

Chronic wasting disease (CWD) is a fatal prion disease found in deer, elk, and moose. Since it was first discovered in the late 1960s, CWD has now spread to at least 25 U.S. states, 2 Canadian provinces, South Korea, Norway, and Finland. Eradication of CWD from areas of endemicity is very unlikely, and additional spread will occur. As the range and prevalence of CWD increase, so will the potential for human exposure to CWD prions. It is currently unknown if CWD poses a risk to human health. However, determining this risk is critical to preventing a scenario similar to that which occurred when mad cow disease was found to be transmissible to humans. In the present study, we used cynomolgus macaque monkeys as a surrogate model for CWD transmission to humans. After 13 years, no evidence for CWD transmission to macaques was detected clinically or by using highly sensitive prion disease-screening assays.
KEYWORDS: cynomolgus macaques, squirrel monkeys, CWD strains, RT-QuIC, prion, cross-species transmission, barrier, chronic wasting disease
ABSTRACT
Chronic wasting disease (CWD) is a fatal prion disease that can infect deer, elk, and moose. CWD was first recognized in captive deer kept in wildlife facilities in Colorado from 1967 to 1979. CWD has now been detected in 25 U.S. states, 2 Canadian provinces, South Korea, Norway, and Finland. It is currently unknown if humans are susceptible to CWD infection. Understanding the health risk from consuming meat and/or products from CWD-infected cervids is a critical human health concern. Previous research using transgenic mouse models and in vitro conversion assays suggests that a significant species barrier exists between CWD and humans. To date, reported epidemiologic studies of humans consuming cervids in areas where CWD is endemic have found no evidence to confirm CWD transmission to humans. Previously, we reported data from ongoing cross-species CWD transmission studies using two species of nonhuman primates as models. Squirrel monkeys (SM) and cynomolgus macaques (CM) were inoculated by either the intracerebral or oral route with brain homogenates from CWD-infected deer and elk containing high levels of infectivity. SM were highly susceptible to CWD infection, while CM were not. In the present study, we present new data for seven CWD-inoculated CM euthanized 11 to 13 years after CWD inoculation and eight additional uninoculated control CM. New and archival CM tissues were screened for prion infection by using the ultrasensitive real-time quaking-induced conversion (RT-QuIC) assay, immunohistochemistry, and immunoblotting. In this study, there was no clinical, pathological, or biochemical evidence suggesting that CWD was transmitted from cervids to CM.
IMPORTANCE Chronic wasting disease (CWD) is a fatal prion disease found in deer, elk, and moose. Since it was first discovered in the late 1960s, CWD has now spread to at least 25 U.S. states, 2 Canadian provinces, South Korea, Norway, and Finland. Eradication of CWD from areas of endemicity is very unlikely, and additional spread will occur. As the range and prevalence of CWD increase, so will the potential for human exposure to CWD prions. It is currently unknown if CWD poses a risk to human health. However, determining this risk is critical to preventing a scenario similar to that which occurred when mad cow disease was found to be transmissible to humans. In the present study, we used cynomolgus macaque monkeys as a surrogate model for CWD transmission to humans. After 13 years, no evidence for CWD transmission to macaques was detected clinically or by using highly sensitive prion disease-screening assays.
INTRODUCTION
Chronic wasting disease (CWD) is a fatal transmissible spongiform encephalopathy (TSE) or prion disease of deer, elk, moose, and, most recently, reindeer. Since it was first recognized in 1967 in Colorado, CWD has now been detected in 25 U.S. states as well as in Canada, South Korea, Norway, and Finland. The spread of CWD has been linked to both human transportation of infected cervids to new locations and natural expansion from infected foci of wild populations (1–3). In areas where CWD is endemic, cervid population declines have been documented, and some models suggest that dramatic declines may occur in the future (for reviews, see references 3 and 4). Unfortunately, eradication of CWD from areas of endemicity is likely impossible due to the long-term stability of infectious prions in the environment, the ease of transmission from animal to animal, and the lack of an effective vaccine or treatment (1–3). As the range and prevalence of CWD increase, so will the potential for human exposure to CWD prions.
The zoonotic transmission of prion disease from animals to humans appears to vary with different species and prion agents. For example, in spite of likely exposures over the last 200 years, there is little evidence of transmission of scrapie from sheep to humans (5–8). However, there is strong evidence for the transmission of bovine spongiform encephalopathy (BSE) from cattle to humans albeit at a very low frequency (9–11). This variability in cross-species transmission raises the question of whether CWD can be transmitted from cervids to humans and, if so, the frequency at which this might occur.
To date, scientists have used several types of laboratory models to test the potential for cross-species transmission of CWD to humans (12–14). Results from seven in vitro experiments using primarily protein-misfolding cyclic amplification (PMCA), real-time quaking-induced conversion (RT-QuIC), and cell-free conversion assays are well summarized in a recent review by Waddell et al. (12). Collectively, the in vitro data suggest that a species barrier exists between cervids and humans, although it may not be 100%. Five studies have used transgenic mice that express human prion protein as models for cross-species transmission. None of the mice that expressed human prion protein developed CWD (15–19). Two epidemiologic studies and nine case studies have also shown no conclusive link of CWD exposure to an increase in human prion disease (12). Transmission to two different species of nonhuman primates (NHP) has also been used to assess the ability of CWD to cross species barriers (20–22). We previously showed that CWD was transmissible to squirrel monkeys (SM), but not to cynomolgus macaques (CM), by either the intracerebral (i.c.) or oral route of infection by 10 years postinoculation (22). Since our last report in 2014, we continued to observe seven remaining CWD-inoculated macaques for onset of disease for three additional years. In the present study, we present data on these seven monkeys using the newly available, highly sensitive RT-QuIC assay, immunohistochemistry (IHC), and immunoblot tests for prion disease. Brain and spinal cord tissues from our previously described CWD-inoculated CM, as well as from newly acquired uninoculated CM, were also tested by using both RT-QuIC and IHC with three different anti-prion protein (PrP) antibodies. Using these new assays, and screening additional tissues, we observed no conclusive evidence of cross-species transmission of CWD to CM.
RESULTS
Clinical observations of CWD-inoculated cynomolgus macaques.
Clinical CWD in cervids presents primarily as a wasting syndrome, with other clinical signs including changes in behavior, polyuria/polydipsia, and excessive salivation (23). In SM, the main presenting sign of CWD was also weight loss, with other clinical signs such as weakness, inactivity, tremors, mild ataxia, and excess salivation, which varied between individuals (21). In our study, CWD-inoculated CM were monitored closely for signs of wasting, neurologic disease, and behavioral changes. Between 6.6 and 10.9 years after CWD inoculation, 5 monkeys developed weight loss; of these monkeys, 4 were confirmed to have diabetes, and 1 had no definitive diagnosis (Table 1). All the other CWD-inoculated monkeys were euthanized for a variety of medical conditions or neurologic signs or electively at the termination of the study at >13 years postinoculation (Table 1). Each euthanized CM was screened for prion disease by using prion disease-specific diagnostic assays, including the RT-QuIC assay for amyloid-seeding activity, examination of brains and spinal cords for neuropathology, and IHC and immunoblotting for the deposition of disease-associated prion protein (PrPSc).
