encoding pectin methylesterase (Tc_contig_36353) and phenylalanine ammonia-lyase (Tc_contig_12036), respectively, are up-regulated in haustoria. GO terms related to oxidation, reduction and proteolysis are significantly enriched in the up-regulated genes at haustoria (FDR <0.05, Supplementary Dataset S6), which is similar to a previous study of the S. album transcriptome (Zhang et al. 2015). In addition, GO terms related to developmental (transcription factor and DNA replication), metabolic (terpene synthase activity, fatty acid metabolic process and flavonoid biosynthetic process) and functional (transporters for various types of carbohydrate as well as sugar alcohol) aspects of haustoria are also enriched (FDR <0.05, Supplementary Dataset S6). On the other hand, GO terms related to secondary cell wall biogenesis and auxin signaling are significantly enriched in up-regulated genes of roots (FDR <0.05, Supplementary Dataset S6). Therefore, our transcriptome data can detect the diverse transcriptional profiles differentiating developmental reprogramming from root cells to haustorial cells even when collected from the field, and this reflects the substantial morphological changes in T. chinense haustoria.
Among the top 10 most significantly up-regulated genes in haustoria, three auxin-related genes [Tc_contig_25082, SHORT INTERNODES/STYLISH (SHI/STY); Tc_contig_8445, PIN-LIKES7 (PILS7); Tc_contig_3701, SUPPRESSOR OF G2 ALLELE SKP1 (SGT1)] and two LATERAL ORGAN BOUNDARIES DOMAIN (LBD) genes (Tc_contig_38475 and Tc_contig_32513) are involved (Fig. 2B;Supplementary Dataset S5). The SHI/STY gene positively regulates the gene expression of the auxin biosynthesis gene YUCCA (YUC) in A. thaliana (Sohlberg et al. 2006, Eklund et al. 2010). PILS is a subfamily of auxin efflux carrier genes and regulates intracellular auxin homeostasis (Barbez et al. 2012). SGT1 functions with heat shock protein 90 (HSP90) to integrate temperature and auxin signaling by regulating the SCFTIR1-mediated auxin response (Gray et al. 2003, Wang et al. 2016). Given that the generation of auxin response maxima at the haustorium apex is the key for haustorial formation in Phtheirospermum japonicum and Triphysaria versicolor (Tomilov et al. 2005, Ishida et al. 2016), auxin response could be one of the shared mechanisms in haustorial formation among parasitic plants. The two up-regulated LBD genes in T. chinense are orthologs of A. thaliana LOB and LBD25, which are closely related paralogs (Kong et al. 2017). Although LOB functions in shoot–organ boundaries (Bell et al. 2012), LBD25 regulates the auxin sensitivity in lateral root formation (Mangeon et al. 2011). In addition, one of the down-regulated genes in haustoria is an ortholog of the KNOTTED-like A. thaliana 6 (KNAT6) gene (Tc_contig_17609), which is known to repress lateral root formation in A. thaliana (Dean et al. 2004). Therefore, as proposed in Yoshida et al. (2016) from transcriptome data of Orobanchaceae under laboratory conditions, the T. chinense haustorium might have evolved through the recruitment of the auxin-responsive lateral root developmental program.
Along with the enrichment of GO terms for VLCFA metabolic processes in haustoria (Supplementary Dataset S6), the genes related to VLCFA biosynthesis [Tc_contig_14102, ACYL-COA OXIDASE 2 (ACX2); Tc_contig_34750, ACYL-COA SYNTHETASE5 (ACOS5); Tc_contig_33689 and Tc_contig_33690, ACOS7] are up-regulated in haustoria (Fig. 2B;Supplementary Dataset S5). VLCFAs are defined as fatty acids with >20 acyl chains in length and biosynthesized through acyl-CoA (Chen et al. 2009). Our transcriptome data indicate the specific regulation of acyl-CoA enzymes by gene expression for the local biosynthesis of VLCFAs in the T. chinense haustoria. In addition to the components of the seed storage triacylglycerols, cuticular and epicuticular lipids and sphingolipids, VLCFAs play a key role in regulation of cell proliferation and differentiation during A. thaliana development (Roudier et al. 2010, Nobusawa et al. 2013). In particular, VLCFAs are involved in polar auxin transport to determine the cell polarity in Arabidopsis lateral root development (Roudier et al. 2010) and regulate the transcription of Aberrant Lateral Root Formation 4, which is a gene in the auxin-responsive lateral root developmental pathway (Shang et al. 2016). Although the function of VLCFAs is unknown in parasitic plant haustoria, trans-specific gene silencing targeting Triphysaria versicolor acetyl-CoA carboxylase, which is essential for VLCFA elongation (Baud et al. 2003), inhibits parasitic plant root growth (Bandaranayake and Yoder 2013).
