

Abstract
Innate lymphoid cells (ILCs) comprise a family of innate immune cells that orchestrate mucosal immune responses: initiating, sustaining, and even curbing immune responses. ILCs are relatively rare (≤1% of lymphocytes in mucosal tissues), lack classical cell-surface markers, and can be divided into 3 subsets (type 1–3 ILCs) based on differences in cytokine production, phenotype, and developmental pathway. Because ILCs can only be identified by combinations of cell surface markers and cytokine production, multicolor flow cytometry is the most reliable method to purify, characterize, and assess the functionality of ILCs. Here, we describe the methods for cell preparation, flow cytometric analysis, and purification of murine ILCs from the lung.
Keywords: Innate lymphoid cells, Respiratory tract, Isolation, Identification
1. Introduction
Regulation of innate immunity at barrier surfaces (e.g. lung, GI tract, skin) is critical to preserving host integrity, thereby preventing inappropriate immune activation and pathology. The immune system must constantly survey its surroundings and discriminate between harmless and potentially harmful materials—a particularly complex task in the mucosa, which is exposed to millions of exogenous stimuli on a daily basis. There, an intricate network of cellular and molecular pathways is employed, which allows the immune system to respond quickly and efficiently to harmful stimuli, while largely ignoring innocuous materials.
Over the last decade, innate lymphoid cells (ILCs) have emerged as a family of hematopoietic effectors and regulators of mucosal immunity [1–3]. These cells bear a strong resemblance to T helper (Th) Th1, Th2, and Th17 cell subsets but lack rearranged, antigen-specific receptors [1–3]. Although they populate nearly every tissue examined, ILCs are preferentially found in mucosal sites [4–6], where they exist as tissue-resident cells that may expand locally during acute inflammation [7]. Additionally, ILC progenitors in the bone marrow can reconstitute the ILC compartment and likely contribute to the renewal of ILCs during chronic inflammation [8].
ILCs guide the reactions of local hematopoietic cells and non-hematopoietic stromal and epithelial cells via production of and responsiveness to an array of soluble mediators, including cytokines and eicosanoids. ILCs function in diverse physiological processes including the organization of lymphoid structures, immune cell recruitment, maintenance of tissue homeostasis, pathogen resistance, metabolic homeostasis, and anti-tumor immunity [6,9–11,1,12–15,5]. Accordingly, deviations in the development, fate or function of ILCs by environmental toxicants may result in immune dysregulation with profound consequences for the host. Because of these important roles and conceivably life-threatening outcomes, it is necessary to consider adverse effects on ILCs when evaluating an agent for immunotoxicity. However, the low relative abundance of ILCs in mouse and human tissues may be a considerable obstacle when cells and/or tissues for immunotoxicity testing are limited.
ILCs are relatively rare [16,11,17], lack classical cell-surface markers, and can be divided into 3 subsets (type 1–3 ILCs) based on differences in cytokine production, phenotype, and developmental pathway [1,18–20,9,10,21–24] (Table 1). Type 1 ILCs, represented by ILC1s and conventional NK (cNK) cells, produce interferon (IFN)-γ, although cNK cells are also capable of producing granzyme and perforin. These two cells can be differentiated by expression of transcription factors (ILC1s are T-bet+Eomes−, while cNK cells are T-bet+Eomes+) and specific surface markers (ILC1s are NK1.1+NKp46+CD49a+CD49b−TRAIL+CD69+CXCR3+CXCR6+, while cNK cells are NK1.1+NKp46+CD49a−CD49b+TRAIL−CD69−CXCR3−CXCR6−). While cNK cells act as the primary cytotoxic lymphoid cells of the immune system, to date, ILC1s appear to function in support of cNK cell activities [25,22]. Therefore, group 1 ILCs contribute to anti-viral immunity and activation of Th1 cells. In contrast, type 2 ILCs seem to be fairly homogenous: ILC2s produce Th2 cell-associated cytokines (IL-4, IL-5, IL-9 and IL-13) in response to stimulation with the cytokines IL-7, IL-25, IL-33, and TSLP, and express several conserved surface markers (IL-7Ra, CD25, ST2, Sca-1, and KLRG1) and transcription factors (GATA-3, RORα, TCF-1, and Notch) that mediate their particular differentiation program and functions [5,4,13]. Therefore, ILC2s can contribute to clearance of infections with extracellular Helminths and can be involved in allergic inflammatory diseases, such as asthma and atopic dermatitis [26]. Type 3 ILCs (comprised of ILC3s and lymphoid tissue inducer (LTi) cells) produce IL-17A and/or IL-22 and share expression of the transcription factor RORγt, but exhibit differential expression of T-bet [1,18–20,25]. The typical markers of the T-bet− LTi cells are NK1.1−NKp46−CD127+CCR6+, while the T-bet+ ILC3s are NK1.1−NKp46+CD127+CCR6+/− [27]. Functionally, ILC3s have been shown to participate in the development of lymphoid tissues, homeostasis and mucosal defense, as well as maintaining memory CD4+ T cells [6,5].
Table 1. Overview of the family of ILCs.