TABLE 1.
CWD-inoculated and normal control cynomolgus monkeysb
| CMa | Gender | CWD source | Route | Total dose (LD50) | Age at euthanization (yr) | Observed period (yr postinfection) | Wt change (%)c | Reason for euthanasia |
|---|---|---|---|---|---|---|---|---|
| 116 | M | WTD | i.c. | 2.0 × 106 | 14.2 | 7.3 | −34 | Diabeticd |
| 144 | F | WTD | i.c. | 2.0 × 106 | 18.0 | 6.6 | −38 | Wasting |
| 128 | M | Elk | i.c. | 3.2 × 105 | 17.1 | 13.4 | 12 | Elective |
| 616 | F | Elk | i.c. | 3.2 × 105 | 15.9 | 7.9 | 12 | Seizurese |
| 609 | F | MD | i.c. | 2.5 × 106 | 11.9 | 4.0 | NA | Aggressionf |
| 135 | M | MD | i.c. | 2.5 × 106 | 16.2 | 13.3 | 50 | Elective |
| 119 | M | WTD | Oral | 1.6 × 109 | 19.6 | 13.3 | 19 | Elective |
| 125 | M | WTD | Oral | 1.6 × 109 | 8.8 | −15 | Diabeticd | |
| 130 | M | Elk | Oral | 2.5 × 108 | 17.6 | 12.4 | 47 | Hemorrhoids |
| 629 | F | Elk | Oral | 2.5 × 108 | 18.5 | 13.1 | 10 | Abdominal pain |
| 121 | M | Elk | Oral | 2.5 × 108 | 17.4 | 10.9 | −30 | Diabeticd |
| 270 | F | MD | Oral | 2.0 × 109 | 8.8 | −33 | Diabeticd | |
| 122 | M | MD | Oral | 2.0 × 109 | 14.7 | 8.1 | 21 | Tremorg |
| 614 | F | MD | Oral | 2.0 × 109 | 19.7 | 11.8 | 39 | Anorexiah |
| 633 | F | NEB | i.c. | NA | 10.9 | 8.1 | Elective | |
| 585 | F | None | NA | NA | 9 | Anemia | ||
| 228 | F | None | NA | NA | 8.2 | Foot injury | ||
| 949 | F | None | NA | NA | 7.9 | Socialization issues | ||
| 82-51 | F | None | NA | NA | 17 | Anorexia | ||
| 161 | F | None | NA | NA | 5.4 | Chronic diarrhea | ||
| 151 | M | None | NA | NA | 10 | NIA tissue bank | ||
| 146 | F | None | NA | NA | 10 | NIA tissue bank | ||
| 27 | F | None | NA | NA | 23.7 | NIA tissue bank |
Results for monkeys in boldface type were not reported in previous studies.
Abbreviations: M, male; F, female; i.c., intracerebral; WTD, white-tailed deer; MD, mule deer; NEB, normal elk brain; NA, not applicable; NIA, National Institute on Aging; LD50, 50% lethal dose.
Weight change was calculated as the percent weight change from the weight at the initiation of the experiment to the final weight taken following euthanasia. Many CM gained weight during the study, possibly contributing to the high incidence of diabetes.
The diabetic monkeys showed a variety of clinical signs, including lethargy, weakness, and intermittent anorexia.
CM616 experienced 2 seizures over a 3-day period and was euthanized. Tumors were discovered at the base of the brain and in the uterus.
CM609 became aggressive toward staff and was euthanized at 4 years postinfection. No previous behavioral issues had been observed.
CM122 developed muscle tremors and intermittent anorexia at around 8 years postinfection. Clinical hypocalcemia (4 mg/dl) was diagnosed and treated successfully with oral calcium supplementation for 4 months.
CM614 was euthanized due to depression and intermittent anorexia, likely due to a concurrent change in animal care personnel.
RT-QuIC analysis of CM brain and spinal cord.
The RT-QuIC assay is an extremely sensitive screening assay for the detection of disease-specific prions (24). Recently, the use of recombinant bank vole (BV) prion protein (rPrPsen) as the RT-QuIC assay substrate was shown to detect numerous different strains of prions from many different species (25). To confirm that the BV substrate worked with CWD, we tested our three original CWD inocula (MD-1, WTD-1, and Elk-1) using serial 10-fold dilutions from 10−4 to 10−8. Elk-1 is shown as an example in Fig. 1A, where the endpoint dilution was 10−8. Similar very high titers were seen with the other two cervid CWD inocula, MD-1 and WTD-1 (data not shown). As a positive control for prion disease in CM, we tested brains from three CM infected with a variant Creutzfeldt-Jakob disease (vCJD) inoculum. Testing of 10-fold dilutions of each vCJD inoculum-infected CM brain showed that brain homogenate dilutions of 10−3, 10−4, 10−5, and 10−6 consistently gave strong positive reactions by 10 h in all three positive-control monkeys. Results for one vCJD-positive CM (CM16999) brain are shown as an example (Fig. 1B). This monkey also had strong positive reactions at a 10−7 brain dilution, but a 10−8 brain dilution reacted more slowly (≥20 h), and the mean fluorescence for four wells remained at only 25% of the maximum for the duration of the experiment, i.e., 50 h.
FIG 1.

RT-QuIC analysis of Elk-1 (CWD inoculum) and vCJD brain from a clinically ill CM. RT-QuIC analysis using BV rPrPsen as a substrate was performed on serial 10-fold dilutions of CWD-infected elk brain (A) and vCJD-infected CM brain (B). Each curve represents the average fluorescence for four wells tested for each individual dilution. Dilutions are shown on the far right of each curve. In panel A, Elk-1 brain dilutions are indicated as ○ for a 10−4 dilution, ■ for 10−5, ▲ for 10−6, ▽ for 10−7, and □ for 10−8, and ● indicates normal deer brain diluted to 10−4 as a negative control. RT-QuIC using the BV rPrPsen substrate was highly sensitive for both CWD and cynomolgus macaque brain infected with vCJD. ND, not detected.
For a more precise analysis of the RT-QuIC results, analysis of the data for individual wells is helpful. When 10−3 brain dilutions from each CM with vCJD were tested, analysis of data from individual wells revealed near-maximal fluorescence values within a 4- to 5-h reaction time (Fig. 2A and B). In contrast, 10−3 dilutions of brain homogenates from 6 uninoculated and 1 normal elk brain-inoculated CM did not have rapid amyloid-seeding reactions, but several wells in some samples showed an increase in fluorescence beginning after 28 h (Fig. 2C and D). Based on the nonspecific reactivity observed after 28 h in the negative-control CM and the rapid reaction times observed in the vCJD-inoculated positive-control CM, we determined that for samples to be considered RT-QuIC positive, they must have a >33% maximal fluorescence signal in 2 or more out of 4 wells prior to the 25-h reaction time.