To dissect the gene interactions in transcriptomic developmental reprogramming further, we constructed an unsigned gene co-expression network using the differentially expressed transcripts. The resultant network has seven major modules including >50 genes determined by the Fast-Greedy modularity optimization algorithm: four modules (M1, M3, M4 and M6) configure a core network connected to two other modules (M2 and M7) on the periphery, as well as one isolated module (M5) (Fig. 3A;Supplementary Dataset S7). GO enrichment analysis using genes from each module indicates that the core network is composed of developmental genes related to the cell cycle and translation, and connects to peripheral modules related to cell wall organization and environmental responses (Fig. 3A;Supplementary Dataset S8). One of the core network modules, the M1 module, has no significant GO terms but retained ACOS7, the VLCFA biosynthesis gene expressed in haustoria, as a highly interconnected hub gene (Fig. 3B). This M1 module has several differentially regulated genes between haustoria and roots, such as the lateral root developmental genes, LBD25 and KNAT6, in addition to another VLCFA biosynthesis gene, ACX2 (Fig. 3B). Notably genes related to lateral root meristem formation, such as SCARECROW-LIKE (SCL) (Tc_contig_18597, Tc_contig_20784, Tc_contig_10626, Tc_contig_21291 and Tc_contig_15072) and WUSCHEL-RELATED HOMEOBOX (WOX) (Tc_contig_2034) (Goh et al. 2016), as well as genes related to auxin signaling, such as PILS6 (Tc_contig_21289) and auxin response factor 9 (ARF9) (Tc_contig_8618) (Barbez et al. 2012, Lavenus et al. 2015), are also involved in the M1 module. Collectively, our network analysis demonstrates that VLCFA biosynthesis genes are strongly associated with the auxin-responsive lateral root developmental pathway, suggesting that VLCFAs and the auxin lateral root developmental pathway could act as a key network module in the developmental reprogramming of T. chinense haustorial formation.
Fig. 3.

Gene regulatory network in developmental reprogramming of T. chinense haustorial formation. (A) Weighted gene co-expression network for the differentially expressed genes in Fig. 2B. Nodes and edges represent genes and co-expression relationships between genes, respectively. Edge width indicates the weight of gene co-expression. The seven modules determined by the Fast-Greedy modularity optimization algorithm are represented by different colored nodes (M1–M7). Selected GO terms enriched in each module (FDR <0.05) are shown, and the detailed results of the GO enrichment analysis are shown in Supplementary Dataset S7. (B) The detailed network structure of M1. The size and color coding of nodes indicate log strength and strength, respectively, whose value sums up the weights of the adjacent edges for each node. Genes related to VLCFA biosynthesis, auxin response and lateral root development are highlighted by the different gray background shading.
Metabolic changes in haustorial formation
In contrast to transcriptome analysis, metabolome analysis data cannot distinguish whether metabolites, or other associated compounds, derived from T. chinense tissues, host tissues or from unrelated environmental materials, even though plant materials were carefully rinsed after collection. Biological replication, as well as comparing parasite transcriptome data, however, allows us to identify the metabolites strongly associated with T. chinense tissues. Our gas chromatography–time-of-flight–mass spectrometry (GC-TOF-MS) analysis detected 460 polar and 595 lipophilic metabolite peaks in whole T. chinense root systems including haustoria and mature roots (Supplementary Datasets S9, S10). In addition, as described below, our field metabolome data show that highly diversified lipophilic (7.1% of all detected metabolites) and polar (17.0%) metabolic profiles are able to distinguish developmental reprogramming from root cells to haustorial cells along with dynamic changes in the transcriptome.
Approximately 80% of the extracted polar metabolites were citric acid and malic acid in T. chinense tissues, except for one root sample showing a high abundance of mannitol (Supplementary Fig. S5). Since accumulation of sugar alcohols, including mannitol, might contribute to the unidirectional flow of water into the parasite from the host, mannose 6-phosphate reductase, a key enzyme in mannitol biosynthesis, has been a potential target for the control of pathogenic parasitic plants such as Orobanche species (Simier et al. 1993, Robert et al. 1999, Delavault et al. 2002, Aly et al. 2009). In our metabolome data, however, other sugar alcohols such as myo-inositol and sorbitol are dominant across tissues rather than mannitol (Supplementary Fig. S5). Our principal component (PC) analysis demonstrates that PC1, which explained 38.5% of variation in polar metabolome datasets, represents tissue-specific metabolite accumulation (Supplementary Fig. S6). An abundance test using an FDR <0.05 shows that 31 and 47 polar metabolites are highly abundant in haustoria and roots, respectively (Supplementary Dataset S9). Notably, hexose was accumulated in haustoria, while citric acid and malic acid were accumulated in roots (Supplementary Dataset S9).
