
Scheme 1.
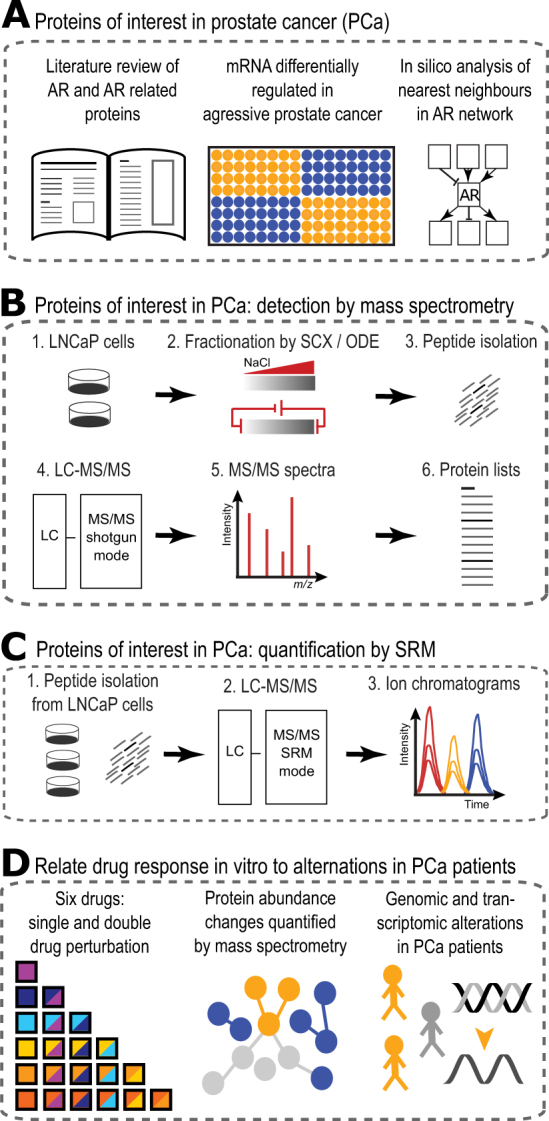
Project overview. a A list of proteins of interest in PCa was generated by extensive literature review, comparison of gene expression data, and in silico analysis of nearest neighbor in androgen receptor (AR) pathway. b To establish which proteins are identifiable in the LNCaP PCa model, cells were grown in a Petri dish and harvested. From the cell pellets the proteome was extracted and extensively fractionated using strong cation exchange (SCX) chromatography and off-gel electrophoresis (OGE). Fractions were purified and peptides identified using LC-MS/MS in discovery (or shotgun) mode. The MS/MS spectra were annotated to obtain a list of proteins identifiable in LNCaP cells. c Quantifying protein abundance following pharmacological treatment was done on undepleted lysate level. Peptides were quantified using targeted proteomics (SRM-MS) and resulting ion chromatograms were analyzed with the software tool Skyline. d Six clinically relevant drugs were chosen and all single plus double drug combinations were added to a PCa model. Proteins served as phenotypic readouts of drug response and were quantified using mass spectrometry in selected reaction monitoring (SRM) mode. Immediate protein abundance changes were analyzed across the dataset identifying a group of proteins consistently upregulated. Publicly available DNA alteration and transcriptomics data from PCa patients was analyzed to link immediate response to drug treatment with adaptations found in PCa disease progression