FIG 2.

RT-QuIC analysis of vCJD-inoculated CM, CWD-inoculated CM, and uninoculated CM using the BV rPrPsen substrate. Four independent wells were tested for each CM, and each well is represented as an individual curve shown with one of four different symbols (○, ▲, ■, and ▽) in each panel. All tested brain samples were diluted to 10−3. (A and B) Positive-control vCJD-inoculated CM. (C) Normal elk brain-inoculated CM. (D) Uninoculated CM. (E and F) Two CWD-inoculated CM. The numbers of wells that reacted were variable between individual CM, but no consistent differences were observed between the uninoculated and CWD-inoculated groups. The vertical line shown at 25 h represents the cutoff time to meet the criteria for a positive reaction (see Results and Materials and Methods for more detail). IP, intraperitoneal; ypi, years postinfection; IC, intracerebral.
RT-QuIC assays were run on tissues from 7 negative-control and 14 CWD-inoculated CM by individuals blind to the inoculation status of the animals. Cerebral cortex was tested for every CM, and brain stem and spinal cord regions (cervical, thoracic, and lumbar) were tested for a subset of CM (Table 2). The two representative CWD-inoculated CM shown in Fig. 2E and F, CM130 and CM614, did not have any positive wells before 25 h and thus were scored negative (Table 2). From 30 to 50 h, many of the CM from both groups had some wells with an increase in fluorescence, but results from uninoculated and CWD-inoculated CM could not be distinguished (Fig. 2C to F and Table 2). Similar results were observed for all the central nervous system (CNS) regions tested, and no CWD-inoculated brain or spinal cord region scored positive (Table 2).
TABLE 2.
RT-QuIC of cynomolgus macaque brain and spinal cord samples using the BV rPrPsen substratea
| Monkey | Inoculum | Route | Yr postinfection | Sample result |
||||
|---|---|---|---|---|---|---|---|---|
| Brain |
Spinal cord |
|||||||
| Cerebral cortex | Brain stem (pons) | Cervical | Thoracic | Lumbar | ||||
| 16999 | vCJD | i.p. | 2.4 | + | TNA | TNA | TNA | TNA |
| 7422 | vCJD | i.p. | 2.4 | + | TNA | TNA | TNA | TNA |
| 7423 | vCJD | i.p. | 2.3 | + | TNA | TNA | TNA | TNA |
| 116 | WTD | i.c. | 7.3 | − | − | − | − | − |
| 144 | WTD | i.c. | 6.6 | − | − | − | − | − |
| 128 | Elk | i.c. | 13.4 | − | − | NT | NT | NT |
| 616 | Elk | i.c. | 7.9 | − | − | − | − | − |
| 609 | MD | i.c. | 4.0 | − | − | − | − | − |
| 135 | MD | i.c. | 13.3 | − | − | − | NT | NT |
| 119 | WTD | Oral | 13.3 | − | − | − | NT | NT |
| 125 | WTD | Oral | 8.8 | − | − | − | − | − |
| 130 | Elk | Oral | 12.4 | − | − | NT | − | NT |
| 629 | Elk | Oral | 13.1 | − | − | − | NT | NT |
| 121 | Elk | Oral | 10.9 | − | − | − | NT | − |
| 270 | MD | Oral | 8.8 | − | − | − | − | − |
| 122 | MD | Oral | 8.1 | − | − | − | − | − |
| 614 | MD | Oral | 11.8 | − | − | − | − | − |
| 633 | NEB | i.c. | 8.1 | − | − | − | − | − |
| 228 | None | NA | − | − | − | − | − | |
| 949 | None | NA | − | − | − | − | − | |
| 585 | None | NA | − | − | − | − | − | |
| 151 | None | NA | − | − | TNA | TNA | TNA | |
| 146 | None | NA | − | − | TNA | TNA | TNA | |
| 27 | None | NA | − | TNA | TNA | TNA | TNA | |
Abbreviations: vCJD, variant Creutzfeldt-Jacob disease; i.p., intraperitoneal; TNA, tissue not available; WTD, white-tailed deer CWD; i.c., intracerebral; NT, not tested; MD, mule deer CWD; NEB, normal elk brain; NA, not applicable.
A subset of CM samples was also tested by using the RT-QuIC assay with another PrP substrate, the hamster 90-231 substrate. This RT-QuIC assay detected high levels of amyloid-seeding activity in the vCJD CM positive-control and cervid-derived CWD samples. However, all CWD-inoculated CM and uninoculated CM tested with the hamster 90-231 substrate were negative, and fluorescence levels remained at baseline levels for >50 h.
Study of neuropathology and PrP staining in brain tissue.
Prion disease can be diagnosed in all species susceptible to infection by various neuropathological features. These features include typical spongiform lesions in gray matter, astrogliosis and microgliosis, and the deposition of PrPSc detectable by IHC. To test for evidence of CWD infection of CM, brains from 8 uninoculated CM, 1 normal elk brain-inoculated CM, and 14 CWD-inoculated CM were analyzed for neuropathology and the deposition of PrPSc. The brain from a clinically ill, CWD-infected squirrel monkey was used as a staining control and provides an example of neuropathology typically found in prion-infected brains (Fig. 3D, F, I, and L). Similar neuropathology was also reported for brain tissues from CM with BSE, vCJD, Kuru, or scrapie (26–28). In contrast, no spongiform degeneration or gliosis was observed in the gray matter of any CWD-inoculated CM (Fig. 3 and Table 3). Three different monoclonal anti-PrP antibodies (3F4, 6H4, and L42) representing different epitopes were used for IHC staining of PrP. PrPsen staining in brain gray matter appeared as a smooth background blush, more prominent in sections stained with 6H4 (Fig. 3G and H) and L42 (not shown) than in sections stained with 3F4 (Fig. 3J and K). This smooth, typical PrPsen staining pattern was the dominant feature of each CM brain. However, many brains from both CWD-inoculated and uninoculated CM had unusual, small focal areas of anti-PrP antibody-specific staining that appeared similar to that of disease-associated PrPSc found in known prion diseases (Table 3). Three different distribution patterns of PrP staining were observed in gray matter: vascular associated (Fig. 4A, C, and D), pericellular (Fig. 4E and F), and parenchymal (Fig. 4I and J). These areas of anti-PrP staining might be focal areas of increased PrPsen expression and aggregation.
FIG 3.

CM and SM brain histopathology. Shown are examples of gray matter regions from the cerebral cortex of an uninoculated CM (A, D, G, and J), CWD-inoculated CM (B, E, H, and K), and a CWD-infected squirrel monkey (C, F, I, and J). (A to C) Tissues were stained by H&E to look for spongiosis/vacuolation and general neuropathology. (D to L) Anti-GFAP IHC was used to detect activated astrocytes (D to F), and anti-PrP antibodies 6H4 and 3F4 were used with IHC to detect PrP deposition (G to L). No pathology, spongiform lesions, or excessive astroglial activation was observed in any of the uninoculated or CWD-inoculated CM brains. PrPsen could be seen in all brains as a smooth brown blush using anti-PrP antibody 6H4 (E and F). The background PrPsen was less noticeable using anti-PrP antibody 3F4 (G and H). The bar shown in panel A is 50 μm and applies to all panels.