The lipophilic metabolome of the T. chinense root systems contains specific metabolites, including, rather unexpectedly, oleanitrile, which is a rare nitrile derived from an 18-carbon (C18) unsaturated fatty acid (Supplementary Dataset S10). Oleanitrile is so rare that it is used as a biomarker of medicinal Dendrobium species (Jin et al. 2016). Approximately 40% and 50% of the lipophilic metabolite profile were fatty acids in roots and haustoria, respectively (Fig. 4A). We detected remarkably diverse fatty acids in T. chinense root systems, such as saturated fatty acids (C9, nonanoic acid; C12, dodecanoic acid; C15, pentadecanoic acid; C16, hexadecanoic acid; C17, heptadecanoic acid; C18, octadecanoic acid; C20, eicosanoic acid; C21, heneicosanoic acid; C22, behenic acid; C23, tricosanoic acid; C24, tetracosanoic acid; and C25, pentacosanoic acid) as well as unsaturated fatty acids (C18, octadecenoic acid, octadecadienoic acid, octadecatrienoic acid; and C22, docosahexaenoic acid) (Fig. 4A;Supplementary Dataset S10). Various VLCFAs were also involved in the T. chinense metabolome profile, consistent with our transcriptome data showing expressed transcripts of VLCFA biosynthesis genes (Fig. 2B). PC analysis in the lipophilic metabolite dataset demonstrates tissue-specific metabolite accumulation (Supplementary Fig. S6), and abundance testing using FDR <0.05 shows that 17 and 25 lipophilic metabolites are highly abundant in haustoria and root, respectively (Supplementary Dataset S10). Among differentially accumulated metabolites, only three lipophilic metabolites are annotated; an unidentified fatty acid that accumulates at high levels in haustoria, and brassicasterol and ethanolamine, both of which accumulate at high levels in roots (Supplementary Dataset S10). To determine the chain length of the unidentified fatty acid, we compared retention indices and mass fragmentation patterns of the detected fatty acid with those of standard compounds that possess various chain lengths of the alkane groups in the saturated fatty acids. The fitted line in the scatter plot of retention index and chain length of each alkane group from C6 to C30 clearly identified that the fatty acid that accumulated in the haustoria was pentacosanoic acid, which is a 25-carbon long chain saturated fatty acid categorized as a VLCFA (Fig. 4B). Although T. chinense root systems contain various fatty acids (Supplementary Dataset S10), the longest chain length of fatty acid was the most highly accumulated in the haustoria (Fig. 4C), which is again consistent with our transcriptome data (Fig. 2B).
Fig. 4.

Lipophilic metabolite profiling of the T. chinense root system. (A) Relative abundance of annotated fatty acids detected in the lipophilic metabolite profiles in T. chinense roots and haustoria. Data presented are the means of four independent biological replicates. (B) Scatter plot of standard compounds of retention index vs. chain length of alkane groups from C6 to C30 saturated fatty acids. R2 is shown. The fitted line identified that the unknown fatty acid accumulated in haustoria was a 25-carbon long chain saturated fatty acid (pentacosanoic acid). (C) The chemical structure of pentacosanoic acid and its abundance (CRMN normalized values) between T. chinense roots and haustoria. (D) Schematic diagram representing recruitment of the lateral root developmental pathway associated with the accumulation of VLCFAs into developmental reprogramming of T. chinense haustorial formation. TF, transcription factor.
Discussion
This study provides, to our knowledge, the first comprehensive view of gene expression and metabolic change in the developmental reprogramming of haustorial formation in parasitic plants in their native environment. Due to the sessile nature of plants, their development is highly plastic, enabling adaptation to their changing environment. A study that is able to target the development of plants in their natural habitat is desirable in order to understand it more thoroughly at the molecular level. Molecular analysis has, however, been challenging in ecological field studies, mostly due to the technological limitations. Recent advances in gene expression analysis, as well as metabolite quantification, have enabled genome-scale capturing of complex biological processes at the molecular level in the field (Alexandersson et al. 2014). This is creating new opportunities in understanding the molecular and environmental complexity of field-based systems, thus shedding light on the ‘black box’ of how plants employ developmental reprogramming for adaptation to natural environmental conditions. In this study, our field-omics plus bioinformatics approach has successfully profiled the transcriptome and metabolome of T. chinense root systems (Supplementary Datasets S2, S3, S9, S10) and detected detailed changes at both the transcript and metabolite levels in the developmental reprogramming of haustorial formation of the root hemi-parasitic plant, T. chinense (Fig. 2B;Supplementary Fig. S6).
Studies of evolutionary developmental biology of plants and animals suggest that gene regulation is key in determining developmental variation and that modification in pre-existing developmental gene regulatory networks is a crucial causal factor driving the innovation of novel morphological traits (Peter and Davidson 2011, Ichihashi et al. 2014). Current comparative transcriptomics in Orobanchaceae revealed that the patterns of gene expression in the haustoria are most similar to those of roots (Yang et al. 2015). This is reflected in the shared function of these highly specialized underground organs for nutrient uptake and transfer. In addition, there are similarities in terms of the developmental and cellular morphology between the formation of haustoria and of lateral roots: auxin accumulates at the initiation sites, cell proliferation is reactivated and cell walls are dramatically remodeled (Yoshida et al. 2016). This suggests a striking similarity in functional and developmental patterns between roots and haustoria, but still cannot define the molecular switch in developmental reprogramming from roots to haustoria. Our field transcriptomics and metabolomics study of T. chinense has sufficient resolution to distinguish the detailed changes in gene expression and metabolite abundance between root and haustoria (Fig. 2B;Supplementary Fig. S6). Furthermore, our statistical tests incorporating network analysis identified the activation of VLCFA biosynthesis and the auxin-responsive lateral root development pathway in haustoria (Figs. 2–4). This allows us to propose that the auxin-responsive lateral root development pathway associated with the accumulation of VLCFAs has been recruited into the developmental reprogramming of T. chinense haustorial formation in their natural environment (Fig. 4D). Since mature root tissues were used as a reference tissue in our experiments comparing the ‘before and after’ of the developmental reprogramming in haustorial formation, the key molecular modification from lateral root to haustorial formation is still unknown. More detailed comparative ‘omics’ targeting the development of lateral root and haustoria in T. chinense should provide the critical genetic change(s) responsible for engineering a lateral root into an haustorium.
