

Abstract
Polycomb Repressive Complex 2 (PRC2) member enhancer of zeste homologue 2 (EZH2) trimethylates histone H3 lysine 27 (H3K27me3), alters chromatin structure and contributes to epigenetic regulation of gene expression in normal and disease processes. Phosphorylation of EZH2 augmented EZH2 oncogenic activity in cancer but observations have been limited to serine 21 (S21) residue by protein kinase B. In addition, phosphorylation of the evolutionarily conserved T372 motif of EZH2 by p38 resulted in EZH2 interaction with Ying Yang 1 and promoted muscle stem cell differentiation. In the present study, we used epithelial ovarian cancer (OC) cells as a model to demonstrate that phosphorylation of EZH2 at T372 by protein kinase A (PKA) induced a dominant-negative EZH2 phenotype, inhibited OC cell proliferation and migration in vitro and decreased ovarian xenograft tumor growth in vivo. Phosphorylation of T372 by PKA enhanced the interaction between EZH2 and signal transducer and activator of transcription 3 (STAT3), and STAT3 binding to pT372-EZH2 reduced cellular levels of pSTAT3 and downregulated interleukin 6 receptor expression in OC. Furthermore, PKA-mediated pT372-EZH2 decreased ATP levels and altered mitochondrial gene expression, resulting in mitochondrial dysfunction and reduced OC cell growth. These findings demonstrate that PKA-mediated T372 phosphorylation reduces oncogenic EZH2 activity and reveal a novel role for pT372 in regulating EZH2 in OC and possibly other cancers.
Keywords: Oncogene, EZH2, Phosphorylation, Protein Kinase A, Epigenetics, Ovarian Cancer
Introduction
The polycomb group (PcG) proteins are important regulators of normal embryonic development and cell fate decisions1. The components of polycomb repressive complex 2 (PRC2) include embryonic ectoderm development (EED) Suppressor of Zeste 12 (Suz12), and Enhancer of Zeste Homolog 2 (EZH2), the catalytic subunit of PRC2. The main PRC-mediated function of EZH2 is primarily to promote transcriptional silencing via histone 3 on lysine 27 trimethylation (H3K27me3), and essential roles of EZH2 in proliferation of normal cells and progression of cancer cells has been demonstrated. In cancer cells, EZH2 has been shown to have both oncogenic and tumor suppressive properties. In addition to its main PRC2 function of transcriptional repression, non-PRC functions of EZH2 via direct binding to major transcription factors (TF) has been widely reported. EZH2 has been shown to act as a co-factor for retinoic acid receptor alpha (RARα)2, nuclear factor kappa B (NF-κB)3, androgen receptor (AR)4, GATA binding factor 4 (GATA4)5, signal transducer and activator of transcription 3 (STAT3)6, and breast cancer associated 1 (BRCA1)7. TF binding altered EZH2 methyltransferase activity and oncogenic EZH2 protein levels as well as EZH2 target gene expression1. Furthermore, EZH2 oncogenic activity, via repression of tumor suppressors p16INK4A and DAB2 interacting protein (DAB2IP), has been demonstrated to contribute to chemotherapy resistance8, 9.
Overexpression of EZH2 in cancer has been widely reported14, and targeting the oncogenic activity of EZH2 in cancer has proven to be a promising therapeutic approach15. In ovarian cancer (OC), the deadliest gynecological malignancy in the United States16, increased expression of EZH2 predicted poor clinical outcome17 and altered EZH2 levels contributed to platinum resistant disease18, 19. EZH2 overexpression enhanced OC cell proliferation20 and increased OC invasion and metastasis21. EZH2 promoted OC tumor angiogenesis17 and inhibited apoptosis. Aberrant EZH2 expression altered microRNA expression22 and OC cell transformation23. Thus, EZH2 is a multifunctional protein with diverse roles in OC pathogenesis.
In addition to TF binding, post-translational modifications altered EZH2 methyltransferase activity, including phosphorylation at serine 21 (S21) by protein kinase B (AKT)10, T372 by p3811 and threonine 345 (T345) by cyclin dependent kinase 1 (CDK1)12. T345 phosphorylation induced EZH2 interaction with the Hox intergenic non-coding RNA (HOTAIR) and X-inactive specific transcript RNA (XIST)13. However, post-translational modifications of EZH2 remain incompletely described and their functional relevance in cancer remains to be fully elucidated.
In order to search for novel binding partners of EZH2, we performed a detailed examination for canonical phosphorylation sites and found T372 to reside in a canonical phosphorylation motif for Protein Kinase A (PKA), an enzyme activated by the second messenger cyclic AMP (cAMP). Intriguingly, we show in ovarian cancer as a model that T372 phosphorylation induced a dominant-negative EZH2 and inhibited OC cell growth and tumorigenesis. Mechanistically, p-T372-EZH2 reduced cellular levels of active signal transducer and activator of transcription 3 (STAT3), reduced intracellular ATP and induced mitochondrial dysfunction in OC cells in vitro and in vivo. Our findings point to a novel role of T372 phosphorylation in fine-tuning the oncogenic properties of EZH2 in cancer and extend the role of this histone methyltransferase in tumor progression.
Results
PKA phosphorylates EZH2 at evolutionarily conserved threonine T372 motif
In order to identify novel EZH2 modifiers, we examined the EZH2 amino acid sequence searching for phosphorylation motifs. Matched consensus (RXXS/T (X is any residue) PKA phosphorylation sites were identified: T350, T371, T372, T392 and T482. To test whether EZH2 and PKA interact in a cellular context, OC cells were treated with the PKA activator forskolin (FSK; 10μM) and endogenous EZH2 and PKA were immunoprecipitated. An increased association of EZH2 and PKAcα was determined (Fig.1A and Supplemental Fig. S1A), and then to identify the interaction domain, GST fusions of EZH2 corresponding to residues 1-173, 173-340, 340-550, and 550-749 were expressed in HEK293 cells. Pull-down assays with anti-PKA antibody demonstrated an interaction between full-length GST-EZH2 as well as GST-EZH2340-550 (Fig. 1B; Supplemental Fig. S1A). Mass spectrometry analysis identified direct phosphorylation of recombinant EZH2 by PKA at residue T372, a site highly conserved in human, mouse, and zebra fish (Fig.1C) and therefore of high biological significance.
Figure 1. EZH2 and PKA interact and PKA phosphorylates EZH2 on T372.