TABLE 3.
Neuropathology and immunohistochemical detection of PrPa
| Monkey | CWD source | Route | Yr postinfection | Result |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Brain |
Spinal cord |
||||||||
| Spongiform lesions | Gliosis | Unusual PrP stainingb | Spongiform lesions | Gliosis | PrP stainingc | ||||
| 116 | WTD | i.c. | 7.3 | − | − | + | NT | NT | NT |
| 144 | WTD | i.c. | 6.6 | − | − | − | NT | NT | NT |
| 128 | Elk | i.c. | 13.4 | − | − | + | NT | NT | NT |
| 616 | Elk | i.c. | 7.9 | − | − | + | − | − | + |
| 609 | MD2 | i.c. | 4.0 | − | − | − | − | − | + |
| 135 | MD | i.c. | 13.3 | − | − | + | − | − | + |
| 119 | WTD | Oral | 13.3 | − | − | + | NT | NT | NT |
| 125 | WTD | Oral | 8.8 | − | − | + | NT | NT | NT |
| 130 | Elk | Oral | 12.4 | − | − | + | − | − | + |
| 629 | Elk | Oral | 13.1 | − | − | + | NT | NT | NT |
| 121 | Elk | Oral | 10.9 | − | − | +d | − | − | + |
| 270 | MD | Oral | 8.8 | − | − | + | − | − | + |
| 122 | MD | Oral | 8.1 | − | − | + | NT | NT | NT |
| 614 | MD | Oral | 11.8 | − | − | +d | NT | NT | NT |
| 633 | NEB | i.c. | 8.1 | − | − | + | − | − | + |
| 228 | None | NA | − | − | + | − | − | + | |
| 949 | None | NA | − | − | + | − | − | + | |
| 585 | None | NA | − | − | + | NT | NT | NT | |
| 82-51 | None | NA | − | − | + | TNA | TNA | TNA | |
| 161 | None | NA | − | − | + | TNA | TNA | TNA | |
| 151 | None | NA | − | − | − | TNA | TNA | TNA | |
| 146 | None | NA | − | − | + | TNA | TNA | TNA | |
| 27 | None | NA | +e | − | − | TNA | TNA | TNA | |
In addition to the data provided here, IHC for PrPSc was also performed on spleen and lymph nodes from all the CWD-inoculated monkeys; no positive PrPSc staining was observed. Colon and ileum samples from most of the orally inoculated CM were also screened by IHC, but no PrPSc was observed. Abbreviations: WTD, white-tailed deer CWD; i.c., intracerebral; NT, not tested; MD, mule deer CWD; TNA, tissue not available.
Unusual PrP staining (PrP staining differing from the typical homogenous PrPsen background seen in uninoculated CM [see Fig. 3 for typical PrP staining and Fig. 4 for examples of unusual PrP staining]).
PrP staining was observed in the gray matter and substantia gelatinosa layer of the spinal cord in both CWD-inoculated and uninoculated CM and did not appear to be disease-specific PrPSc.
These two monkeys also had PrP staining, as shown in Fig. 5.
Negative-control CM27 had widespread white matter vacuolation; this monkey was 23.7 years old and had renal failure.
FIG 4.

Immunohistochemical staining of CM brain using anti-PrP and -GFAP antibodies. Shown are examples of a normal elk brain-inoculated CM (A, B, E, G, and I) and a CWD-inoculated CM (C, D, F, H, and J). We tested three monoclonal anti-PrP antibodies (3F4, 6H4, and L42). All antibodies gave similar staining results, but only 3F4 is shown. Three patterns of aggregated PrP staining were observed: perivascular (A, C, and D), pericellular (E and F) and parenchymal (I and J). In all cases, aggregated PrP staining was rare and affected only small, focal regions of the brain. Panels G and H show minimal anti-GFAP staining and a lack of astrocytic activation in the same brain regions shown in panels E and F, respectively. A no-primary-antibody control (NP) is shown in panel B to demonstrate primary antibody staining specificity. The bar shown in panel A is 25 μm and applies to all panels.
A rare, fourth distribution pattern was observed in two CM that had been orally inoculated with CWD. In these monkeys, PrP staining was observed as clusters of aggregated stain in a spherical pattern, with the center of the lesion having the strongest and most dense staining (Fig. 5A and B). Clusters ranged from 25 to 500 μm in diameter and covered an area of the caudate nucleus of approximately 3 by 5 mm in CM121. The second monkey with a similar staining pattern (CM614) had only 3 small clusters localized to the frontal cortex. Importantly, in the PrP-stained areas seen in CM121 and CM614 and in the areas of unusual staining in numerous CM described above, there was never an association with gray matter vacuolation by hematoxylin and eosin (H&E) staining or an increase in the amount of astroglia (Fig. 4G and H and 5C and D). Such features are often located in proximity to PrPSc deposits in prion-infected brain tissue.
FIG 5.
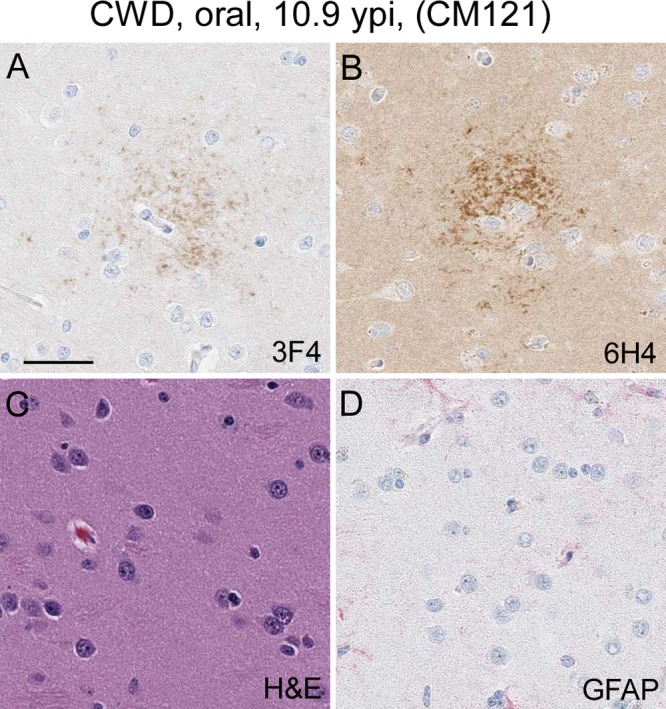
Immunohistochemical and H&E staining of the caudate nucleus from CM121. (A and B) Anti-PrP staining with the 3F4 antibody (A) and anti-PrP staining with the 6H4 antibody (B) show a cluster of PrP staining (brown). Several additional similar clusters were observed in the caudate nucleus of this monkey. (C) H&E staining of the same region depicted in panels A and B. No spongiform lesions were observed. (D) Minimal anti-GFAP staining and a lack of astrocytic activation in the same brain regions shown in panels A and B. The bar shown in panel A is 25 μm and applies to all panels.