Our field-omics also identified the uniqueness in the gene expression and metabolic prolife of the T. chinense root system. The essential oil from the S. album tree is represented by a mixture of sesquiterpenes, which are biosynthesized by a series of terpene synthases (Jones et al. 2011, Srivastava et al. 2015). Four genes annotated as santalene synthases are highly expressed in T. chinense haustoria (Tc_contig_26963, Tc_contig_20619, Tc_contig_32744 and Tc_contig_5980; Supplementary Dataset S5). A recent study showed that sesquiterpenes function as biologically active agents from ectomycorrhizal fungi to reprogram plant root architecture (Ditengou et al. 2015). Although we could not detect any metabolites related to santalene in the T. chinense root system, except for pi-Eudesmol (Supplementary Dataset S10), probably due to the volatile nature of these compounds, the compounds produced by the santalene synthases might have a specific function in T. chinense haustoria. In addition, the T. chinense root system has a high proportion of fatty acids, including rare compounds such as oleanitrile (Fig. 4A;Supplementary Dataset S10). Pentacosanoic acid is highly abundant in haustoria by the expression of VLCFA biosynthesis genes (Figs. 2B, 4C). Because terpenoids and fatty acid-related compounds are well known to be derived from acetyl-CoA, our ‘omics’ data suggest that the Santalales lineage, including T. chinense, might have evolved a unique method of regulation of acetyl-CoA metabolism. Thus, the lineage-specific regulatory machinery in acetyl-CoA metabolism might be involved in pre-conditioning of the genetic background to initiate the haustorial developmental reprogramming through the recruitment of the auxin-responsive lateral root development pathway associated with the accumulation of VLCFAs. Comparative ‘omics’ across evolutionarily independent lineages of parasitic plants could assess the specificity in the Santalales lineage, providing further insights into the convergent evolution of developmental reprogramming in plant parasitism.
Materials and Methods
Plant materials and sample collection
Thesium chinense is a perennial herb that has branched stems up to 60 cm in length. Thesium chinense can parasitize a diverse range of species from many plant families, and disturbs grasslands and riversides. It develops narrow linear leaves and has greenish-white, cylindrical flowers that are approximately 2 mm long (Fig. 1A). Samples were collected near the Kizu River, Kyoto Prefecture, Japan on February 3, 2015 and September 16, 2016 (Fig. 1A). Eight sections of 30×30×20 (depth) cm3 turf, containing at least two T. chinense plants and their hosts, such as A. capillaris, E. curvula and L. juncea, were randomly collected and then seven and four biological replicates made from pooled tissues of all plants were prepared for transcriptome and metabolome analyses, respectively. In order to minimize any surface tissue contamination, tissue samples were carefully collected using hand sectioning with a razor blade and rinsed using 70% ethanol or tap water for transcriptome and metabolome analyses, respectively, immediately frozen in liquid nitrogen and stored at −80 °C until sample preparation.
Histological observations
Field-collected samples were washed with tap water to remove soil. Safranin-O staining of whole haustoria was performed as previously described (Yoshida and Shirasu 2009). For histological sections, haustoria and infected host roots were separated and fixed with FAA solution (10% formaldehyde, 5% acetic acid, 50% ethanol) under vacuum for 15 min followed by incubation at room temperature for 2 h. Samples were subjected to an ethanol dehydration series and embedded into Technovit 7100 resin (Heraeus Kulzer GmbH) according to the manufacturer’s instruction. Solidified resin blocks were mounted on wood blocks with Technovit 3040 (Heraeus Kulzer GmbH), then sectioned using a microtome (Leica, RM2135) to 4 μm thickness. Sections were stained with 0.1% Safranin-O (Sigma-Aldrich) and counterstained with 0.1% Fast Green (Wako Chemical) on APS-coated glass slides (Matsunami Glass).
Transcriptomic and bioinformatics analysis
RNA-seq libraries were prepared from the collected tissues using a custom high-throughput method (Kumar et al. 2012). These RNA-seq libraries with seven biological replicates each were sequenced in a single lane of an Illumina HiSeq4000 platform at the Beijing Genome Institute (BGI) and reads were generated in a 100 bp paired-end format according to the manufacturer’s instructions (Illumina Inc.). RNA-seq read data have been deposited in the NCBI Sequence Read Archive (SRA) database under accession No. SRP114897. Seven biological replicates were used for de novo assembly, but six biological replicates were used for differential expression analysis, as one replicate was an outlier based on multidimensional scaling plots.
Pre-processing of reads involved quality trimming (removal of low-quality reads with a Phred score of >30 and trimming of low-quality bases from the 3' ends of the reads) and removal of adaptor/primer contamination and duplicated reads using custom Perl scripts (Kumar et al. 2012). The pre-processed reads were sorted into individual samples based on barcode sequences, and the barcodes were trimmed using the Fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). To remove host transcripts, the reads from T. chinense haustoria and roots were mapped to the host de novo assembled transcriptome, which was generated using the CLC Genomics Workbench version 8.0.1 with a word size of 64. Reads that did not map to host transcriptomes were extracted to use for subsequent transcriptome assembly. The CLC Genomics Workbench with a word size of 64 was also used for de novo assembly of the T. chinense tissue transcriptome. In order to remove redundant contigs, CD-HIT-EST from the CD-HIT suite, a clustering program based on similarity thresholds with a sequence similarity threshold of 95% and word size of eight, was used (Huang et al. 2010). The prediction of likely coding sequences from the clustered contigs was performed using TransDecoder (https://transdecoder.github.io), and bacterial and virus genome contamination was removed using DeconSeq (Schmieder and Edwards 2011). To construct a phylogenetic tree of PHYA orthologs, the amino acid alignment of angiosperm PHYA was obtained from a previous study (Mathews 2010). Conserved amino acid sequences of PHYA were aligned to construct a Neighbor–Joining tree using Clustal X verison 2.1 (Larkin et al. 2007). Bootstrap value were calculated from 1,000 replications.