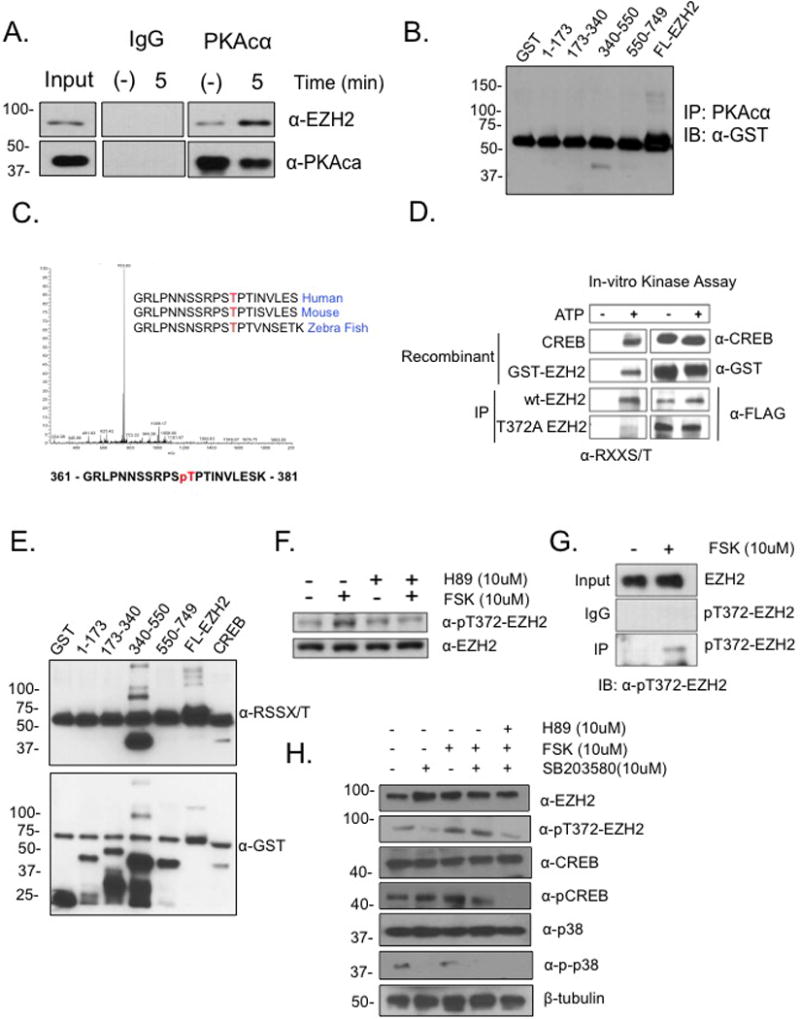
(A) Serum starved A2780p cells were treated with forskolin (FSK; 10μM) for 5 min and endogenous PKA was IP’d and blotted for PKA and EZH2 (western blot was cropped because of different exposure times between IP and Input). (B) GST-tagged truncations of EZH2 (AA residues, 1-173, 173-340, 340-550, 550-749) were expressed in e. coli (DH5α) and blotted with anti-GST. The prominent band at 55kDa is IgG. (C) In vitro kinase assay with recombinant EZH2 (50ng) and PKA (100ng) was performed in the presence of ATP (1mM) and analyzed by LC MS/MS mass spectrometry. MS/MS spectrum shows phosphorylation of ThrT372. (D) In vitro kinase assay was performed with recombinant CREB and purified GST-EZH2 plus IP’d FLAG- EZH2 and FLAG-T372A from HEK293 cells. Anti-RXXS/T antibody was used for immunoblotting (E) In vitro kinase assay was performed with recombinant PKAcα incubated with GST-truncations and blotted with anti-GST. The prominent band seen at 55kDa is IgG. (F) Serum starved HEK293 cells were pre-treated with PKA inhibitor H89 (10μM) for 24 hrs and then treated with or without FSK (10μM) for 24 hrs and resolved by western blot using antibody raised against phosphorylated Thr-372 (pT372-EZH2). Western blot was cropped because of different exposure times between IP and Input. (G) Serum starved HEK293 cells were treated with FSK (10μM) and pT372 EZH2 was immunoprecipitated using anti-pT372-EZH2 (western blot was cropped due to use of different exposure times between IP and Input). Representative data of at least three biological experiments unless otherwise indicated. (H) Serum starved HEK293 cells were pre-treated with PKA inhibitor H89 (10μM), p38 inhibitor SB203580 (10μM) for 24 hrs and then treated with or without FSK (10μM) for 24 hrs resolved by western blot using antibody raised against phosphorylated Thr-372 (pT372-EZH2), EZH2, CREB, pCREB, p38, p-p38, and β-tubulin. Western blot was cropped because of different exposure times between IP and input.
To validate and further assess EZH2 phosphorylation at residue T372, an in vitro kinase assay with GST-EZH2, immunoprecipitated FLAG-tagged EZH2 and FLAG-T372A was done. Strong signals from both GST-EZH2 and immunoprecipitated wild-type (Wt)-EZH2 were detected by a phospho-specific anti-RXXS/T antibody, in contrast to the markedly reduced signal and diminished phosphorylation at T372A by PKA (Fig. 1D). These results were further examined using an in vitro kinase assay with the anti-RXXS/T antibody, and phosphorylation of both full length GST-EZH2 and the 340-550 fragment was detected (Fig. 1E). Next, a phospho-specific antibody generated against T372 (pT372) detected in vivo phosphorylation after FSK (10μM) treatment and PKA inhibitor H89 (10μM) reduced T372 phosphorylation (Supplemental Fig. S1B; Fig. 1F). In addition, anti-pT372 pulled down a greater amount of EZH2 from cells treated with FSK vs. control (Fig. 1G), confirming that PKA phosphorylates EZH2.
As mentioned above, phosphorylation of T372 by p38 in response to upstream signals induced by pro-inflammatory cytokines11 or stress inducing conditions such as oxidative stress is possible. Therefore, to examine whether the in vivo phosphorylation we observed could be due to p38 activity, we utilized a chemical inhibitor of p38 (SB203580; 10μM) in the presence and absence of FSK (10μM) and H89 (10μM). We observed T372 phosphorylation in the presence of SB203580 (Fig. 1H), suggesting that PKA can phosphorylate EZH2 irrespective of p38 activity. Because EZH2 phosphorylation could also alter binding to other PRC2 partners, GFP-EZH2, phospho-null mutant GFP-T372A and phospho-mimetic mutant GFP-T372E were immunoprecipitated; however, no effect on the interaction between modified EZH2 and Suz12, Eed1-4 or Ring1A was observed (Supplemental Fig. S2A), and nuclear localization of EZH2 was not altered (Supplemental Fig. S2B). Taken together, these results demonstrated that PKA-mediated phosphorylation of EZH2 at T372 and had no effect on the overall composition of the PRC2 complex.
EZH2 phosphorylation at T372 reduces ovarian cancer cell proliferation, migration and tumor formation
To study the biological function of pT372-EZH2, we performed in vitro and in vivo OC cell-based assays using Wt-EZH2 and a phosphomimetic T372E-EZH2. Compared to Wt-EZH2, expression of T372E-EZH2 reduced (p<0.05) OC cell proliferation (Fig. 2A) and migration (Fig. 2B). Next, tumor-forming capacity of Wt-EZH2 and T372E-EZH2 was measured using OC cells injected subcutaneously in Balb c nu/nu mice. Tumor volume was greater (p<0.05) in OC cells expressing Wt-EZH2 compared to control OC cells, and compared to Wt-EZH2, phosphomimetic T372E-EZH2 inhibited (p<0.05) ovarian tumor size (Fig. 2C). Having observed an anti-tumor effect of pT372, we measured pT372-EZH2 levels in normal ovarian surface epithelium (NOSE), human embryonic kidney (HEK293) cells as well as primary high-grade serous ovarian tumor samples. Expression of both Wt- and pT372-EZH2 was observed in NOSE and HEK293 cells, but only Wt-EZH2 and oncogenic pT345 EZH212, 13 expression was detected in primary ovarian tumors, and expression of pT372-EZH2 in these primary tumor samples was very low (Fig. 2D, E; pT345/pT372 ratio; Supplemental Fig. 3A (Tumors 5-9)).
Figure 2. Effect of T372 phosphorylation on proliferation, migration, and tumor formation.
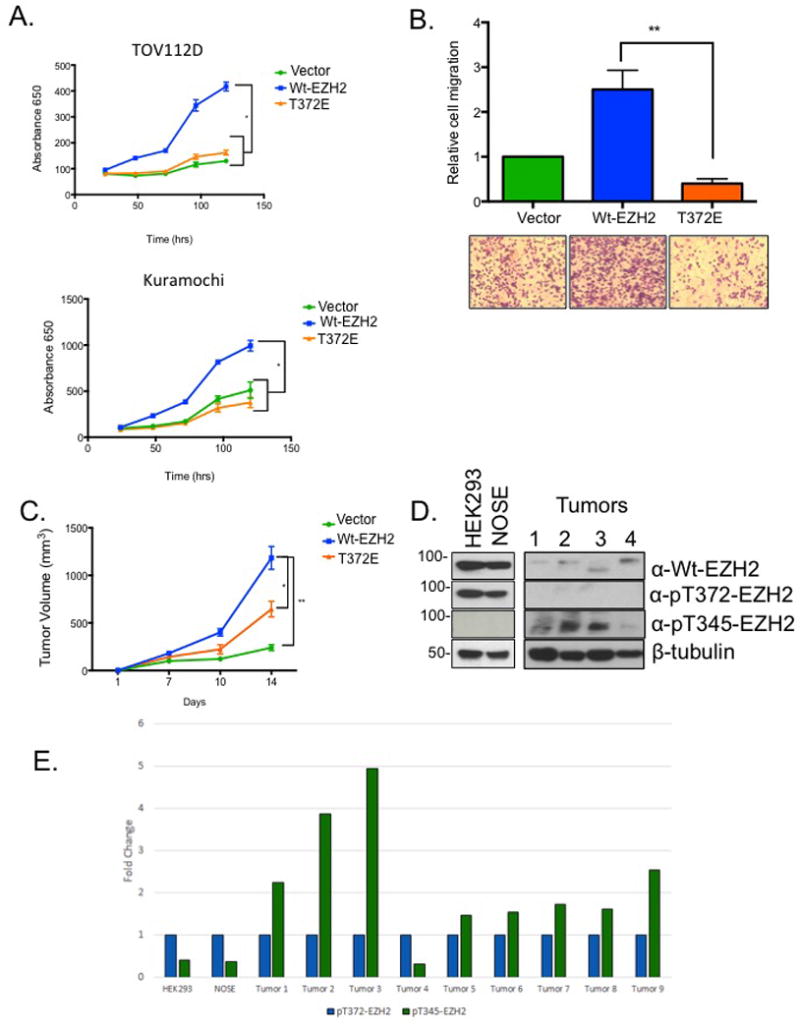
(A) TOV112D and Kuramochi cells were reverse transfected with either GFP, Wt-EZH2, or EZH2-T372E and proliferation was measured at indicated times with MTT assay (B) TOV112D cells expressing GFP, Wt-EZH2, or EZH2-T372E were seeded into migration chambers and cell migration was analyzed 16 hrs later. (C) Ovarian cancer cells expressing FLAG, FLAG-EZH2, and FLAG-T372E were injected subcutaneously into nude mice (2 × 106 cells per mouse). Tumor volume was measured 4 weeks post injection. (D) Western blot showing endogenous levels of EZH2, pT372, pT345 and B-Tubulin in normal cells (NOSE, and HEK293) as well as patient high grade serous ovarian tumor samples (Tumors 1-4). Representative data of at least three biological experiments unless otherwise indicated. (E) Results of densitometry illustrating the relative levels pT345-EZH2 to pT372-EZH2 in normal cells (NOSE, and HEK293) and patient tumor samples (high grade serous ovarian cancers 1-9).
T372 phosphorylation alters EZH2-mediated global mitochondrial gene expression and mitochondrial function
To begin to examine the mechanism underlying the effect of pT372-EZH2 on OC cell proliferation and migration, we examined global gene expression changes of OC cells ectopically expressing Wt-, T372E- or T372A-EZH2 or GFP using RNA-sequencing. Overall gene expression patterns were markedly altered by expression of T372E-EZH2 compared to controls (Fig. 3A; Supplemental Tables S2-S5). Moreover, expression of genes associated with oxidative phosphorylation (OxPhos), mitochondrial dysfunction, and DNA damage checkpoint response genes were significantly (FDR<0.05) altered by T372E-EZH2 (Fig. 3B; Supplemental Tables S7-S11). Compared to T372A-EZH2, T372E-EZH2 reduced expression of mitochondrial genes associated with normal mitochondrial function (e.g., MT-ND4, 5, 6; CO1, 2, 3 and ATP6, 8) and interleukin 6 receptor (IL6R), a STAT3 target gene (Fig. 3C; Supplemental Table S5). Furthermore, overexpression of T372E-EZH2 reduced (P<0.05) expression of mitochondrial genes MT-CO1, MTCO3 and MTND5 compared to Wt-EZH2 in OC cells (Fig. 3D). Next, we examined the relationship between T372 phosphorylation and mitochondrial function by ectopically expressing T372E-EZH2 in OC cells. We observed reduced protein levels of mitochondrial gene MT-ATP8 and Hypoxia Inducible Factor 1-alpha (HIF-1α) compared to vector and Wt-EZH2 controls, whereas expression of mitochondrial transcription factor A (TFAM) remained unchanged (Fig 3E). To examine mitochondrial function, we measured total intracellular ATP in OC cells overexpressing EZH2-T372E and observed reduced (p<0.05) ATP levels compared to Wt-EZH2 (Fig. 3F). Furthermore, the observed effects on mitochondrial gene expression appeared to be indirect, as EZH2 localization to mitochondria was not detected by western blotting or immunofluorescence (data not shown). These results suggested that pT372-EZH2 altered expression of mitochondrial genes and consequently mitochondria biogenesis and cellular energetics.
Figure 3. Effect of EZH2-T372 phosphorylation on gene expression.
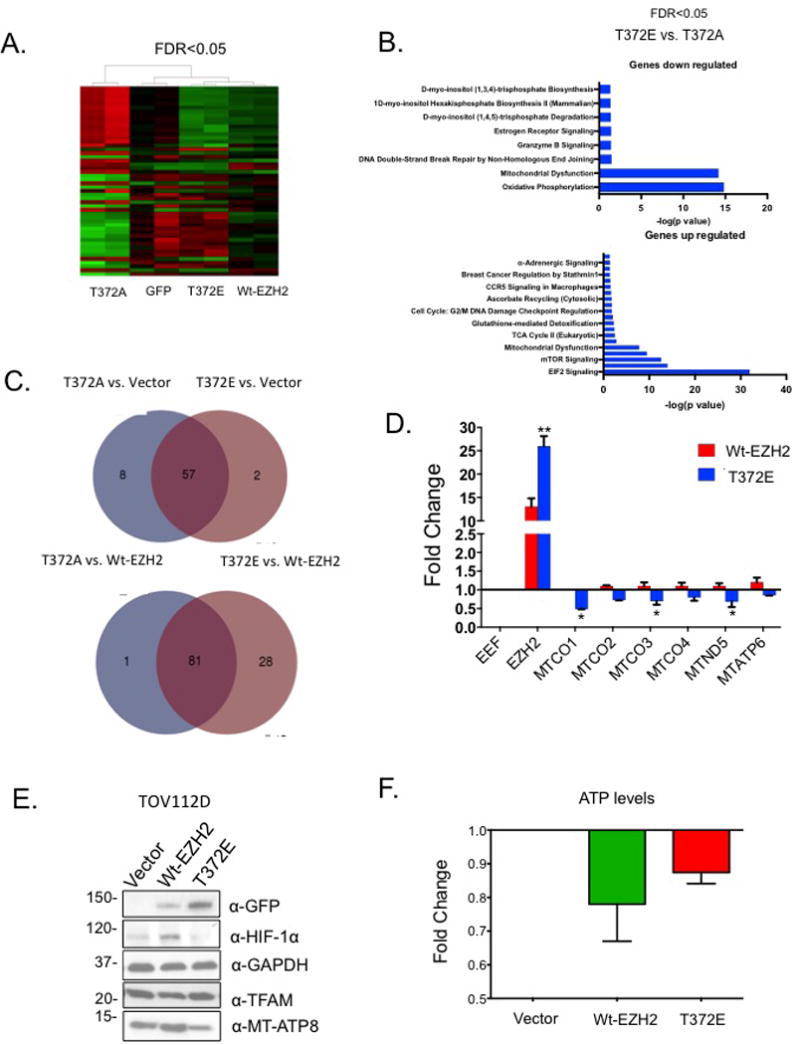
(A) Heatmap of TOV112D cells overexpressing GFP, Wt-EZH2, EZH2-T372A or EZH2-T372E. Red and green represent upregulation and downregulation of genes (FDR<0.05; RNA-seq data). (B) RNA-sequencing analysis of T372A vs. T372E and corresponding Ingenuity® Pathway Analysis. (C) Overlap of gene expression changes in EZH2 mutants vs. vector cells or EZH2 mutants vs. Wt-EZH2. The overlapping regions represent the same genes that are either up-or-down regulated. (D) TOV112D cells were transfected with GFP-EZH2, or GFP-T372E. 48 hrs post-transfection, RNA was isolated, and changes in mitcohondrial gene expression were measured using qRT-PCR. (E) Western blot analysis of MT-ATP8 and HIF-1α in TOV112D cells transfected with GFP, Wt-EZH2 or T372E. (F) TOV112D cells transfected with GFP, Wt-EZH2 or mutant and 48 hrs later intracellular ATP was measured. Representative data of at least three biological experiments unless otherwise indicated.
EZH2-T372 phosphorylation alters STAT3 activation and EZH2-STAT3 interaction
Given that EZH2 directly interacts with STAT3 and stimulates STAT3 tyrosine phosphorylation and activation6, we hypothesized that EZH2 phosphorylation at T372 may alter STAT3 activation. Expression of T372E in TOV112D and Kuramochi OC cells decreased pSTAT3 (Fig. 4A), indicating that T372-phosphorylation inhibited EZH2-mediated STAT3 phosphorylation. Expression levels of known STAT3 target genes including IL6R were altered by EZH2 mutants T372A and T372E (Supplemental Table S2), and IL-6 secretion by OC cells was reduced (P<0.05) by ectopic expression of Wt- and T372E-EZH2 (Fig. 4B). Levels of H3K27me3 on EZH2 target genes (IL6, EPC2, MYC, NANOG, and SMYD3) were reduced by ectopic expression of T372E-EZH2 (Fig. 4C), further supporting the observation of decreased binding of mutant T372E-EZH2 to target sites in the genome. In addition, EZH2 inhibitor GSK126 or FSK reduced IL6R levels in OC cells (Fig 4C) while increasing pSTAT3, suggesting that reduced IL6R expression was not due to the histone methyltransferase (HMT) activity of EZH2 nor was it due to increased STAT3 phosphorylation. Interestingly, total H3K27me3 decreased after serum starvation but increased post-FSK treatment (Fig. 4D), suggesting activation of PKA (indicated by pCREB induction) increased H3K27me3 even under serum starvation, a phenomenon that needs further exploration but is beyond the scope of this study.