To further complicate the situation, another type of brown pigment deposit was present in every CM brain tested. This material was associated primarily with blood vessels and meningeal surfaces and often located near the Purkinje cell layer of the cerebellum, and it was observed in all IHC slides (including no-primary-antibody controls) and in H&E-stained slides (Fig. 6A to D). Lipofuscin or hemosiderin aggregates were suspected, so brain tissue was stained with Prussian blue (PB) for the detection of iron that would be present in hemosiderin deposits. The positive PB staining pattern was identical to the brown pigment deposition pattern seen previously, strongly suggesting that the brown pigment was hemosiderin (Fig. 6E).
FIG 6.

Hemosiderin pigment deposition in CM brain. All panels show a similar region of cerebellum from CWD-inoculated CM270. Staining methods are shown in the lower left corner of each panel. (A to D) Brown pigments (yellow arrows) were observed in slides stained with 3F4 (A), no-primary-antibody controls (B), and H&E-stained sections (C and D) of brain tissues examined from all the CWD-inoculated and uninoculated CM. (E) Prussian blue iron staining (blue) on adjacent sections confirmed that the pigment was hemosiderin. Hemosiderin pigment deposition was common near Purkinje cells, often near blood vessels (red arrow). The bar shown in panel A is 25 μm and applies to all panels.
Study of neuropathology and PrP staining in spinal cord.
Cervical, thoracic, and lumbar spinal cord sections from a subset of negative-control CM and CWD-inoculated CM were stained for PrP using anti-PrP antibodies (3F4, 6H4, or L42) and for astrocytes using anti-glial fibrillary acid protein (anti-GFAP) antibody and were stained by H&E to look for general pathology. Spinal cord white matter had much lower levels of PrPsen staining than did gray matter. PrP staining in gray matter regions of the dorsal and ventral horns was most prominent by using antibody 6H4 or L42 compared to 3F4 (Fig. 7). Within the dorsal horn of both uninoculated and CWD-inoculated CM, we observed PrP staining, which varied from a smooth, homogenous brown pattern to a more irregular, punctate pattern in portions of the substantia gelatinosa. PrP staining was also present in the nerve roots adjacent to the spinal cord in both uninoculated and CWD-inoculated CM (Fig. 7F to H). This staining was most likely due to normal PrP. For the spinal cord, uninoculated and CWD-inoculated CM showed indistinguishable staining with anti-GFAP (Fig. 8). No spongiform lesions, gliosis, or other abnormal pathology was observed in any of the spinal cord regions examined (Table 3). The lack of any pathological differences in spinal cords between CWD-inoculated CM and negative-control CM provided no evidence for positive transmission of CWD to these CM.
FIG 7.

Detection of PrP in spinal cords and nerve roots of CM using anti-PrP antibody (6H4). Spinal cord regions (cervical, thoracic, and lumbar) were screened for PrP deposition in 6 CWD-inoculated CM, 2 uninoculated CM, and 1 normal elk brain-inoculated CM. (A to E) Examples of thoracic spinal cord from one uninoculated CM (A), a normal elk brain-inoculated CM (C), and three CWD-inoculated CM (B, D, and E). PrP staining (brown) was similar for all CM. The strongest staining was located in the substantia gelatinosa region of the dorsal horn (blue arrow). (F and H) PrP staining (purple arrows) was also observed in nerve roots adjacent to the spinal cord. (G) The same nerve region depicted in panel F, without antibody 6H4 being applied (no primary ab). The bar shown in panel A is 1 mm and applies to panels A to E; the bar shown in panel F is 50 μm and applies to panels F to H.
FIG 8.

Detection of astroglia in CM spinal cord using anti-GFAP antibody. Shown are an uninoculated CM (A, C, and D) and a CWD-inoculated CM (B, E, and F). (A and B) Typical astrocyte staining in spinal cord. (C to F) Higher magnifications show astrocyte staining in white matter (C and E) and gray matter (D and F), and squares drawn in panels A and B show the location of the magnified regions. No differences between CWD-inoculated and uninoculated CM were seen in astrocyte density, distribution, or cellular morphology. The bar in panel A is 1 mm and applies to panels A and B. The bar in panel C is 50 μm and applies to panels C to F.
Analysis of protease-resistant PrP by immunoblotting.
To screen the CM tissues for the presence of proteinase K (PK)-resistant PrP (PrPres), we performed immunoblot analyses on proteinase K-treated tissue homogenates of brain, spleen, and lymph nodes (Table 4). Several regions of each brain were tested for each CM, including the cerebral cortex, thalamus, cerebellum, claustrum, and brain stem. Tissues from each CM were tested as soon as possible following euthanasia (Fig. 9A and B). In addition, cerebral cortex samples from each CWD-inoculated CM were all retested together at the end of the experiment to facilitate comparisons (Fig. 9C and D). No positive signals were observed for any of the CM tissues screened (Fig. 9 and Table 4).
TABLE 4.
Cynomolgus monkey tissues tested for PrPSc by immunoblotting using anti-PrP antibody 3F4a
| Monkey | CWD source | Route | Yr postinfection | Tissue(s) screened | Result |
|---|---|---|---|---|---|
| 116 | WTD | i.c. | 7.3 | Cortex, CC, thalamus, claustrum, cerebellum, BS, spleen, LNs | Negative |
| 144 | WTD | i.c. | 6.6 | Cortex, thalamus, claustrum, cerebellum, BS | Negative |
| 128 | Elk | i.c. | 13.4 | Cortex, claustrum, BS, spleen | Negative |
| 616 | Elk | i.c. | 7.9 | Cortex, claustrum, spleen, LN | Negative |
| 609 | MD | i.c. | 4.0 | Cortex, thalamus, cerebellum, cervical SC, spleen | Negative |
| 135 | MD | i.c. | 13.3 | Cortex, claustrum, BS, spleen | Negative |
| 119 | WTD | Oral | 13.3 | Cortex, claustrum, BS, spleen | Negative |
| 125 | WTD | Oral | 8.8 | Cortex, claustrum, thalamus, cerebellum, BS, spleen, LNs | Negative |
| 130 | Elk | Oral | 12.4 | Cortex, claustrum, thalamus, cerebellum, BS, HS | Negative |
| 629 | Elk | Oral | 13.1 | Cortex, claustrum, thalamus, cerebellum, BS, spleen | Negative |
| 121 | Elk | Oral | 10.9 | Cortex, thalamus, cerebellum, spleen, LNs | Negative |
| 270 | MD | Oral | 8.8 | Cortex, thalamus, claustrum, cerebellum, BS, spleen, LNs | Negative |
| 122 | MD | Oral | 8.1 | Cortex, claustrum, spleen, LN | Negative |
| 614 | MD | Oral | 11.8 | Cortex, claustrum, thalamus, BS, cerebellum, spleen, tongue, ileum | Negative |
| 633 | NEB | i.c. | 8.1 | Cortex, claustrum, spleen, LN | Negative |
| 228 | None | NA | Cortex | Negative | |
| 949 | None | NA | Cortex | Negative |
Abbreviations: WTD, white-tailed deer CWD; i.c., intracerebral; MD, mule deer; NEB, normal elk brain; CC, corpus callosum; BS, brain stem; LN, lymph node; SC, spinal cord; HC, hippocampus.