For annotation, homology searches were performed against the NCBI non-redundant database using BLASTX with an e-value threshold of 1e-5 with Blast2go (Gotz et al. 2008). Prediction of gene ontology was performed by Trinotate version 2.0 (https://trinotate.github.io). Sequence comparison against The Arabidopsis Information Resource 10 (TAIR10) was performed using BLASTX with an e-value threshold of 1e-5.
For differential expression analysis, RNA-seq reads for each tissue were mapped to the final T. chinense contigs using the CLC Genomics Workbench with length fraction 0.8 and similarity 0.9. The count data (paired reads were count as two reads) were filtered such that there was at least one count per million in at least half of the samples and then normalized using the trimmed mean of M-values method in the Bioconductor package EdgeR version 3.16.5 (Robinson et al. 2010, McCarthy et al. 2012). The normalized reads were then used for the differential expression analysis between T. chinense haustoria and roots using EdgeR. The lists of differentially expressed contigs (FDR <0.05) are presented in Supplementary Dataset S5. Differentially expressed genes were then analyzed for enrichment of GO terms at a 0.05 FDR cut-off compared with all expressed genes (goseq Bioconductor package) (Young et al. 2010). In addition, the differentially expressed genes were also used for gene co-expression analysis using the weighted gene correlation network analysis (WGCNA) package version 1.60 (Langfelder and Horvath 2008). The soft-thresholding power was chosen based on a scale-free topology with fit index 0.8. An adjacency matrix with the selected soft-thresholding power was calculated and transformed into a topological overlap matrix. Using the topological overlap matrix, network properties such as strength and modularity were calculated and the network was visualized using the igraph package version 1.0.1 (Csardi and Nepusz 2006).
To confirm the reliability of the transcriptome results, RT–PCR was performed for two differentially expressed transcripts. mRNA was extracted directly from T. chinense haustoria and roots using the method in our previous study (Townsley et al. 2015). cDNA from the mRNA was prepared using the SuperScript™ III First-Strand Synthesis System (Thermo Fisher Scientific) according to the manufacturer’s protocol with oligo(dT)20. The cDNA was used for amplification by PCR using the primer pairs: 5'-CGAAGGCTTGGTTCGTATGT-3' and 5'-TAGTCCAGCTCTCCGCATTT-3' for Tc_contig_20619, and 5'-TGCTCGCGAACATATTGAAG-3' and 5'-CGCACAACATTGAGAAGGAA-3' for Tc_contig_32744. The coefficient of variation, which is the SD/mean expression value, was calculated for each gene in our transcriptome datasets to identify genes with the most stable expression across samples (Czechowski et al. 2005). Among them, Tc_contig_2828 amplified with the primers 5'-CTGGTTCCGTCACTTTGGTT-3' and 5'-ACCTCCAGGATCACCTTCCT-3' was used as internal control.
Metabolome analysis
The lipophilic and polar metabolites were extracted from the collected tissues with four biological replicates each in an extraction mixture comprised of chloroform : methanol : milliQ water (1 : 3 : 1 by vol.), following the addition of milliQ water to fractionate the lipophilic and polar phases. Metabolite profiling of the extracted metabolites in the lipophilic and polar phases was performed separately using GC-TOF-MS (Kusano et al. 2007a, Kusano et al. 2007b, Kusano et al. 2011). Briefly, the raw data of detected metabolites were transferred from the ChromaTOF software in the NetCDF format to the MATLAB software, version 2011b (MathWorks). The chromatograms were processed as described previously (Jonsson et al. 2005) and then normalized with the cross-contribution compensating multiple standard normalization algorithm (Redestig et al. 2009). Metabolite identification was performed as described previously (Kusano et al. 2007a). Different chain lengths of the fatty acids (C6, C7, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17, C18, C19, C20, C21, C22, C24, C26, C28 and C30, Supplementary Table S1) were used as standard compounds for GC-TOF-MS analysis.
For PC analysis, the abundance profiles of the metabolome were log10-transformed, and then PC values were calculated using the R stats package, prcomp function. As a differential abundance test, the Welch test was performed between T. chinense haustoria and roots with the FDR method of Benjamini and Hochberg (1995). All basic statistical functions were performed in R (R Core Team 2017) and visualized in the package ggplot2 (Wickham 2009).
Author Contributions
Y.I., K.S. and K.S conceived and co-ordinated the study. Y.I. designed the experiments. Y.I. carried out developmental observation, transcriptomics and all bioinformatics. M.K. and M.K. carried out metabolome analysis. K.S. collected plant materials from the field. S.Y. supported functional annotation for de novo assembly. T.W. carried out histological sectioning. K.K. and A.S. prepared tissue samples for transcriptome and metabolome analysis and performed RT–PCR. Y.I. wrote the manuscript with input from the other authors. All authors read and approved the final manuscript.