Characteristics, localization, and function of ILC subsets. Innate lymphoid cells (ILCs) are defined by select transcription factors and their capacity to produce effector cytokines. ILCs are activated by subset-specific cytokine signals. The cytokines produced by ILCs induce important physiological responses; however, ILC-derived cytokines can also provoke immunopathology.
| ILC subset | ILC lineage | Major stimulating cytokine | Signature cytokine produced | Function | Transcription Factors | Localization | Disease associations |
|---|---|---|---|---|---|---|---|
| ILC1 | ILC1 NK cell |
IL-12 IL-15 IL-18 |
IFN-γ IFNγ, TNFα, perforin, granzymes |
Intracellular pathogens (viruses, bacteria, parasites) Viral infections, tumor surveillance |
T-bet Eomes |
Skin, intestine, lymphoid tissues, thymus, liver | Colitis, Crohn’s Crohn’s |
| ILC2 | iILC2 nILC2 |
IL-25 IL-33 TSLP |
IL-5, IL-9, IL-13, amphiregulin | Extracellular parasites, wound healing, metabolic homeostasis | RORα, Bcl-11b | Spleen, liver, lung, intestine, skin, bone, adipose tissue, brain | Asthma & Allergy, Colitis |
| ILC3 | LTi ILC17 ILC22 |
IL-1β IL-23 IL-7 |
LTα/β, IL-17, IL-22 IL-17, IFNγ IL-22 |
Lymphoid tissue formation & repair, bacterial & fungal infections Extracellular bacterial & fungal infections, autoimmunity Homeostasis of epithelial immunity |
RORγt, AhR, Notch, RUNX1, TCF-1, GATA-3 | Lymphoid tissues, intestine, lung, skin, liver, tonsils | Colitis, COPD, ARDS, Asthma |
The functionality of ILCs is dependent on their microenvironment or local cytokine milieu. In general, murine ILCs lack pattern recognition receptors, which are broadly expressed by other innate immune cells for the detection of pathogen-associated molecular patterns. Instead, ILCs react to exogenous stimuli indirectly by sensing myeloid or epithelial cell derived cytokines, alarmins, and inflammatory mediators. In turn, ILCs promote immune responses by secreting soluble factors such as cytokines and other peptides, such as IFN and TNF (ILC1s), IL-5, IL-9, and IL-13 (ILC2s), and IL-17, IL-22, and GM-CSF (ILC3s) [6]. Although ILCs act largely by secreting soluble mediators, some molecules expressed by ILCs require cell-cell contact for effector mechanisms. At present, the regulatory and inhibitory pathways that control ILC responses are not well understood.
Improvements in the identification and isolation of ILCs has led to a recent explosion of studies, yet many questions remain surrounding their development, regulation, and function in homeostasis and disease. In this chapter, we focus on the basic methods for the identification and isolation of ILCs from the murine lung, which play key roles in regulating lung inflammation during allergic respiratory disease, influenza infection, helminth infection, chronic obstructive pulmonary disease, and other pulmonary disease states [6,5] (Figure 1). The protocols described below can be applied to total ILCs, as well as individual subsets of ILCs from the lungs of wild-type or transgenic mice.
Figure 1.

Typical workflow to isolate and identify total ILCs from the murine lung.
2. Materials
C57Bl/6 or other mouse strain expressing CD90.2 (e.g. Thy1.2)
Reagents and equipment for AVMA approved method of euthanasia
Dissection tools: alcohol wipes, dissecting pins and board, 2 pairs of fine serrated forceps, fine scissors (sharp-blunt tips), 2 pairs of fine curved hemostats (Fine Science Tools)
Single edge razor blades (VWR)
100mm sterile petri dishes
15 and 50mL conical tubes (BD Falcon)
Collagenase D (Roche)
DNAse I (Roche)
RPMI 1640 medium (Corning)
Sodium pyruvate (Atlanta Biologicals)
Fetal bovine serum (FBS, Atlanta Biologicals)
HEPES (Atlanta Biologicals)
β-mercaptoethanol (Sigma-Aldrich)
Centrifuge
Tissue Dissociation
lung digestion medium: complete RPMI (cRPMI) containing 2mg/mL collagenase D + 0.02mg/mL DNase I
cRPMI-1640: with L-glutamine, 10% FBS, 25mM HEPES, 10mM sodium pyruvate, 50μM β-mercaptoethanol, 50 μg/ml gentamicin.
70μm sterile cell filters (BD)
1mL syringes (BD)
Cell separation
Fico/Lite-LM (Mouse) (Atlanta Biologicals)
CD90.2 Enrichment
CD90.2 microbeads (Miltenyi Biotec)
MACS columns & magnets (Miltneyi Biotec)
40μm sterile cell filters (BD)
PBS (pH 7.2) containing 0.5% bovine serum albumin (BSA) and 2 mM EDTA
Multicolor flow cytometry
5mL round-bottom polystyrene test tube, with cell strainer snap cap
Multicolor flow cytometer
Compensation beads (Thermo Fischer Scientific)
Anti-CD16/CD32 blocking reagent (Tonbo Biosciences)
Directly conjugated antibodies tailored to the specific flow cytometer and previously titrated to optimize concentrations (Biolegend, BD Biosciences, eBiosciences/Affymetrix, Tonbo Biosciences)
Fixable Live-dead discriminator (Tonbo Biosciences or Thermo Fischer Scientific)
3. Methods
Isolation of murine lung & preparation of single cell suspension
This protocol describes a general method for isolating leukocytes from the murine lung. First, lung tissue is mechanically dissociated and digested in medium containing collagenase D plus DNase I. Second, the single cell suspension is subjected to Ficoll gradient centrifugation to isolate lymphocytes from red blood cells and debris. After washing, the single cell suspension is ready for flow cytometric analysis, enrichment, cell sorting, and culture, etc.