FIG 9.

Immunoblot screening for PrPres in CWD-inoculated CM tissues. CWD-inoculated CM were euthanized at many different times throughout the 13.4-year study. Tissues from each individual CM were screened soon after euthanasia and again as a group at the conclusion of the study. All tissues shown were treated with proteinase K prior to testing. CWD-infected squirrel monkey brain (SM) was used as a positive control in each immunoblot. Lanes used for the molecular weight markers are marked with “mw.” Approximate molecular weights (in thousands) are shown at the left of each immunoblot. Immunoblots were probed with anti-PrP antibody 3F4 (A and B) and with anti-PrP antibody L42 (C and D). (A and B) Examples from two individual CM. (A) Lanes 3 to 10 show tissues from CM121 inoculated orally with CWD. Lanes: 3, cortex; 4, thalamus; 5, cerebellum; 6, spleen; 7, axillary lymph node; 8, cervical lymph node; 9, inguinal lymph node; 10, mesenteric lymph node. (B) Lanes 2 to 8 are tissues from CM130 inoculated orally with CWD, and lanes 9 to 10 are tissues from normal elk-inoculated CM633. Lanes: 2, ventral cerebral cortex; 3, hippocampus; 4, cerebellum; 5, brain stem; 6, dorsal cerebral cortex; 7, thalamus; 8, claustrum; 9, claustrum; 10, thalamus. (C) Cerebral cortex samples from 6 CM inoculated i.c. with CWD and 2 naive controls. (D) Cerebral cortex samples from 8 CM inoculated i.c. with CWD. Individual monkey identifications are provided across the top of panels C and D. In all blots, the SM positive-control lanes contained 0.24 mg and 0.36 mg of tissue equivalents, respectively. Experimental tissue lanes in panels A, C, and D contained 0.72 mg tissue equivalents per lane, and those in panel B contained 1.2 mg tissue equivalents per lane.
DISCUSSION
Several human prion diseases, including sporadic CJD (sCJD), vCJD, and BSE, have been transmitted to CM previously (9, 27, 29, 30), suggesting that CM might be a good surrogate primate species to predict human susceptibility to CWD. Previously, we reported that 7 of 14 CM inoculated with CWD by the i.c. or oral route were negative for prion disease after observation for 4.0 to 8.8 years (22). The present paper reports the final data on the remaining 7 CM that were not euthanized prior to the previous report in 2014. These animals were observed for 10.9 to 13.4 years and were euthanized for a variety of clinical issues or electively at the termination of the experiment. No CM was found to be positive for CWD based on the evaluation of brain and spinal cord for spongiosis and gliosis as well as testing for PrPSc deposition by IHC and by immunoblotting. In addition, tissues from these CM as well as the CM reported in 2014 were tested for PrP amyloid-seeding activity using the highly sensitive RT-QuIC test. The results of the RT-QuIC assay for all 14 of these CM were negative, and CWD-inoculated CM could not be distinguished from 7 negative-control CM who were not exposed to CWD. Thus, in this study, no clinical, pathological, or biochemical evidence suggested that CWD was transmitted from cervids to CM.
The long observation time and advanced age of some of the animals in this experiment posed potential problems for the analysis and diagnosis of prion disease in this cohort of CM. In fact, analysis of brain and spinal cord for PrPSc by IHC showed evidence of some unusual PrP deposits suggestive of the disease-associated form of PrP, PrPSc. However, these deposits were quite rare, and most were seen in both uninoculated and CWD-inoculated CM. The deposits stained with 3 different anti-PrP monoclonal antibodies, suggesting that they were indeed PrP, but their detection in uninoculated CM strongly implied that they were not PrPSc (Fig. 4). The detection of PrP staining unique to two orally inoculated CM was a concern (Fig. 5). However, in these two monkeys and all the other monkeys with PrP staining, there was no evidence of gliosis or vacuolation in the region of the PrP deposits (Fig. 4G and H and 5C and D), as is often found in prion disease brain tissue. In addition, the sensitive RT-QuIC test for PrP amyloid-seeding activity was negative with these samples, as was standard immunoblotting done for the detection of PK-resistant PrP. Thus, there was no supporting evidence for CWD infection of these two CM from either clinical observations or biochemical tests.
Additional experiments attempting to transmit CWD to CM were done by another group from Canada and Germany. Although their results have not yet been reported, oral presentations and abstracts have stated that they may have seen positive transmission of CWD to some CM. IHC staining of PrP in spinal cords of CWD-inoculated CM was the main disease-specific feature reported (31). However, in our present study, we saw similar PrP staining by IHC in spinal cords of both uninoculated and CWD-inoculated CM (Fig. 7). Therefore, we did not regard our data on this observation to be evidence for CWD infection of CM.
Prion disease transmission to SM and CM has been studied previously by several groups using both human- and animal-derived prion agents. Comparing these known patterns of transmission may be helpful for understanding which monkey model would be more predictive of prion disease transmission to humans. Human prion diseases, including sCJD and vCJD, as well as the closely related disease BSE, were shown previously to readily infect both CM and SM (28, 30, 32, 33). Similar experiments using sheep- or rodent-adapted scrapie agents gave slightly different results. SM were highly susceptible to scrapie with rather short incubation periods (1 to 3 years) (34, 35), and CM were susceptible to scrapie in only 2 of 3 reports, both following much longer incubation periods (28, 34, 36). In studies of the transmission of CWD to SM and CM, results differed for these two species. SM were susceptible to CWD (22), while CM were not, as shown here and reported by another group as an experiment in progress (28). It is not clear why CM and SM models give such different results with different prion agents. However, known human prion disease susceptibility appears to correlate more closely with the susceptibility pattern of CM than with that of SM.
Based on paleontological and DNA data, the evolutionary divergence between SM and CM is believed to have occurred 40 million to 50 million years ago (37, 38), so differences in disease susceptibility should not come as a surprise. However, PrP, which is a key protein influencing the species barrier to prion transmission, has only a 5-amino-acid-residue differences between SM and CM (not counting the extra octapeptide repeat seen in some SM) (21, 39). Any of the five differing residues might explain the differences in CWD susceptibility between SM and CM (14, 40–42). Compared to human PrP, CM and SM have nearly equal numbers of amino acid residue differences (21, 39). Due to the strong influence of single-amino-acid substitutions within PrP, it remains difficult to predict which of the two primate models better predicts the susceptibility of humans to CWD based on amino acid sequence alone. Nevertheless, there are no data suggesting that humans are similar to SM in susceptibility to CWD. In fact, SM are considered by some researchers to be similar to bank voles in their profile of broad susceptibility to diverse prion agents.