Supplementary Data
Supplementary data are available at PCP online.
Funding
This work was supported by the RIKEN Special Postdoctoral Researchers Program; the Ministry of Education, Culture, Sports, Science and Technology, Japan [Grant-in-Aid for Young Scientists B; grant No. 15K18589]; Japan Science and Technology Agency (JST) [PRESTO, JPMJPR15Q2] to Y.I., and KAKENHI [15H05959 and 17H06172] to K.S.
Supplementary Material
Acknowledgements
We thank Dr. Tsuneo Hakoyama and Dr. Keiko Yonekura-Sakakibara for their critical comments on this project.
Disclosures
The authors have no conflicts of interest to declare.
Glossary
Abbreviations
- ACOS
ACYL-COA SYNTHETASE
- ACX2
ACYL-COA OXIDASE 2
- FDR
false discovery rate
- GC-TOF-MS
gas chromatography–time-of-flight–mass spectrometry
- GO
Gene Ontology
- KNAT6
KNOTTED-like Arabidopsis thaliana 6
- LBD
LATERAL ORGAN BOUNDARIES DOMAIN
- NCBI
National Center for Biotechnology Information
- PC
principal component
- PHYA
phytochrome A
- PILS
PIN-LIKES
- RNA-seq
RNA-sequencing
- RT–PCR
reverse transcription–PCR
- SHI/STY
SHORT INTERNODES/STYLISH
- VLCFA
very long chain fatty acid
Glossary
Footnote
- The sequences reported in this paper have been deposited in the Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/) (accession No. SRP114897)
References
- Alexandersson E., Jacobson D., Vivier M.A., Weckwerth W., Andreasson E. (2014) Field-omics—understanding large-scale molecular data from field crops. Front. Plant Sci. 5: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly R., Cholakh H., Joel D.M., Leibman D., Steinitz B., Zelcer A., et al. (2009) Gene silencing of mannose 6-phosphate reductase in the parasitic weed Orobanche aegyptiaca through the production of homologous dsRNA sequences in the host plant. Plant Biotechnol. J. 7: 487–498. [DOI] [PubMed] [Google Scholar]
- Baldovini N., Delasalle C., Joulain D. (2011) Phytochemistry of the heartwood from fragrant Santalum species: a review. Flavour Fragr. J. 26: 7–26. [Google Scholar]
- Bandaranayake P.C., Yoder J.I. (2013) Trans-specific gene silencing of acetyl-CoA carboxylase in a root-parasitic plant. Mol. Plant Microbe Interact. 26: 575–584. [DOI] [PubMed] [Google Scholar]
- Barbez E., Kubes M., Rolcik J., Beziat C., Pencik A., Wang B., et al. (2012) A novel putative auxin carrier family regulates intracellular auxin homeostasis in plants. Nature 485: 119–122. [DOI] [PubMed] [Google Scholar]
- Barkman T.J., McNeal J.R., Lim S.H., Coat G., Croom H.B., Young N.D., et al. (2007) Mitochondrial DNA suggests at least 11 origins of parasitism in angiosperms and reveals genomic chimerism in parasitic plants. BMC Evol. Biol. 7: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud S., Guyon V., Kronenberger J., Wuilleme S., Miquel M., Caboche M., et al. (2003) Multifunctional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 33: 75–86. [DOI] [PubMed] [Google Scholar]
- Bell E.M., Lin W.C., Husbands A.Y., Yu L., Jaganatha V., Jablonska B., et al. (2012) Arabidopsis lateral organ boundaries negatively regulates brassinosteroid accumulation to limit growth in organ boundaries. Proc. Natl. Acad. Sci. USA 109: 21146–21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57: 289–300. [Google Scholar]
- Chang M., Lynn D.G. (1986) The haustorium and the chemistry of host recognition in parasitic angiosperms. J. Chem. Ecol. 12: 561–579. [DOI] [PubMed] [Google Scholar]
- Chen M., Mooney B.P., Hajduch M., Joshi T., Zhou M., Xu D., et al. (2009) System analysis of an Arabidopsis mutant altered in de novo fatty acid synthesis reveals diverse changes in seed composition and metabolism. Plant Physiol. 150: 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. (2006) The igraph software package for complex network research. InterJ. Complex Syst. 1695. [Google Scholar]
- Czechowski T., Stitt M., Altmann T., Udvardi M.K., Scheible W.R. (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean G., Casson S., Lindsey K. (2004) KNAT6 gene of Arabidopsis is expressed in roots and is required for correct lateral root formation. Plant Mol. Biol. 54: 71–84. [DOI] [PubMed] [Google Scholar]
- Delavault P., Simier P., Thoiron S., Veronesi C., Fer A., Thalouarn P. (2002) Isolation of mannose 6-phosphate reductase cDNA, changes in enzyme activity and mannitol content in broomrape (Orobanche ramosa) parasitic on tomato roots. Physiol. Plant. 115: 48–55. [DOI] [PubMed] [Google Scholar]
- Ditengou F.A., Muller A., Rosenkranz M., Felten J., Lasok H., van Doorn M.M., et al. (2015) Volatile signalling by sesquiterpenes from ectomycorrhizal fungi reprogrammes root architecture. Nat. Commun. 6: 6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostálek T., Münzbergová Z. (2010) Habitat requirements and host selectivity of Thesium species (Santalaceae). Bot. J. Linn. Soc. 164: 394–408. [Google Scholar]
- Eklund D.M., Staldal V., Valsecchi I., Cierlik I., Eriksson C., Hiratsu K., et al. (2010) The Arabidopsis thaliana STYLISH1 protein acts as a transcriptional activator regulating auxin biosynthesis. Plant Cell 22: 349–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh T., Toyokura K., Wells D.M., Swarup K., Yamamoto M., Mimura T., et al. (2016) Quiescent center initiation in the Arabidopsis lateral root primordia is dependent on the SCARECROW transcription factor. Development 143: 3363–3371. [DOI] [PubMed] [Google Scholar]
- Gotz S., Garcia-Gomez J.M., Terol J., Williams T.D., Nagaraj S.H., Nueda M.J., et al. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36: 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray W.M., Muskett P.R., Chuang H.W., Parker J.E. (2003) Arabidopsis SGT1b is required for SCF(TIR1)-mediated auxin response. Plant Cell 15: 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Niu B., Gao Y., Fu L., Li W. (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26: 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y., Aguilar-Martinez J.A., Farhi M., Chitwood D.H., Kumar R., Millon L.V., et al. (2014) Evolutionary developmental transcriptomics reveals a gene network module regulating interspecific diversity in plant leaf shape. Proc. Natl. Acad. Sci. USA 111: E2616–E2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y., Mutuku J.M., Yoshida S., Shirasu K. (2015) Transcriptomics exposes the uniqueness of parasitic plants. Brief. Funct. Genomics 14: 275–282. [DOI] [PubMed] [Google Scholar]
- Ishida J.K., Wakatake T., Yoshida S., Takebayashi Y., Kasahara H., Wafula E., et al. (2016) Local auxin biosynthesis mediated by a YUCCA flavin monooxygenase regulates haustorium development in the parasitic plant Phtheirospermum japonicum. Plant Cell 28: 1795–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q., Jiao C., Sun S., Song C., Cai Y., Lin Y., et al. (2016) Metabolic analysis of medicinal Dendrobium officinale and Dendrobium huoshanense during different growth years. PLoS One 11: e0146607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel D.M., Losner-Goshen D. (1994) The attachment organ of the parasitic angiosperms Orobanche cumana and O. aegyptiaca and its development. Can. J. Bot. 72: 564–574. [Google Scholar]
- Jones C.G., Moniodis J., Zulak K.G., Scaffidi A., Plummer J.A., Ghisalberti E.L., et al. (2011) Sandalwood fragrance biosynthesis involves sesquiterpene synthases of both the terpene synthase (TPS)-a and TPS-b subfamilies, including santalene synthases. J. Biol. Chem. 286: 17445–17454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson P., Johansson A.I., Gullberg J., Trygg J., A J., Grung B., et al. (2005) High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based metabolomic analyses. Anal. Chem. 77: 5635–5642. [DOI] [PubMed] [Google Scholar]
- Kong Y., Xu P., Jing X., Chen L., Li L., Li X. (2017) Decipher the ancestry of the plant-specific LBD gene family. BMC Genomics 18: 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijt J. (1969) The Biology of Parasitic Flowering Plants. University of Califoria Press, Berkeley, CA. [Google Scholar]
- Kumar R., Ichihashi Y., Kimura S., Chitwood D.H., Headland L.R., Peng J., et al. (2012) A high-throughput method for Illumina RNA-Seq library preparation. Front. Plant Sci. 3: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano M., Fukushima A., Arita M., Jonsson P., Moritz T., Kobayashi M., et al. (2007a) Unbiased characterization of genotype-dependent metabolic regulations by metabolomic approach in Arabidopsis thaliana. BMC Syst. Biol. 1: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano M., Fukushima A., Kobayashi M., Hayashi N., Jonsson P., Moritz T., et al. (2007b) Application of a metabolomic method combining one-dimensional and two-dimensional gas chromatography-time-of-flight/mass spectrometry to metabolic phenotyping of natural variants in rice. J. Chromatogr. B 855: 71–79. [DOI] [PubMed] [Google Scholar]
- Kusano M., Tabuchi M., Fukushima A., Funayama K., Diaz C., Kobayashi M., et al. (2011) Metabolomics data reveal a crucial role of cytosolic glutamine synthetase 1;1 in coordinating metabolic balance in rice. Plant J. 66: 456–466. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lavenus J., Goh T., Guyomarc’h S., Hill K., Lucas M., Voss U., et al. (2015) Inference of the Arabidopsis lateral root gene regulatory network suggests a bifurcation mechanism that defines primordia flanking and central zones. Plant Cell 27: 1368–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeon A., Bell E.M., Lin W.C., Jablonska B., Springer P.S. (2011) Misregulation of the LOB domain gene DDA1 suggests possible functions in auxin signalling and photomorphogenesis. J. Exp. Bot. 62: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews S. (2010) Evolutionary studies illuminate the structural–functional model of plant phytochromes. Plant Cell 22: 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Chen Y., Smyth G.K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40: 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobusawa T., Okushima Y., Nagata N., Kojima M., Sakakibara H., Umeda M. (2013) Synthesis of very-long-chain fatty acids in the epidermis controls plant organ growth by restricting cell proliferation. PLoS Biol. 11: e1001531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen Z., Deng Y., Saeed M.K., Dai R., Ahamad W., Yu Y.H. (2007) Antiinflammatory and analgesic activities of Thesium chinense Turcz extracts and its major flavonoids, kaempferol and kaempferol-3-O-glucoside. Yakugaku Zasshi 127: 1275–1279. [DOI] [PubMed] [Google Scholar]
- Peter I.S., Davidson E.H. (2011) Evolution of gene regulatory networks controlling body plan development. Cell 144: 970–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Redestig H., Fukushima A., Stenlund H., Moritz T., Arita M., Saito K., et al. (2009) Compensation for systematic cross-contribution improves normalization of mass spectrometry based metabolomics data. Anal. Chem. 81: 7974–7980. [DOI] [PubMed] [Google Scholar]
- Robert S., Simier P., Fer A. (1999) Purification and characterization of mannose 6-phosphate reductase, a potential target for the control of Striga hermonthica and Orobanche ramosa. Funct. Plant Biol. 26: 233–237. [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F., Gissot L., Beaudoin F., Haslam R., Michaelson L., Marion J., et al. (2010) Very-long-chain fatty acids are involved in polar auxin transport and developmental patterning in Arabidopsis. Plant Cell 22: 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R., Edwards R. (2011) Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6: e17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang B., Xu C., Zhang X., Cao H., Xin W., Hu Y. (2016) Very-long-chain fatty acids restrict regeneration capacity by confining pericycle competence for callus formation in Arabidopsis. Proc. Natl. Acad. Sci. USA 113: 5101–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simier P., Fer A., Renaudin S. (1993) Identification of the main osmotically active solutes in the unstressed and water-stressed root-hemiparasitic angiosperm Thesium humile and its host Triticum vulgare. Funct. Plant Biology 20: 223–230. [Google Scholar]
- Sohlberg J.J., Myrenas M., Kuusk S., Lagercrantz U., Kowalczyk M., Sandberg G., et al. (2006) STY1 regulates auxin homeostasis and affects apical–basal patterning of the Arabidopsis gynoecium. Plant J. 47: 112–123. [DOI] [PubMed] [Google Scholar]
- Srivastava P.L., Daramwar P.P., Krithika R., Pandreka A., Shankar S.S., Thulasiram H.V. (2015) Functional characterization of novel sesquiterpene synthases from Indian Sandalwood, Santalum album. Sci. Rep. 5: 10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens P.F. (2001. onwards) Angiosperm Phylogeny Website. Version 12, July 2012 [and more or less continuously updated since].
- Suetsugu K. (2015) Seed dispersal of the hemiparasitic plant Thesium chinense by Tetramorium tsushimae and Pristomyrmex punctatus. Entomol. Sci. 18: 523–526. [Google Scholar]
- Suetsugu K., Kawakita A., Kato M. (2008) Host range and selectivity of the hemiparasitic plant Thesium chinense (Santalaceae). Ann. Bot. 102: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomilov A.A., Tomilova N.B., Abdallah I., Yoder J.I. (2005) Localized hormone fluxes and early haustorium development in the hemiparasitic plant Triphysaria versicolor. Plant Physiol. 138: 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley B.T., Covington M.F., Ichihashi Y., Zumstein K., Sinha N.R. (2015) BrAD-seq: Breath Adapter Directional sequencing: a streamlined, ultra-simple and fast library preparation protocol for strand specific mRNA library construction. Front. Plant Sci. 6: 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Zhang Y., Kieffer M., Yu H., Kepinski S., Estelle M. (2016) HSP90 regulates temperature-dependent seedling growth in Arabidopsis by stabilizing the auxin co-receptor F-box protein TIR1. Nat. Commun. 7: 10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2009) ggplot2: Elegant Graphics for Data Analysis. Springer Publishing Company, Berlin. [Google Scholar]
- Yang Z., Wafula E.K., Honaas L.A., Zhang H., Das M., Fernandez-Aparicio M., et al. (2015) Comparative transcriptome analyses reveal core parasitism genes and suggest gene duplication and repurposing as sources of structural novelty. Mol. Biol. Evol. 32: 767–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Cui S., Ichihashi Y., Shirasu K. (2016) The haustorium, a specialized invasive organ in parasitic plants. Annu. Rev. Plant Biol. 67: 643–667. [DOI] [PubMed] [Google Scholar]
- Yoshida S., Shirasu K. (2009) Multiple layers of incompatibility to the parasitic witchweed, Striga hermonthica. New Phytol. 183: 180–189. [DOI] [PubMed] [Google Scholar]
- Young M.D., Wakefield M.J., Smyth G.K., Oshlack A. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Berkowitz O., Teixeira da Silva J.A., Zhang M., Ma G., Whelan J., et al. (2015) RNA-Seq analysis identifies key genes associated with haustorial development in the root hemiparasite Santalum album. Front. Plant Sci. 6: 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Liu B., Guo Q., Song L., Chen L., Wang C. (2016) Construction of a haustorium development associated SSH library in Thesium chinense and analysis of specific ESTs included by Imperata cylindrica. Biochem. Syst. Ecol. 64: 46–52. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.