Figure 4. T372 phosphorylation alters STAT3 and mitochondria activation.
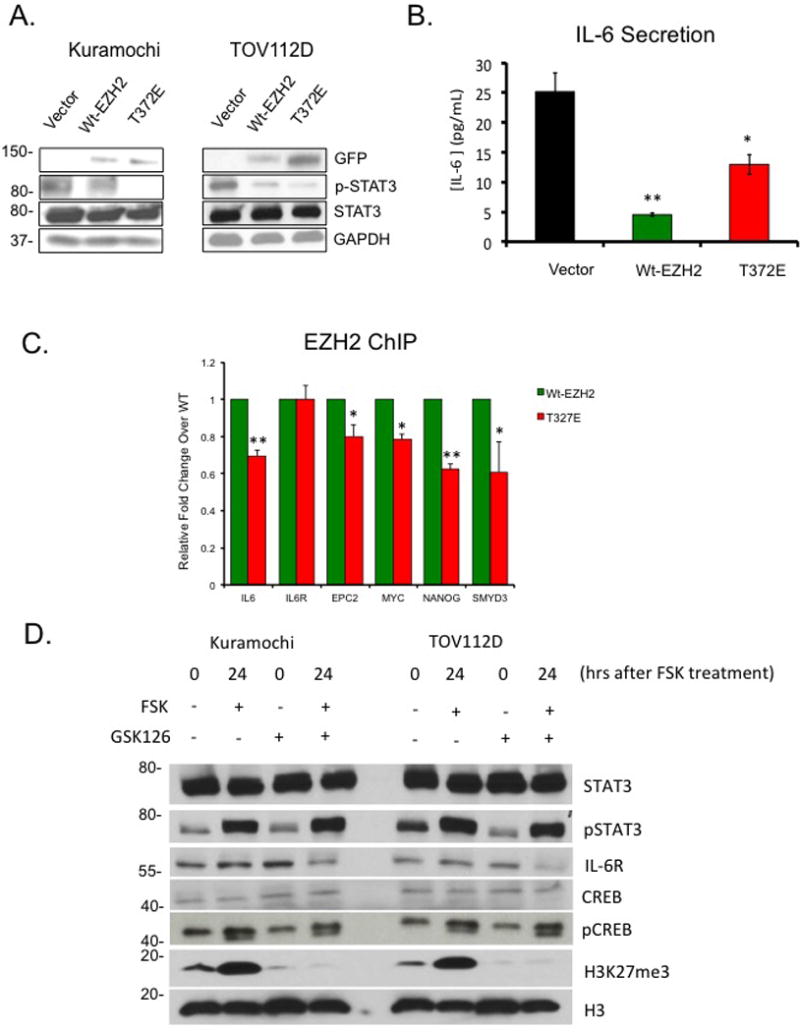
(A) Western blot analysis of STAT3 and pSTAT3 in TOV112D and Kuramochi cells transfected with GFP, Wt-EZH2, or T372E. (B) IL-6 ELISA in TOV112D cells transfected with GFP, Wt-EZH2 or T372E. (C) Chromatin immunoprecipitation (ChIP) analysis of H3K27me3 enrichment at IL6, IL6-R, EPC2, MYC, NANOG, and SMYD3 loci in TOV112D cells overexpressing Wt-EZH2 or T372E-EZH2. Quantification is representative of three independent experiments. (D) Western blot analysis of CREB, pCREB, H3K27me3, H3, IL6-R, STAT3, and pSTAT3 in TOV112D and Kuramochi cells serum starved and then treated with and without GSK126 (10μM) and FSK (10μM). Representative data of at least three biological experiments unless otherwise indicated.
In order to detect the extent of phosphorylation in a cellular setting, we measured endogenous pT372 levels in a panel of OC cell lines compared to NOSE and HEK293 cells. Low pT372 expression relative to total Wt-EZH2 was observed in most of the OC cell lines (except Kuramochi) compared to NOSE and HEK293 cells (Fig. 5A), similar to the very low levels observed in the patient primary tumors (Fig. 2D). pSTAT3 levels were higher in all cell lines compared to pT372-EZH2 relative to NOSE cells as a control (Fig. 5A), demonstrating an inverse relationship of this mark and the malignant phenotype with STAT3 activation. As STAT3-EZH2 interaction was shown to increase STAT3 methylation and phosphorylation6, we observed increased STAT3-EZH2 (Fig. 5B), demonstrating that T372 modification alters EZH2 binding to STAT3. In addition, a significant decrease (approximately 50%) in total H3K27me3 levels was seen in response to ectopic expression of either Wt-EZH2 or T372E-EZH2 in TOV112D cells (Fig. 5C).
Figure 5. T372 phosphorylation corresponds to non-oncogenic cells and tissues.
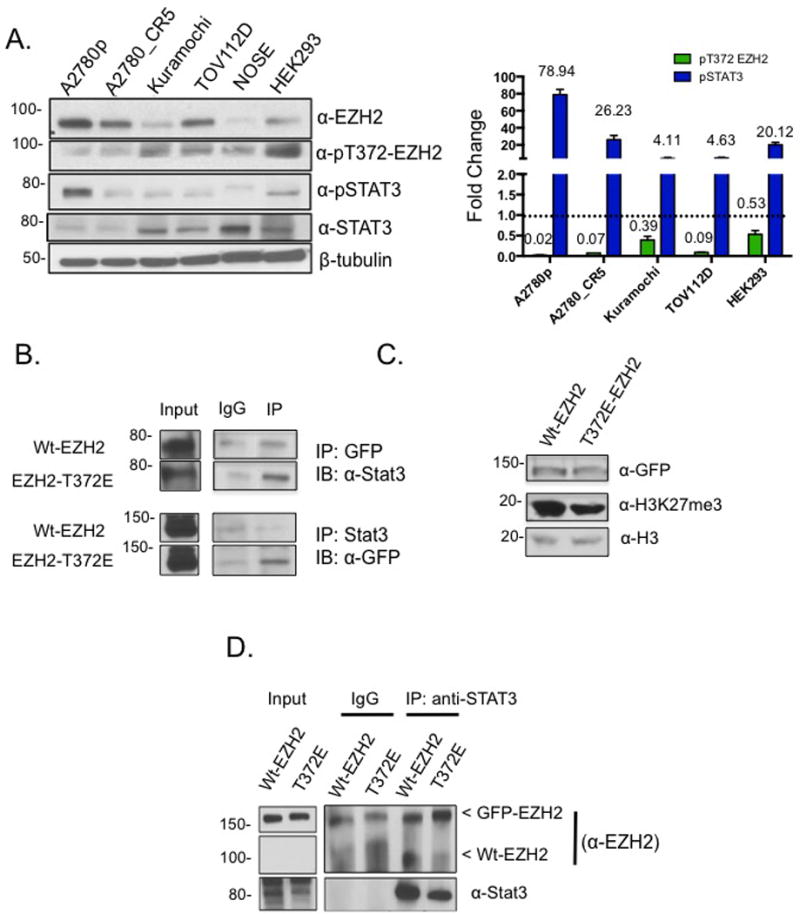
(A) Western blot and densitometry showing endogenous levels of EZH2, pT372 EZH2, STAT3, pSTAT3 in ovarian cancer cell lines and normal cells (NOSE, normal ovarian surface epithelium; HEK293). Relative densitometry was analyzed by dividing pT372-EZH2 by Wt-EZH2, and pSTAT3 by Wt-STAT3. (B) GFP and STAT3 IP from TOV112D cells expressing Wt-EZH2 and T372E-EZH2 and blotted with anti-EZH2 and anti-STAT3 antibodies. (C) Western blot showing levels of H3K27me3 in TOV112D cells overexpressing either Wt-EZH2 or EZH2-T372. (D) GFP and STAT3 IP from TOV112D cells expressing Wt-EZH2 and T372E and blotted with anti-EZH2 and anti-STAT3 antibodies (western blots were cropped due to different exposure times between IP and Input). One gel represents two forms of EZH2, GFP-EZH2 (~150kDa) that is ectopically expressed and the endogenous EZH2 (~85kDa). Representative data of at least three biological experiments unless otherwise indicated.
To test the hypothesis that increasing T372E-EZH2-STAT3 interaction resulted in tight binding and competition for endogenous STAT3, serving as a mode of inhibiting STAT3 activation, GFP was immunoprecipitated from TOV112D cells expressing Wt-EZH2 and T372E-EZH2. We observed decreased endogenous EZH2 and increased GFP-tagged EZH2 (Fig. 5D), demonstrating that T372E-EZH2 competed for endogenous cellular levels of STAT3.
Discussion
Aberrant EZH2 expression and activity correlates with aggressive cancers24, including OC24, and targeting EZH2 or modified EZH2 has recently been shown as a viable therapeutic strategy in cancer24, 25. However, as secondary EZH2 mutations in cancer have been reported24, 26, a better understanding of EZH2, its interacting partners and novel post-translational modifications of EZH2 are needed in order for the clinical potential of EZH2-based therapy to be fully realized. Here we report that PKA interacts with and phosphorylates EZH2 on T372, resulting in mitochondrial dysfunction and attenuating EZH2 tumor promoting activity. The mechanism underlying this observation includes an altered EZH2-STAT3 signaling network by STAT3 activation and altered mitochondrial gene expression and cellular energetics. Our results in OC cells extend a recent observation on tumor suppressive function for EZH2 via cell fate switching observed in glioblastoma cells to an undifferentiated stem cell-like27, methylating NOTCH 1 target genes in T-cell acute lymphoblastic leukemia28 and more recently in OC cells29.
Post-translational modifications that alter EZH2 activity and inhibit the interaction of EZH2 with subunits of the PRC2 complex and other signaling enzymes30 have been demonstrated. Phosphorylation of EZH2 on S21 by AKT has been shown to reduce EZH2 methyltransferase activity10 by reducing EZH2s binding to H3, which further correlates with reduced oncogenic properties of breast cancer cells. In addition, T345 phosphorylation of EZH2 by CDK1 and CDK2 drives an oncogenic phenotype that includes increased cancer cell proliferation, migration and spheroid formation12. EZH2-P-T345 has also been shown to enhance EZH2 interaction with the hox intergenic long non-coding RNA (HOTAIR)13, and we have shown that targeting EZH2 and HOTAIR contributes to synthetic lethality in OC31. Here we report an inverse relationship between T372 phosphorylation and OC, with high levels of P-T372 in non-cancer cell lines and low (or absent) P-T372 levels in OC cell lines as well as primary tumor samples (Figs. 2D and 5A). Similarly to AKT phosphorylation that alters EZH2 binding to H3, we hypothesize that PKA phosphorylation of T372-EZH2 alters the binding kinetics of EZH2, based on its ability to compete for STAT3 binding compared to Wt-EZH2 (Fig. 5D).
Recently, p38 was reported to phosphorylate T372-EZH2 during myogenic differentiation, by repressing Pax7 and allowing for satellite cell differentiation11. As p38 MAPK is activated by inflammatory cytokines11, p38-mediated phosphorylation of EZH2 reveals an important regulatory role for EZH2 in response to cytokines and stress inducing conditions. PKA normally is activated by adenylate cyclase (AC) which can be activated by G-Protein binding ligands like adrenaline, and epinephrine. In our work, we artificially activate PKA using forskolin, an activator of AC. Here we show that activation of AC increases intracellular cAMP that activates PKA-EZH2 axis altering EZH3-STAT3 interaction and their target genes. cAMP plays an important regulatory role across many diseases, and our model (Supplemental Fig. S4) includes tumor suppressor activity due to phosphorylation of EZH2 by PKA.
As a function for PKA in cellular energetics and OxPhos in cancer has been reported32, our findings support a new mechanism linking PKA to EZH2 and STAT3 in regulating IL6-R levels and mitochondrial gene expression and activity (Supplemental Fig. S4), extending PKA’s role in regulating mitochondrial function33, 34. Moreover, changes in IL6-R expression appear to be independent of HMT activity, as co-inhibition of EZH2 and induction of PKA activity reduced IL6-R levels (Fig. 4E), agreeing with previous findings linking P-T372-EZH2 to inflammation11. It is worth noting that PKA-EZH2 interaction could regulate basic mitochondrial functions in primary cells, as upregulation of PKA activity has been observed in primary cultures of myocytes35, hepatocytes36, and adipocytes37. In addition, coupling of EZH2 activity and mitochondrial function and cellular energetics has been recently reported in immune responses38, and our findings link EZH2 phosphorylation on T372 with mitochondrial activity in cancer cells.
Based on our findings, we suggest a role for T372 phosphorylation in “fine-tuning” the oncogenic properties of EZH2 in cancer and a new mechanism for the regulation of intracellular ATP by the EZH2-PKA-STAT3 axis. By reducing mitochondrial gene expression, non-canonical roles of EZH2, independent of H3K27me34, may contribute to the newly described tumor suppressor properties of EZH227, 39. Furthermore, targeting non-canonical EZH2 interactions, such as EZH2-STAT3 in tumors with elevated PKA activity or increased PT372-EZH2 might be a novel therapeutic strategy in OC and other cancers.
Materials and Methods
Cell lines, patient tumors, culture conditions and reagents
Epithelial ovarian cancer cell lines Kuramochi, TOV112D and A2780 were authenticated by ATCC and determined to be free of any mycoplasma contamination. Epithelial ovarian carcinomas (stage 3-4, high grade serous) were obtained from the Indiana University Simon Cancer Center tumor bank according to institutional guidelines. Cisplatin (CDDP) was purchased from Calbiochem (Billerica, MA), etoposide (ETOP), forskolin (FSK) and H89 were purchased from Santa Cruz Biotech. (Santa Cruz, CA). The EZH2 inhibitor (EZH2i) GSK126 was purchased from Biovision Inc. (Milipitas, CA).
Cell proliferation and cell migration assays
The quantity of viable cells was calculated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described40. Cells were assessed in Boyden chamber assays as described in Supplemental Materials and Methods.
Transfection and survival assays
Expression vectors (300ng) were transfected using Turbofect transfection reagent (Thermo scientific). Cells were then treated with indicated concentrations of CDDP or ETOP for 3 hrs, and fresh media was added and indicated assays were performed as we have described41.
Caspase 3/7 cleavage, ATP, cAMP, and ROS assays
2 × 104 cells were reverse transfected with indicated vectors and cAMP and ATP (Promega, Cat # G8090), and ROS (Promega, Cat # G8090) was detected with a luminometer as described previously40 and see Supplemental Materials and Methods.
Mouse xenograft experiments
All animal studies adhered to ethical regulations and protocols approved by the Institutional Animal Care and Use Committee of Indiana University. To assess tumorigenicity of cells, cultured A2780 OC cells expressing indicated expression constructs were washed with 1x PBS trypsinized and counted with trypan blue, re-suspended in 1:1 PBS/matrigel (BD Bioscience) and 2×106 cells were injected subcutaneously into the left flank of 3- to 4- week-old female nude athymic mice (BALB/c-nu/nu; Harlan, Indianapolis, IN), as described40. Engrafted mice (n=6 per group) were inspected three times per week for tumor appearance by visual observation and palpation. Tumor length (l) and width (w) were measured biweekly using digital calipers and tumor volume (v) was calculated as v=½ × l × w2 as described26. No randomization was used and no animals were excluded from the final data. The investigator measuring tumor size was blinded to the treatment groups. Mice were sacrificed when tumor diameter reached 2 cm or at the end of study.
Immunoprecipitation assays
Cells were grown to 80-90% confluence in a 10cm culture plate and transfected with 15 μg of indicated expression vectors, lysed 48 hrs later, and immunoprecipitated with indicated antibodies overnight at 4°C (See Supplemental Table S1 and Supplemental Materials and Methods).
Chromatin Immunoprecipitation (ChIP) assays
TOV112D cells ectopically expressing either Wt-EZH2 or T372E-EZH2 were cross-linked (1% formaldehyde) and dynabeads (Life Technologies) coupled to the anti-H3K27me3 antibody was used to immunoprecipitate sheared chromatin. After reversing the crosslinks, DNA was purified, and standard curve of ChIP input DNA was prepared. Enrichment was calculated by using qPCR to compare the level of the target region in each sample to the mean of negative control genomic regions. Primers designed for the specified genomic regions (Integrated DNA Technologies, Coralville, Iowa) amplified a single product from input DNA based on a single melting peak and the sequences can be found in (Supplemental Table S1). Each ChIP DNA sample was assayed for the levels of negative control regions.
Luciferase assays
Luciferase activity was analyzed using the Dual Luciferase Reporter Assay System (Promega, Madison, WI) as previously described40 and see Supplemental Materials and Methods.
RNA extraction and quantitative RT-PCR (qPCR)
RNA was extracted using RNeasy kit (Qiagen, Venlo, Limburg), cDNA was prepared using MMLV RT system (Promega), and qPCR) was performed with primers for indicated genes and EEF1A as the endogenous control (Supplemental Table S1) as we have described42.
Immunoblot analysis
Total cell lysate was prepared with RIPA lysis buffer and blotting was performed as described previously43 using antibodies listed (Supplemental Table S1).
Recombinant protein expression and mutagenesis
The GST-tagged EZH2 fragments (amino-acid residues 1-173, 173-340,340-550, 550-741) cloned into pGEX-4T-1 vector (GE Healthcare) were kindly provided by Dr. H. Huang (University of Minnesota). Mutagenesis of GFP and FLAG tagged EZH2 constructs were generated using Quickchange II mutagenesis kit (Agilent Technologies, Cat# 200521) using primers listed on (Supplemental Table S1).
RNA-sequencing analysis
Stranded whole transcriptome RNA-seq was performed as we have described40. Illumina NextSeq 500 at the Indiana University Center for Genomics and Bioinformatics. Demultiplexing was performed by CASAVA v1.8.2 and trimming was accomplished with Trimmomatic v0.22 with additional trimming by fastx_clipper v0.0.13.2. Read mapping was performed by tophat2 v2.0.6 to the human genome hg19 (UCSC) with Gencode annotation v13 allowing no more than two mismatches. Pathway analysis was performed using Ingenuity Pathway Analysis (Qiagen).
Monoclonal antibody generation
Monoclonal antibody against phosphorylated EZH2 at the Thr 372 residue was raised by immunizing mice with phosphorylated (GRLPNNSSRPS[Tp]PTINVLESKDT) or unphosphorylated (GRLPNNSSRPSTPTINVLESKDT) human EZH2 peptide (Thermo scientific). A detailed protocol for selecting positive clones can be found in Supplemental Materials and Methods.
Statistical analysis
All data are presented as mean values ± SD of at least three biological experiments unless otherwise indicated. IC50 values for CDDP were determined by Prism 6 (GraphPad Software, San Diego, CA), using logarithm normalized sigmoidal dose curve fitting.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Fang Fang and Jay Pilrose for assistance with animal studies, Dr. Scott Michaels, Dr. Doug Rusch and Aaron Buechlein (Center for Genomics and Bioinformatics, Indiana University) for RNA-seq data analysis, Dr. Haojie Huang for EZH2-pGEX-4T-1 vectors (University of Minnesota) and Dr. Jian Jian (UC Davis) for PGL2-3x-SIEGAS STAT3 reporter plasmid. We thank Dr. Heather O’Hagan and Dr. Peter Hollenorst (Medical Sciences, Indiana University School of Medicine) for critically reading the manuscript and offering valuable suggestions. This work was made possible by funding from the National Cancer Institute (Awards CA13001 and CA182832), The V Foundation for Cancer Research Translational Grant, Walther Cancer Foundation (Indianapolis, IN), and the Doane and Eunice Dahl Wright Fellowship (Medical Sciences Program, Indiana University).
Footnotes
Disclosure of Potential Conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nature Reviews Cancer. 2016 doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- 2.Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Viré E, et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer cell. 2007;11:513–525. doi: 10.1016/j.ccr.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RM, et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Molecular cell. 2011;43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes & development. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, et al. BRCA1 is a negative modulator of the PRC2 complex. The EMBO journal. 2013;32:1584–1597. doi: 10.1038/emboj.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes & development. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-[kappa] B. Nature medicine. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cha T-L, Zhou BP, Xia W, Wu Y, Yang C-C, Chen C-T, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 11.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, et al. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell stem cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nature cell biology. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaneko S, Li G, Son J, Xu C-F, Margueron R, Neubert TA, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes & development. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koppens M, Van Lohuizen M. Context-dependent actions of Polycomb repressors in cancer. Oncogene. 2016;35:1341–1352. doi: 10.1038/onc.2015.195. [DOI] [PubMed] [Google Scholar]
- 15.Copeland R, Moyer M, Richon V. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene. 2013;32:939–946. doi: 10.1038/onc.2012.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RG, Barzi A, et al. Colorectal cancer statistics, 2017. CA: a cancer journal for clinicians. 2017;67:177–193. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 17.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, et al. Regulation of tumor angiogenesis by EZH2. Cancer cell. 2010;18:185–197. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer research. 2006;66:5582–5591. doi: 10.1158/0008-5472.CAN-05-3575. [DOI] [PubMed] [Google Scholar]
- 19.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer biology & therapy. 2010;10:788–795. doi: 10.4161/cbt.10.8.12913. [DOI] [PubMed] [Google Scholar]
- 20.Garipov A, Li H, Bitler BG, Thapa RJ, Balachandran S, Zhang R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Molecular Cancer Research. 2013;11:360–369. doi: 10.1158/1541-7786.MCR-12-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Cai KQ, Godwin AK, Zhang R. Enhancer of zeste homolog 2 (EZH2) promotes the proliferation and invasion of epithelial ovarian cancer cells. Molecular cancer research. 2010 doi: 10.1158/1541-7786.MCR-10-0398. molcanres.0398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Hou L, Huang Y. EZH2-specific microRNA-98 inhibits human ovarian cancer stem cell proliferation via regulating the pRb-E2F pathway. Tumor Biology. 2014;35:7239. doi: 10.1007/s13277-014-1950-9. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto Y, Ning G, Howitt BE, Mehra K, Wu L, Wang X, et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. The Journal of pathology. 2016;238:519–530. doi: 10.1002/path.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nature medicine. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nature medicine. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2015 doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vries NA, Hulsman D, Akhtar W, de Jong J, Miles DC, Blom M, et al. Prolonged Ezh2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell reports. 2015;10:383–397. doi: 10.1016/j.celrep.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 28.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic Inactivation of the PRC2 Complex in T-Cell Acute Lymphoblastic Leukemia. Nature medicine. 2012;18:298. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardenas H, Zhao J, Vieth E, Nephew KP, Matei D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget. 2016;7:84453–84467. doi: 10.18632/oncotarget.11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell reports. 2014;8:204–216. doi: 10.1016/j.celrep.2014.05.045. [DOI] [PubMed] [Google Scholar]
- 31.Özeş AR, Wang Y, Zong X, Fang F, Pilrose J, Nephew KP. Therapeutic targeting using tumor specific peptides inhibits long non-coding RNA HOTAIR activity in ovarian and breast cancer. Scientific Reports. 2017;7 doi: 10.1038/s41598-017-00966-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papa S, Rasmo DD, Technikova-Dobrova Z, Panelli D, Signorile A, Scacco S, et al. Respiratory chain complex I, a main regulatory target of the cAMP/PKA pathway is defective in different human diseases. FEBS letters. 2012;586:568–577. doi: 10.1016/j.febslet.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 33.Feliciello A, Gottesman ME, Avvedimento EV. cAMP-PKA signaling to the mitochondria: protein scaffolds, mRNA and phosphatases. Cellular signalling. 2005;17:279–287. doi: 10.1016/j.cellsig.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 34.García-Bermúdez J, Sánchez-Aragó M, Soldevilla B, del Arco A, Nuevo-Tapioles C, Cuezva JM. PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H+-ATP synthase. Cell reports. 2015;12:2143–2155. doi: 10.1016/j.celrep.2015.08.052. [DOI] [PubMed] [Google Scholar]
- 35.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 36.Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nature cell biology. 2011;13:668–675. doi: 10.1038/ncb2228. [DOI] [PubMed] [Google Scholar]
- 37.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, et al. PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. The EMBO journal. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Sheng C, Liu D, Yao C, Gao S, Song L, et al. Enhancer of zeste homolog 2 is a negative regulator of mitochondria-mediated innate immune responses. The Journal of Immunology. 2013;191:2614–2623. doi: 10.4049/jimmunol.1203143. [DOI] [PubMed] [Google Scholar]
- 39.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nature medicine. 2012;18:298–303. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Özeş A, Miller D, Özeş O, Fang F, Liu Y, Matei D, et al. NF-κB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene. 2016 doi: 10.1038/onc.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72:2197–2205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic Targeting of Ovarian Cancer Stem Cells. Cancer research. 2014;74:4922–4936. doi: 10.1158/0008-5472.CAN-14-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2010;30:1082–1097. doi: 10.1038/onc.2010.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.