Experiments using transgenic mice expressing nonmouse PrP previously indicated that PrP itself appears to be the most important factor in determining the barrier to cross-species prion infection (14, 40–42). Therefore, transgenic mice expressing human PrP might be an important tool to assess the susceptibility of humans to CWD. CWD transmission studies using such mice have been attempted by several laboratories (15–19). Transmission and infection were not clearly demonstrated in any of these studies. However, there are many types of mice expressing human PrP at different levels and with different codon 129 genotypes, and the best combination for successful CWD transmission might not yet have been tested. It is unlikely that background non-Prnp mouse genes would block CWD infection of transgenic mice because transgenic mice expressing cervid PrP are in fact susceptible to CWD (19, 43, 44).
The existence of different strains of CWD infectivity was demonstrated previously (45–48), and such strains might be a confounding variable in assessments of the susceptibility of humans or related animal models to CWD. Possibly, human susceptibility to CWD might be restricted to a subset of CWD strains. Therefore, a variety of different CWD sources should be tested in animal models pertinent to human susceptibility to reduce the chance of overlooking a strain capable of human infection.
Setting aside the above-mentioned concerns about CWD strains and the validity of CM as a surrogate primate for human susceptibility, the present study found no evidence for the transmission of CWD to CM using a broad range of data, including clinical, pathological, and biochemical observations. Despite the observation of several suspicious types of PrP staining by IHC, this evidence was not definitive due to the presence of similar lesions in uninoculated and CWD-inoculated CM as well as a lack of gliosis or vacuolation. Similarly, biochemical tests using RT-QuIC and immunoblotting were also unable to distinguish uninoculated from CWD-inoculated CM, thus also supporting the conclusion that no transmission had occurred.
MATERIALS AND METHODS
Ethics statement.
All CWD-inoculated monkeys and six of the nine uninfected control monkeys were housed at the Rocky Mountain Laboratory (RML) in an AAALAC-accredited facility in compliance with guidelines provided by the Guide for the Care and Use of Laboratory Animals (49). Archival tissues from three uninoculated CM were provided by the National Institute on Aging, in cooperation with the Wisconsin National Primate Research Center, Madison, WI, USA. Experimentation with and housing of CM followed RML Animal Care and Use Committee-approved protocols 2002-01, 2006-30, 2008-05, 2011-21, 2014-016, 2014-047, and 2016-058.
Experimental design, inoculations, and clinical observations.
Cynomolgus monkeys were inoculated in 2003 and 2004 by either the i.c. or oral route with one of three different pools (MD-1, WTD-1, or Elk-1) of brain homogenates derived from CWD-infected deer and elk (Table 1). Details of CWD inoculum infectivity titers and inoculation methods and schedules were described previously (21). Briefly, six CM were injected intracerebrally with 500 μl of brain homogenate containing 5 mg of CWD-positive cervid brain. Oral challenges of the nine CM were performed by oral gavage using a rubber gastric tube. Doses were given at 2- to 6-day intervals for a total of 5 doses. Each dose contained 0.8 g of brain homogenate for a total of 4 g of CWD-infected cervid brain containing up to 2.0 × 109 infectious doses, as measured by a bioassay in transgenic mice expressing mule deer PrP (21). As a negative control, one CM (CM633) was inoculated i.c. with a normal elk brain homogenate. Following inoculations, CM were housed in individual cages together in one room. Monkeys could see, smell, and interact with each other. Toys, music, television, and a variety of treats were provided as stimulation. Tissue samples were also collected from other negative-control CM as follows: 5 uninoculated CMs housed at RML in a different building from the CWD study CM (animals 228, 949, 585, 82-51, and 161) and 3 uninoculated CM provided by the National Institute on Aging, Bethesda, MD, in cooperation with the NHP tissue bank, Wisconsin National Primate Research Center, Madison, WI (animals 151, 146, and 27). Prion disease-positive control brain tissues were obtained from 3 vCJD-infected CM (animals 16999, 7422, and 7423) from Luisa Gregori at the FDA (50).
CM housed at RML were observed twice daily by animal care staff and as needed by clinical staff veterinarians. In addition, CM were observed 1 to 2 times per week by prion investigators for assessment of overall health, with attention being given to behavioral changes and/or the onset of neurologic signs consistent with prion infection. Over the 13.4-year course of the study, 3 prion investigators shared the responsibility for these observations. Having a consistent group of observers was critical, as each monkey displayed unique behaviors and personalities. Investigators remained in close communication with care staff when behavioral changes or health concerns arose. Physical exams and blood chemistry analyses were performed on each monkey each year to assess general health. Additional exams and diagnostic tests were run as needed for particular individuals. Following euthanasia of each monkey, tissues were collected for prion-specific screening tests, including RT-QuIC, IHC, and immunoblot analysis.
RT-QuIC.
RT-QuIC reactions were performed as described previously, using recombinant bank vole prion protein (BV rPrPsen) as the substrate (residues 23 to 230 [methionine at residue 109]; GenBank accession no. AF367624) (25). PrP from bank voles has proven to be very good at detecting PrP amyloid-seeding activity by RT-QuIC assays in a wide variety of species, including deer and elk with CWD (25). Briefly, sample brains were homogenized to 10% (wt/vol) in phosphate-buffered saline (PBS). Homogenate supernatants were then collected following a 1-min clearance step at 2,000 × g. Samples were then 10-fold serially diluted in 0.05% SDS (sodium dodecyl sulfate; Sigma)–PBS–N2 (catalog no. 17502-048; Gibco) to yield brain tissue concentrations of 10−3. Four independent wells were tested for each monkey brain or spinal cord sample. Two-microliter sample volumes were added to reaction wells of a black 96-well, clear-bottom plate (Nunc) containing 98 μl of RT-QuIC reaction mix, resulting in final concentrations of 0.001% SDS, 10 mM phosphate buffer (pH 7.4), 300 mM NaCl, 0.1 mg/ml BV rPrPsen, 10 μM thioflavin T (ThT), and 1 mM EDTA. The plate was then sealed with a plate sealer film (Nunc) and incubated at 42°C in a BMG Labtech FLUOstar Omega plate reader with a repeating protocol of 1 min of shaking (700 rpm, double orbital) and 1 min of rest throughout the indicated incubation times. ThT fluorescence measurements (450-nm ± 10-nm excitation and 480-nm ± 10-nm emission; readings were collected from the bottom of the plate) were taken every 45 min. Data were normalized to the percentage of maximum fluorescence obtained from the strongest signal by a positive-control sample tested in the same run. In the experiments shown here, vCJD CM16999 brain at a 10−3 dilution was used as the positive control. To distinguish RT-QuIC-positive and -negative results, we established criteria based on testing of negative- and positive-control samples. Positive controls included three vCJD-inoculated CM and three cervid-derived CWD pools. Negative controls included 1 normal deer brain, 6 uninoculated CM, and 1 CM inoculated with normal elk brain. To be scored positive in this assay, samples had to show >33% of the maximal fluorescence signal obtained for the strongest positive-control signal (described above) in 2 or more out of 4 wells prior to the 25-h reaction time (Fig. 1 and 2).
RT-QuIC reactions using recombinant hamster 90-231 prion protein (GenBank accession no. K02234) were performed under conditions identical to those described above except that the running temperature of the plate was 50°C. Since no uninoculated or CWD-inoculated CM had measurable fluorescence above baseline levels prior to 50 h, they were all scored negative.
Immunohistochemistry.
Tissues were removed and placed into 10% neutral buffered formalin for 3 to 5 days. Following fixation, half-brains were cut into 4 to 6 blocks at even increments, representing different levels from midline to lateral aspects of the brain. These sections representing different brain regions were all stained as independent samples for each monkey. For spinal cords, tissues were blocked to obtain a 1- to 2-cm-long sagittal section and multiple cross sections on each slide. All other tissue types were processed for routine cross sections and longitudinal sections of the tissue. Tissues were then processed by dehydration and embedding in paraffin. Sections were cut by using a standard Leica microtome, placed onto positively charged glass slides, and air dried overnight at room temperature. On the following day, slides were heated in an oven at 60°C for 20 min. Neuropathology was assessed on hematoxylin and eosin (H&E)-stained sections. H&E staining was performed according to the manufacturer's (Shandon) instructions, with hematoxylin incubation for 12 min and eosin incubation for 4 min.
All IHC, deparaffinization, antigen retrieval, and staining procedures were performed on the automated Discovery XT staining system (Ventana Medical Systems). “No-primary-antibody” controls were run on a subset of slides for each CM for each antibody detection system.
For staining of prion protein using monoclonal antiprion antibody 3F4, 6H4, or L42, antigen retrieval was done by using the Discovery XT system with the extended CC1 protocol (cell conditioning buffer containing Tris-borate-EDTA [pH 8.0], incubated for 44 min at 100°C). This antigen retrieval step does not destroy normal cellular, non-disease-associated PrP (PrPsen), and a background level of PrPsen staining can be seen by using 6H4 or L42 with the protocols described below. In comparison, 3F4 staining (described below) did not show a strong PrPsen background. The 3F4 antibody (Laboratory of Persistent Viral Diseases tissue culture 3F4 supernatant, lot no. 2-18-04) was used at a dilution of 1:5 in antibody dilution buffer (catalog no. ADB250; Ventana), applied for 60 min at 37°C. The 6H4 antibody (catalog no. ID01-010; Prionics) was used at a 1:10,000 dilution in antibody dilution buffer, applied for 2 h at 37°C. The L42 antibody (R-Biopharm) was used at a 1:400 dilution in antibody dilution buffer, applied for 2 h at 37°C. The secondary antibody for all reactions was biotinylated goat anti-mouse IgG (BioGenex ready-to-use supersensitive rabbit link), applied for 32 min at 37°C. For all anti-PrP staining, staining was completed by using a DABMap detection kit and hematoxylin counterstain.
For GFAP staining, antigen retrieval was done by using the Discovery XT system with the mild CC1 protocol (cell conditioning buffer containing Tris-borate-EDTA [pH 8.0], with incubation for 12 min at 100°C). The anti-GFAP antibody was used at a dilution of 1:3,500 in antibody dilution buffer, applied for 16 min at 37°C. The secondary antibody was biotinylated goat anti-rabbit IgG, as described above, applied for 16 min at 37°C. Staining was completed by using a RedMap detection kit and hematoxylin counterstain.
For the detection of microglia, polyclonal rabbit anti-Iba1 antiserum was used. This antiserum was generated by the immunization of rabbits with a 14-amino-acid peptide from the C terminus of the Iba1 protein, as described previously (51), and was a generous gift from John Portis. For Iba1, antigen retrieval was done by using the Discovery XT system standard CC1 protocol (cell conditioning buffer containing Tris-borate-EDTA [pH 8.0] for ∼44 min at 100°C). Anti-Iba1 was used at a 1:2,000 dilution and applied for 40 min at 37°C. The secondary antibody used was biotinylated goat anti-rabbit IgG (BioGenex ready-to-use supersensitive rabbit link) and was applied for 40 min at 37°C. Staining was completed by using a RedMap detection kit and hematoxylin counterstain.
Sections stained with H&E, 3F4, 6H4, L42, Iba1, and GFAP were scanned with an Aperio ScanScope XT instrument (Aperio Technologies, Inc.) and analyzed and photographed by using Aperio Imagescope software.
Immunoblotting for detection of PrP.
Tissue was homogenized in 1× PBS as a 20% (wt/vol) tissue homogenate using a mini-bead beater for 45 s on the homogenization setting. Aliquots were stored at −20°C. For the detection of proteinase K (PK)-resistant PrP (PrPres), samples were treated with PK at 50 μg/ml. Briefly, 20 μl of a 20% homogenate from each sample was adjusted with a solution containing 100 mM Tris-HCl (pH 8.3), 1% Triton X-100, 1% sodium deoxycholate, and 50 μg/ml PK in a total volume of 31 μl. Samples were incubated for 45 min at 37°C. All PK digestions were stopped by the addition of 2 μl of 100 mM Pefabloc (Roche Diagnostics), and the reaction mixture was placed on ice for 5 min. An equal volume of 2× Laemmli sample buffer (Bio-Rad, Hercules, CA) was added, and the samples were boiled for 5 min. Samples were frozen at −20°C until needed for gel electrophoresis. At this time, they were thawed, reboiled for 5 min, electrophoresed on a 16% Tris-glycine SDS-polyacrylamide gel electrophoresis (PAGE) gel (Life Technologies, CA), and blotted onto polyvinylidene difluoride (PVDF) membranes using a 7-min transfer on an iBlot device (Life Technologies). Immunoblots were probed with anti-PrP antibody 3F4 at a 1:500 to 1:3,000 dilution or L42 at a 1:3,000 dilution. The secondary antibody used was peroxidase-conjugated rabbit anti-mouse IgG (Sigma) at a 1:5,000 to 1:80,000 dilution, depending on the lot used. Protein bands were visualized by using either an enhanced chemiluminescence (ECL) or SuperSignal West Femto detection system according to the manufacturer's instructions (Thermo Scientific).
ACKNOWLEDGMENTS
We thank Suzette Priola, Cathryn Haigh, and James Carroll for critical reviews of the manuscript; Nancy Kurtz and Dan Long for assistance with histology preparation; Andrew MacLean and Dana Scott for hemosiderin pigment interpretation; Jamie Lovaglio, Dana Scott, Greg Saturday, Jennifer Hayes, and Tracy Cope for additional normal CM tissues; Don Gardner and RMVB staff for assistance with CM inoculations, animal husbandry, and health care; Luisa Gregori from the FDA, Division of Emerging and Transfusion-Transmitted Diseases, for vCJD-infected CM brain material; