

Abstract
Aim:
The aim of this paper was to present a 65 year old female patient with chronic heart disease, surgically treated for congenital heart defect type Tetralogy of Fallot.
Case report:
In the sixth year of life the patient underwent palliative Potts anastomosis surgery which created an anastomosis between the left pulmonary artery and the descending aorta. Total correction was made in 34 years of life, six months after catheterization, which indicated malignant pulmonary hypertension. She is regularly followed up by the cardiologists and receives daily therapy. The present state of the patient is satisfactory with cardiomegaly, light left ventricular dysfunction, moderate mitral and tricuspid regurgitation, pulmonary arterial hypertension, and aneurysmatic dilatation of left pulmonary artery as well as atrial fibrillation.
Conclusion:
The intense development of cardiology and cardiac surgery in the USA in children and adults over the last fifty years has led to the extension and improvement of the quality of life.
Keywords: Tetralogy of Fallot, clinical course, treatment
1. INTRODUCTION
La Maladie Bleue (although originally described by Niels Stensen), described by Louis Arthur Etienne Fallot in 1888 is a physiological anatomical entity that is now defined as Tetralogy of Fallot (ToF) (a large ventricular septal defect (VSD) overriding aorta, right ventricular outflow obstruction and right ventricular hypertrophy (2)). Tetralogy of Fallot represents the most common cyanogenic defect with an incidence of 3 to 5 per 10000 live births (1), and represents the first cyanogenic defect that was successfully surgically treated (first palliative surgery was performed in 1945 and complete correction surgery was performed in 1954). The aim of this paper was to present a 65-year-old patient with ToF, showing complexity of surgical and pharmacological treatment, taking into account the development of cardiology and cardiac surgeryin more than half of century.
2. CASE REPORT
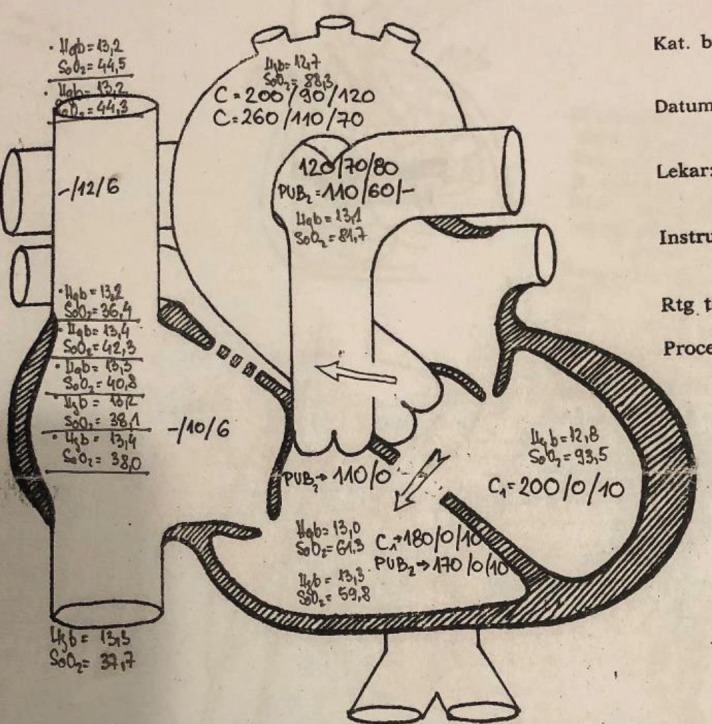
When the patient was in the age of toddler, cyanosis was observed, with occasional squat after a long walk and with legs bent along stomach during sleep. In 1958, at the age of six, a palliative surgery with Potts anastomosis was performed.She was monitored by the cardiologists, and after the deterioration in the third decade of life after catheterization (in May 1987, Sremska Kamenica), she was advised to perform complete correction in Houston, Texas, USA. She had malignant pulmonary hypertension due hyperkinetic situation at the level of L-Rventricleat about 53mmHg (gradient at the level of aorta and pulmonary artery (Figure 1.) While waiting for total correction, catheterization was repeated (in November 1987), where the dominant L-R shunt was confirmed, but without the fixation of pulmonary hypertension (saturation in right ventricle 61%, saturation in pulmonary artery 83%).In addition, function of Potts shunt was confirmed, but with development of second degree aortic regurgitation, with the hypertrophic wall of the right ventricular tract, which due to stenosis filled right ventricle retrogradely and pulmonary artery anterogradely. Infundibulectomy (excision of hypertrophied ventricular septal myocardium encroaching on the ventricular outflow tract and valvuloplasty of pulmonary valve) was performed in Houston.From birth to total correction, there was also an atrial septal defect which was closed. The closure of the wide ventricular septal defect was performed too, with the aortic retraction in the left heart ventricle. On the operating table, significant left ventricular hypertrophy was detected, primarily of the left ventricle with the global hypokinesis. After the surgery she was treated with amiodarone and digoxin. She is regularly monitored by the cardiologists, but she developed left ventricular dysfunction with persistence of pulmonary arterial hypertension and rhythm disorders (eight years ago with permanent atrial fibrillation and right bundle branch block). In addition, moderate regurgitation on both control valves, as well as characteristic mild stenosis and regurgitationwas registered at ultrasound check-up visits.The aneurysmatic dilatation of the main branch of the left pulmonary artery was found, and she was treated with ACE inhibitor (ramipril), beta blocker (carvedilol), lower doses of diuretics and antiarrhythmic drugs (amiodarone), and with regard to symptomatic association with fibrillation, warfarin was included. 4 years ago, amiodarone is excluded, and the cardiotonic are included again.The ejection fraction of the left ventricle is maintained at about 50%. Occasionally, liver failure, of lower or higher stage, and mild renal insufficiency occurs. Although hemodynamics and physiology of the heart are endangered, the condition of the patient is relatively stable in 65 years of life.
Figure 1. Catheterization Laboratory, Sremska Kamenica–(TT 60kg, TC 174cm) PCW-40mmHg, AP 110mmHg, in infundibulum 110 mmHg, right ventricle 170 mmHg (gradient 60 mmHg), cardiac index 1.54–taken samples for saturation on the gradient at left ventricle. Right ventricle and aorta- pulmonary artery level (left to right shunt at the level of membrane septum about 53%, at aorta- pulmonary artery level about 73%).
3. DISCUSSION
Congenital heart defect with incidence of approximately 1%, in most cases require surgical treatment. ToF is normally operated until the sixth month of life, with total correction, rarely with palliationwhich will result with total correction in 6 months to 2 years. The aforementioned Potts anastomosis as well as Waterston’s anastomosis (descending aorta and the tree or branch of the pulmonary artery) are no longer used because they have been replaced by Blalock-Taussig anastomosis (one branch of the subclavian artery or carotid artery is separated and connected with the pulmonary artery thus making connection with systemic and pulmonary circulation). Total correction is not common to be performed so late due to the high risk of surgery.Hemodynamics of the right-left shunt usually does not go with PAH, it is actually characteristic of left-right shunt in progression. Probably bidirectional shunt that was initially right-left, and at age 34 of life left-right, did not lead to the fixation of PAH, so the operation was possible.On the other hand, complications of postoperative ToF (residual hemodynamic disorders–pulmonary insufficiency, right ventricular outflow tract (RVOT) obstruction, pulmonary valve or annular stenosis, stenosis of the branch of the pulmonary artery, RVOT aneurysms, tricuspid regurgitation, right ventricular dysfunction, ventricular septal defect, left ventricular dysfunction; conduction abnormalities–supraventricular tachycardia, ventricular tachycardia, ventricular extrasystoles, complete AV block) often overlap.In countries where cardiac surgery is developed, there is an increasing number of adult patients with deficiencies completely corrected. The complete surgical correction consists of closing the ventricular septal defect and solving the obstruction on the RVOT and the pulmonary artery. The contemporary approach to treatment is aimed at reducing residual lesions and late complications of the disease. It consists in an early complete correction, with a tendency to minimize the number of palliative surgery interventions.Surgery is most often performed between 6 and 18 months of life. If ToF is not surgically treated 11% of patients would not have experienced 20 years, 6% 30 years, and 3% 40 years. The quality of life of operated patients is very bad, with markedly decreased exercise tolerance. The far-reaching results of patients with surgically treated ToF are very good, with actual survival of about 85%, 30 years after surgery (1). Second author has had two cases of ToF in the later years of life, which without surgeryhad relatively well tolerated hemodynamics of the heart (52 and 60 years of life). In literature there is also a case where first diagnosis was madeat age 61, where due to hypertensive retinopathy (because of multiple hypertension, left-right shunt was decreased) cyanosis was barely noticeable (4).
4. CONCLUSION
It would be interesting to compare this clinical course with the natural evolution of ToF. Two issues of GUCH (grown-up congenital heart disease) and standardization of diagnostic- therapeutic relations is being addressed, and also the issue of uncritical obsession with the rapid radical surgical treatment of most CHDs, and especially the Tetralogy of Fallot, which clinically and anatomically has few usual characteristics and pathoanatomically has multiple and individual characteristics. Also the age of optimal correction is shifted to young infant and neonatal age.The frequency, diversity of clinical presentation, including treatment of complications requires complex treatment in children, and especially in adults. Mentioned report of catheterization and surgery from 1987, makes us proud followers of the honourable and progressive cardiology.
Declaration of patient consent:
Authors certify that they have obtained patient consent form.
Conflict of interest:
none declared.
Authors contribution:
All authors gave substantial contribution to the conception or design of article. They revised it critically and gave final approval of the version to be published.
REFERENCES
- 1.Fallot ELA. Contribution à l’anatomie pathologique de la maladie bleue (cyanose cardiaque) Marseille médical. 1888;25:77–93. [PubMed] [Google Scholar]
- 2.Monaco M, Williams I. Tetralogy of Fallot: fetal diagnosis to surgical correction. Minerva Pediatr. 2012 Oct;64(5):461–470. [PubMed] [Google Scholar]
- 3.Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet Journal of Rare Diseases. Orphanet J Rare Dis. 2009 Jan 13;4:2. doi: 10.1186/1750-1172-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann A, Gunthardt J, Gatzi H, et al. A 63 year old man with uncorrected tetralogy of Fallot. Z Kardiol. 1995;84:1039–1042. [PubMed] [Google Scholar]