-
Euthanize mice and remove lungs from chest cavity.
Note: To ensure that blood-derived ILCs are not present in the cell preparation, lungs can be perfused with cold phosphate buffered saline prior to removal. -
Place dissected lung lobes in a petri dish on ice and mechanically dissociate tissue with sterilized razor blades or dissection scissors into fragments of tissue ~1mm in size.
Note: Tissues should be kept on ice, unless otherwise noted. Transfer tissue into a 24-well tissue culture plate containing 2mL/lung of lung digestion medium.
Incubate plate at 37°C for 30 minutes, gently agitating every 10 minutes.
Push digested tissue and supernatant through a 70μm cell strainer using the plunger end of a 1mL syringe to make a single cell suspension.
Wash cell strainer with 5–10mL of complete RPMI-1640 medium.
Centrifuge cells at 4°C for 10 minutes at 1500rpm.
Discard supernatant (pour off, do not vacuum) and re-suspend cells in 5mL cRPMI.
Transfer cell suspension to a 15mL conical.
Using a glass Pasteur pipette, slowly layer 5mL (room temperature) Fico-Lite beneath the single cell suspension.
Centrifuge cells at 4°C for 30 minutes at 2000rpm (select no brake and minimal acceleration settings on the centrifuge).
Carefully collect the cells at the interface and transfer to a 15mL conical (Figure 2).
Wash cells with 5–10mL of complete RPMI-1640 medium.
Centrifuge cells at 4°C for 10 minutes at 1500rpm.
Discard supernatant and re-suspend cells in 3mL recommended buffer for magnetic enrichment (see below) or flow cytometric analysis (see below).
Figure 2.
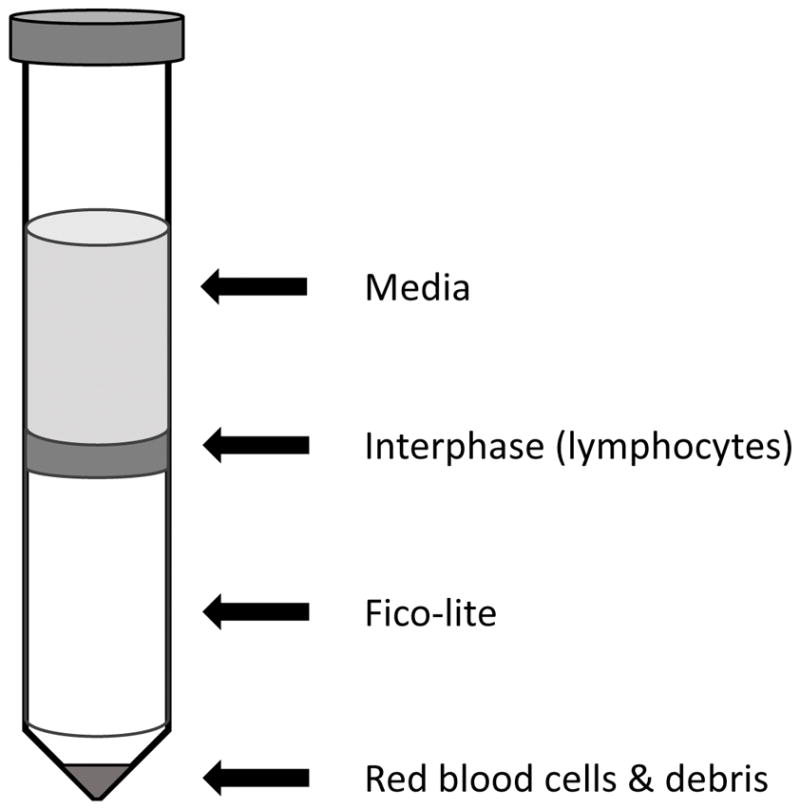
Typical aspect of the lymphocyte-rich interphase between medium and Fico-lite after centrifugation.
Magnetic enrichment of CD90.2+ cells
This protocol describes the enrichment of CD90.2+ cells from a single cell suspension of digested lung tissue. First, the CD90.2+ cells are magnetically labelled with CD90.2 microbeads. Then, the cell suspension is loaded onto a MACS column, which is placed in the magnetic field of a MACS separator. The magnetically labelled CD90.2+ cells are retained within the column allowing unlabeled CD90.2− cells to pass through. After removing the column from the magnetic field, the CD90.2+ cells can be eluted as the positively selected cell fraction. The purity of the CD90.2+ cells should be assessed prior to proceeding to flow cytometric analysis and/or cell sorting. If necessary, it is possible to increase purity and/or yield of CD90.2+ cells, by enriching the eluted CD90.2− and CD90.2+ fractions over additional magnetic columns. After washing, the single cell suspension is ready for flow cytometric analysis or sorting.
Determine total cell number
Re-suspend cells in 90μL recommended buffer (PBS/0.5% BSA/2mM EDTA) per 107 total cells. Scale up as appropriate.
Add 10μL of CD90.2 MicroBeads per 107 total cells
Mix well and incubate for 15 minutes in the dark in the refrigerator (2–8 °C).
Wash cells by adding 1–2mL of buffer per 107 cells
Centrifuge cells at 4°C for 10 minutes at 1500rpm
Discard supernatant
Re-suspend ≤ 108 cells in 500μL of buffer.
Proceed to magnetic separation with an appropriate column, based on the number of total cells and the expected number of CD90.2+ cells retained on the column.
Place column in the magnetic field and rinse with 3mL of recommended buffer.
Pass cells through a 40μm sterile filter to remove cell clumps which may clog the column.
Apply cells onto the column.
Collect the flow through (CD90.2− fraction) containing unlabeled cells.
Wash column with 3mL of recommended medium.
Collect the flow through (CD90.2− fraction) and set aside on ice.
Remove column from the magnetic separator and place it on a suitable collection tube.
Pipette 5mL of recommended buffer onto the column.
Immediately flush out the magnetically labeled cells by firmly pushing the plunger into the column.
Check the purity of CD90.2+ cells within the CD90.2− and CD90.2+ fractions to determine the efficiency of the enrichment.
To increase the purity and/or yield of CD90.2+ cells, repeat the magnetic separation procedure on the CD90.2− and CD90.2+ fractions as described above.
Determine the cell number for the CD90.2− and CD90.2+ fractions.
-
Continue to flow cytometric analysis and/or purification of ILCs (see below).
Note: ILCs may be enriched by negative selection as well, depleting labeled cells of known lineages (macrophages, dendritic cells, red blood cells, and T, B, and NK cells) using a cocktail of biotinylated antibodies as described in Table 2 and a similar microbead-based selection protocol.
Table 2. Antibody Staining Panel to identify ILCs.
This table shows a typical staining strategy to identify bulk ILCs and discriminate between ILC2s and ILC3s in the lungs of C57Bl/6 wild-type mice using a 3 laser, 10 color BD FACSAria II cytometer. FcR-blocked single cell suspensions from the lungs should be used for unstained, single stained, and fluorescence minus one (FMO) controls. An appropriate live dead discriminator (e.g. prodidium iodide (PI)) is used for all samples to exclude non-viable cells. Lineage monoclonal antibodies (mAbs) include: CD3ε, CD5, CD11c, CD19, NK1.1, Gr-1, TER119 and gd TCR. Bulk ILCs are gated from the live (PI-negative) small/non-granular (FSClowSSClow) leukocytes (CD45+) as CD90.2+CD127+CD11b−. ILC2s and ILC3s can be discriminated from one another based upon differential expression of CD25, IL-33Rα, CD4, and CCR6. mAbs should be used at their individual, optimally titrated dilution in PBS containing 2% FBS, 2mM EDTA, and 0.1% sodium azide. Note: Expression of these markers may vary by mouse strain and in response to stimuli; therefore, individual titration is highly recommended for optimal results. With minimal modifications, this staining strategy can be applied to other immune tissues. See Figure 4 for example gating strategy.
| Gate | Antibody | clone | Conjugation |
|---|---|---|---|
| Lineage negative | CD3ε | 145-2C11 | PerCP Cy5.5 |
| CD11c | N418 | PerCP Cy5.5 | |
| CD11b | M1/70 | PerCP Cy5.5 | |
| CD5 | 53-7.3 | PerCP Cy5.5 | |
| CD19 | 1D3 | PerCP Cy5.5 | |
| NK1.1 | PK136 | PerCP Cy5.5 | |
| Gr-1 | RB6-85C | PerCP Cy5.5 | |
| TER119 | TER-119 | PerCP Cy5.5 | |
| gd TCR | GL3 | PerCP Cy5.5 | |
| ILCs | CD45 | 30-F11 | APC-Cy7 |
| CD90.2 | 53-2.1 | BV510 | |
| CD127 | A7R34 | PE-Cy7 | |
| ILC2s | CD25 | PC61.5 | PE |
| IL-33Rα | DIH9 | BV421 | |
| ILC3s | CD4 | RM4-5 | FITC |
| CCR6 | 29-2L17 | AF647 |
Example of an enrichment using CD90.2 microbeads
CD90.2+ cells were isolated from a mouse lung digestion using CD90.2 microbeads, two consecutive LS columns, and a MACS separator. Cells were fluorescently stained with CD90.2 APC (Biolegend). Cell debris and dead cells were excluded from the analysis based on side scatter and live dead discriminator fluorescence (Hoechst, Thermo Fisher Scientific). The frequency of live, CD90.2+ cells was analyzed from a sample of the single cell suspension pre-enrichment (0.23% ± 0.008), as well as the CD90.2− fraction (0.03% ± 0.004) and CD90.2+ fractions after the first (4.82% ± 0.31) and second magnetic enrichment columns (45.68% ± 1.63) (Figure 3A). A representative overlay shows the relative frequency of live, CD90.2+ cells from each of the cell fractions (Figure 3B).
Figure 3.

Typical enrichment of CD90.2+ cells from lung digests. (A) The frequency of live, CD90.2+ cells was analyzed from the single cell suspension pre-enrichment, the CD90.2−fraction, and the CD90.2+ fraction after the first and second magnetic enrichment columns. (B) A representative overlay shows the frequency of CD90.2+ cells before magnetic separation (light grey line), the CD90.2− cells (dark grey line), and the CD90.2+ cells (black line).
Flow cytometry & cell sorting
This protocol describes the analysis of a single cell suspension from digested lung tissue or a single cell suspension of magnetically enriched CD90.2+ cells from the murine lung for the presence of ILCs by multicolor flow cytometry (CD90.2+ cell enrichment is not required but may be desirable). First, 3 × 106 cells are re-suspended in PBS containing 2% FBS, 2mM EDTA, and 0.1% sodium azide plus purified anti-CD16/CD32 blocking reagent. Cells are then fluorescently labelled for 30 minutes at 4°C in the dark with a panel of previously optimized, directly conjugated monoclonal antibodies. After careful washing, the single cell suspension is ready for flow cytometric analysis and/or sterile sorting. Care must be taken to include unstained, single stained, and fluorescence minus one (FMO) controls when working with rare immune cell populations. An intermediate level of flow cytometry experience is required to setup, perform compensation, and modify the suggested antibody panels to suit individual hardware configurations for the identification of ILC subsets. In contrast, an advanced level of flow cytometry experience is required to setup and sort ILCs.
Centrifuge 3 to 5 × 106 cells at 4°C for 10 minutes at 1500rpm, and discard supernatant.
Wash cells by adding 1mL of PBS per 106 cells.
Re-suspend cells in 1mL of PBS (azide and protein-free) per 106 cells.
-
While vortexing, add 1μL of appropriate live dead fixable Ghost Dye (Tonbo Biosciences) per mL of PBS.
Note: Cells labeled with Ghost Dyes are compatible with intracellular staining protocols without any loss of fluorescence intensity. Incubate cells at 4°C for 30 minutes in the dark.
-
Wash cells by adding 1mL of PBS containing 2% FBS, 2mM EDTA, and 0.1% sodium azide per 106 cells.
Note: Washing with a protein containing buffer removes unreacted live dead discriminator dye prior to staining with fluorescent antibodies. Centrifuge cells at 4°C for 10 minutes at 1500 rpm, and discard supernatant.
Wash pellet by repeating steps 6 & 7.
-
Re-suspend cells in 50μL staining buffer (PBS containing 2% FBS, 2mM EDTA, and 0.1% sodium azide plus purified anti-CD16/CD32 blocking reagent diluted 1:100) per 106 total cells and set aside on ice.
Note: Alternatively, cells may be re-suspended in 100μL of 2.4G2 hybridoma cell culture supernatant to block Fc Receptors. Prepare master mix using directly conjugated antibodies diluted in staining buffer and stain at 4°C for 30 minutes in the dark.
Wash cells by adding 1mL of staining buffer per 106 cells.
Centrifuge cells at 4°C for 10 minutes at 1500 rpm, and discard supernatant.
Wash pellet by repeating steps 11 & 12.
Re-suspend cells in 0.5mL of staining buffer and filter cells through a 35μm filter cap 5mL test tube prior to flow cytometric analysis.
-
Keep cell suspension on ice in the dark prior to analysis or cell sorting.
Note: It is highly recommended to include a viability dye when working with a single cell suspension from digested lung tissue or a single cell suspension of enriched CD90.2+ cells.Note: It is highly recommended to use compensation beads and to prepare unstained, single-stained, and fluorescence minus one (FMO) controls when analyzing ILCs.
Example gating scheme identifying bulk ILCs in the murine lung
A single cell suspension from digested lung tissue or a single cell suspension of enriched CD90.2+ cells were fluorescently labelled with a panel of directly conjugated antibodies (Table 2). Cell debris and dead cells were excluded from the analysis based on side scatter and fixable live dead discriminator fluorescence (Ghost Dye red 710, Tonbo Biosciences). Using multicolor flow cytometry, ILCs are identified based upon a combination of markers denoting live, CD45+, lineage negative cells (lacking CD3, CD5, CD11b, CD11c, CD19, Gr-1, γδTCR, TER119, and NK1.1) which express CD90.2, CD127, and CD25 [24,28–30]. Lastly, in the lung, ILC3s may be distinguished from the predominant ILC2 subset based upon differential expression levels of ST2 and CD4, as well as CCR6 (Figure 4). For cell sorting, typical yield from a single, naïve murine lung is approximately 5,000–8,000 total ILCs. Following induction of inflammation in models of allergy or infection or expansion of ILCs in situ (see below), this yield increases 2–5 fold, depending upon the stimulus.
Figure 4.

Staining strategy to identify bulk lung ILCs (CD90.2+CD45+Lineage−CD127+CD11b−). Lung ILC2s are identified as ST2+CD4−, whereas ILC3s are ST2−CD4+/-. Cells were first gated for live propidium iodide (PI) negative, singlet leukocytes. See Table 2 for antibody panel.
Complementary Protocols
A. Expansion of ILCs in situ
A major obstacle to the study of ILCs is their relative paucity compared to other lymphoid cells and plasticity between subsets [31]. ILCs react to myeloid or epithelial cell derived cytokines, alarmins, and other inflammatory mediators during injury and repair, resulting in the expansion and activation of ILCs [32,33]. Consequently, it should be possible to experimentally manipulate specific subsets of ILCs present in the lungs depending on the soluble mediator delivered. This system allows for a selective rise in ILC subsets in order to obtain sufficient quantities of cells for analysis, thus reducing the numbers of mice necessary to complete the desired experiments.
This protocol describes the intranasal (i.n.) delivery of 100ng/mouse of recombinant (rm) IL-33 or IL-1β once a day for four consecutive days to expand either ILC2s or ILC3s in vivo.
Anesthetize mice using isoflurane vaporizer system.
Remove animal from induction chamber.
Confirm the absence of reflexes on the footpad.
-
Working quickly, hold the mouse vertical by the scruff of the neck and gradually release 30μl of the inoculum into the nostrils (~15ul in each nostril) with the help of a micropipette. This should cause an increase in the breathing rate.
Note: Adjust the rate of release so as to allow the mouse to inhale the inoculum without forming bubbles. Hold the mouse in the hanging position for ~1–2 minutes until its breathing gradually returns to normal.
Place animal in recovery area and monitor until fully recovered.
On day 5 following the start of treatment, euthanize the animals and purify or analyze ILC populations as described above.
B. Depletion of ILCs in situ
Because ILCs do not express a specific lineage marker and there are currently no conventional ILC knockout mice, ILC depletion via anti-CD90 is often used in research to investigate the essential functions of ILCs [5,11]. Some criticize the use of anti-CD90.2 as a tool because anti-CD90 may elicit the depletion of other cells (e.g. T cells and neurons). Ideally, ILC research would utilize a mouse model that ensures specific deletion of individual subsets of ILCs to fully elucidate the relative contributions of ILCs to the etiology of immune-mediated diseases. When such a tool becomes available, it will be incorporated into common experimental procedures.
This protocol describes the depletion of ILCs from the murine lung using 0.5mg of anti-CD90.2 (clone 30H12) monoclonal antibody i.p. at days −3, 0 and 2.
Note: Based on the model used, monoclonal antibody treatment can continue for longer periods of time; however, mice should be carefully monitored for effects on cell types other than ILCs in this case.
Disinfect top of multi-dose vial with 70% alcohol and gauze.
Draw up, into the syringe and needle, the amount of pre-warmed solution to be administered.
-
Gently remove animal from the cage and restrain appropriately in the head-down position. Identify anatomical landmarks in order to inject into the appropriate area of the abdomen.
Note: Typically the injection site will be in the animal’s lower right quadrant of the abdomen to avoid damage to the urinary bladder, cecum and other abdominal organs.Note: Tilt the mouse with its head slightly toward the ground so that its head is lower than its hind end. This allows the abdominal viscera to shift cranially and minimize accidental puncture of abdominal organs at site of injection. -
Insert needle to the depth in which the entire bevel is within the abdominal cavity (in fat animals, almost the entire length of the needle length may need to be inserted but in smaller mice, only about 1/2 the needle length may need to be inserted).
Note: Pull back on the plunger to ensure negative pressure prior to injecting. If there is negative pressure, proceed with the injection - depress the plunger until the solution has been fully administered.Note: Do not allow the needle to move around inside the abdominal cavity. Pull the needle straight out and place the syringe/needle directly into a sharps container without recapping. Use a new needle and syringe for each animal.
-
Place the animal back into its cage and observe for any complications.
Note: The volume to be injected should be the lowest volume possible and not exceed the current AVMA recommended guidelines.Note: All substances for injection should be sterile since contamination can cause infection and irritation at the site of injection and cause clinical illness in the animals and affect research results.Note: Warm substances to room or body temperature since injection of cold substances can cause discomfort and drop in body temperature.
C. Adoptive transfer of ILCs
To investigate ILC functions in vivo, researchers can take advantage of the ability to adoptively transfer relatively pure subsets of magnetically enriched and sterile-sorted ILCs. Adoptive transfer of specific subsets of ILCs into Rag2−/−IL-2rγ−/− mice (which lack all ILCs, in addition to lacking all T and B cells) can be used to demonstrate essential functions of ILCs. Additionally, adoptive transfer of CD90.1+ ILCs into CD90.2+ hosts can be used to demonstrate trafficking and distribution of ILCs during immune responses.
Example of adoptive transfer of ILC2s promoting lung inflammation
A group of 5 CD45.1-expressing C57BL/6 mice were treated for 5 days with 300ng recombinant murine IL-33 delivered intraperitoneally for in situ expansion of ILC2s. On day 6 of treatment, the mice were euthanized and the lungs were collected. Single cell suspensions from the lungs were stained for cell sorting, and CD45.1+Lineage (CD3/ CD4/CD5/CD11b/CD11c/CD19/NK1.1)−CD90.2+CD25+CD127+IL-33R+ ILC2s were isolated. Then, 1×105 ILC2s were transferred intravenously to Rag2−/−cγ−/− CD45.2-expressing mice that lack ILC2s, and this was repeated every 7 days for a total of 4 transfers. The day after the first transfer, transferred Rag2−/−cγ−/− mice, untransferred Rag2−/−cγ−/− mice, and control Rag2−/− mice that have ILC2s were infected with 500 L3 larvae of the helminth parasite Nippostrongylus brasiliensis, which drives chronic type 2 inflammation in the lung that persists after all parasites are expelled. On day 32 following infection, type 2 inflammation was evaluated in the lung of all mice. Transferred ILC2s could be identified by flow cytometry in the Rag2−/−cγ−/− recipient lung (Figure 5A). Rag2−/− mice that have ILC2s had an infection-induced increase in levels of the type 2 cytokine IL-5 in the lung homogenate, while Rag2−/−cγ−/− mice did not mount a significant IL-5 response. In contrast, Rag2−/−cγ−/− mice that received transferred ILC2s had increased levels of the type 2 cytokine IL-5 in the lung (Figure 5B).
Figure 5. Transferred ILC2s drive type 2 inflammation in the lung of ILC2-deficient mice.

CD45.1-expressing C57BL/6 mice were treated for 5 days with 300 ng recombinant murine IL-33. On day 6, CD45.1+ Lineage(CD3/CD4/CD5/CD11b/CD11c/CD19/NK1.1)−CD90.2+CD25+ CD127+IL-33R+ ILC2s were sort-purified, and 1×105 ILC2s were transferred intravenously to Rag2−/−cγ−/− CD45.2-expressing mice every 7 days for 4 transfers. The day after the first transfer, transferred Rag2−/−cγ−/− mice, untransferred Rag2−/−cγ−/− mice, and Rag2−/− mice were infected with 500 N. brasiliensis L3 larvae. On day 32 following infection, (A) transferred ILC2s could be identified by flow cytometry in the Rag2−/−cγ−/− lung, and (B) IL-5 levels were measured by ELISA in the lung homogenate. Data are representative of 2 experiments, n = 2–4/group in each experiment; mean±sem.
4. Notes
It is important to note that the methods and gating strategies described herein cover the identification, expansion in situ, isolation and ex vivo culture of ILCs from naïve C57Bl/6 mice. The expression of intracellular cytokines and cell surface molecules on ILCs is likely to change in response to exogenous stimuli and across transgenic strains of mice. Therefore, it is imperative to not only use wild-type ILCs as a control to define the gating strategy and optimize the protocol, but to select the appropriate antibody based on the mouse strain utilized in the study. For example, anti-NK1.1, which is used to detect and exclude NK cells from mice on a C57Bl/6 background, is not expressed in mice on a Balb/c background. In this instance, DX5 would be the antibody of choice. At present, the best method for identification of ILCs remains detailed multi-color analysis of those lineage negative cells that express the lymphoid associated markers CD90.2, CD127, and/or CD25. Intracellular staining for transcription factors may then distinguish ILC2s from ILC3s. Similarly, intracellular staining for cytokines may then link ILC subsets with functional capabilities. Another method is to sort purify an ILC subset of interest, followed by ex vivo manipulation to interrogate its function.
For the basic lung digestion protocol with collagenase plus DNase, it is critically important to keep dissected tissues on ice at all times during processing, except where specifically noted otherwise. RBC lysis is recommended when fluorescently labelling ILCs from digested lung tissue immediately following harvest, although this step is not compatible with the CD90.2 magnetic enrichment. The Fico-Lite gradient is not required, though it does significantly decrease the amount of debris and epithelial cells in the lung preparations. All cells should be blocked for Fc receptor before staining.
It is extremely important to filter single cell suspensions before running flow cytometry, because clumping and excess debris are common amongst tissue-derived cell suspensions. If large amounts of debris are present, samples may require reduced flow rates when acquiring data. It is also imperative to assess cell viability, as debris and non-viable cells will make positive identification of ILCs nearly impossible.
While it is important to design the antibody staining panel to suit individual hardware configurations and target ILCs, it is perhaps more important to carefully titrate antibodies before analysis, keeping in mind spectral overlap and the need to use “bright” fluorochromes for markers that are difficult to detect.
An alternative method to identifying transcription factors and/or cytokines by intracellular staining of ILCs is to utilize transgenic mice that express fluorescent cytokine and/or transcription factors.
Lastly, similar methods can used to isolate and analyze human ILCs. However, surface markers differ somewhat between human and murine ILCs. Notably, human ILCs do not express appreciable levels of CD90 (see ref [31]).
5. Conclusions and Perspectives
Although research on ILCs and their related cytokines is still in its infancy, significant progress has been made toward elucidating their functions as regulators of immunity, inflammation, and tissue homeostasis. ILCs are present in nearly all tissues examined (albeit in very small numbers), yet substantially enriched in mucosal tissues. It is now clear that these cells play important roles in diverse physiological responses: resistance to pathogens, regulation of chronic inflammation, tissue remodeling, cancer, and promoting metabolic homeostasis [6].
ILCs do not express rearranged antigen receptors, but do exhibit a functional diversity similar to T cells. With the exception of FoxP3+ regulatory T cells, ILC counterparts have been identified for each T cell subsets (e.g. Th1, Th2, Th17/22). Moreover, ILCs express and developmentally depend upon expression of key transcription factors that control a cytokine profile similar to that of their corresponding T cell counterpart. The functionality of ILCs is dependent on their microenvironment or local cytokine milieu. Because ILCs do not express T or B cell receptors, they can be identified in the lineage negative gate when performing flow cytometric analysis. ILCs can be identified in mice by analyzing the lineage-negative cells for combinations of different cell surface molecules. The challenge in working with ILCs lies in the fact that research has not (yet) discovered a single cell surface molecule that can be used to distinguish these cells by either flow cytometry of immunohistochemistry. Therefore, the best method remains detailed multi-color analysis of those lineage negative cells that express the lymphoid associated markers CD90.2, CD127, and/or CD25 to successfully identify and isolate ILCs for further manipulation. Depending on the tissue of interest, mouse strain, exogenous stimuli, antibody/fluorochrome combination, one may require different combinations of these marker to positively identify subsets of ILCs. For example, in the analysis of ILC2s, it is important to use anti-ST2 antibody to detect IL-33 responsive cells; whereas in the analysis of ILC3s, it is important to include Sca-1, CD117 (c-kit), and CCR6. Future efforts to elucidate the tissue distribution and molecular mechanisms underlying the functions of ILCs in triggering immunity, inflammation and tissue repair will result in a more comprehensive view of how ILCs regulate immunological and physiological processes.
References
- 1.Yazdani R, Sharifi M, Shirvan AS, Azizi G, Ganjalikhani-Hakemi M. Characteristics of innate lymphoid cells (ILCs) and their role in immunological disorders (an update) Cell Immunol. 2015;298(1–2):66–76. doi: 10.1016/j.cellimm.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol. 2013;13(2):75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 3.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature immunology. 2011;12(1):21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 4.Mackay LK, Kallies A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol. 2017;38(2):94–103. doi: 10.1016/j.it.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Tait Wojno ED, Artis D. Emerging concepts and future challenges in innate lymphoid cell biology. The Journal of experimental medicine. 2016;213(11):2229–2248. doi: 10.1084/jem.20160525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nature immunology. 2016;17(7):765–774. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 7.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350(6263):981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508(7496):397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual review of immunology. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 10.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature immunology. 2011;12(1):21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 11.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nature medicine. 2014;20(1):54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marashian S, Mortaz E, Jamaati H, Alavi-Moghaddam M, Kiani A, Abedini A, Garssen J, IMA, Velayati A. Role of Innate Lymphoid Cells in Lung Disease. Iranian journal of allergy, asthma, and immunology. 2015;14(4):346–360. [PubMed] [Google Scholar]
- 13.Lai DM, Shu Q, Fan J. The origin and role of innate lymphoid cells in the lung. Military Medical Research. 2016;3:25. doi: 10.1186/s40779-016-0093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Withers DR. Innate lymphoid cell regulation of adaptive immunity. Immunology. 2016;149(2):123–130. doi: 10.1111/imm.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo Y, Jeong D, Chung DH, Kim HY. The roles of innate lymphoid cells in the development of asthma. Immune network. 2014;14(4):171–181. doi: 10.4110/in.2014.14.4.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, Michel E, Reuter S, Renauld JC, Arnold-Schild D, Schild H, Buhl R, Becher B. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS One. 2011;6(7):e21799. doi: 10.1371/journal.pone.0021799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol. 2011;23(3):159–163. doi: 10.1093/intimm/dxr001. [DOI] [PubMed] [Google Scholar]
- 18.Hwang YY, McKenzie AN. Innate lymphoid cells in immunity and disease. Adv Exp Med Biol. 2013;785:9–26. doi: 10.1007/978-1-4614-6217-0_2. [DOI] [PubMed] [Google Scholar]
- 19.Huntington ND, Carpentier S, Vivier E, Belz GT. Innate lymphoid cells: parallel checkpoints and coordinate interactions with T cells. Curr Opin Immunol. 2016;38:86–93. doi: 10.1016/j.coi.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Mjosberg J, Bernink J, Peters C, Spits H. Transcriptional control of innate lymphoid cells. European journal of immunology. 2012;42(8):1916–1923. doi: 10.1002/eji.201242639. [DOI] [PubMed] [Google Scholar]
- 21.Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends in immunology. 2010;31(11):407–413. doi: 10.1016/j.it.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41(3):354–365. doi: 10.1016/j.immuni.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortez VS, Robinette ML, Colonna M. Innate lymphoid cells: new insights into function and development. Current opinion in immunology. 2015;32:71–77. doi: 10.1016/j.coi.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 25.McKenzie AN, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41(3):366–374. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nature immunology. 2013;14(6):536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 27.Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. 2017;150(3):265–275. doi: 10.1111/imm.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumoutier L, de Heusch M, Orabona C, Satoh-Takayama N, Eberl G, Sirard JC, Di Santo JP, Renauld JC. IL-22 is produced by gammaC-independent CD25+ CCR6+ innate murine spleen cells upon inflammatory stimuli and contributes to LPS-induced lethality. Eur J Immunol. 2011;41(4):1075–1085. doi: 10.1002/eji.201040878. [DOI] [PubMed] [Google Scholar]
- 29.Eberl G. Development and evolution of RORgammat+ cells in a microbe’s world. Immunol Rev. 2012;245(1):177–188. doi: 10.1111/j.1600-065X.2011.01071.x. [DOI] [PubMed] [Google Scholar]
- 30.Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, Willis C, Budelsky A, Farber DL, Artis D. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–1323. doi: 10.1038/mi.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 32.Kim CH, Hashimoto-Hill S, Kim M. Migration and Tissue Tropism of Innate Lymphoid Cells. Trends Immunol. 2016;37(1):68–79. doi: 10.1016/j.it.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng H, Jin C, Wu J, Zhu S, Liu YJ, Chen J. Guards at the gate: physiological and pathological roles of tissue-resident innate lymphoid cells in the lung. Protein Cell. 2017 doi: 10.1007/s13238-017-0379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]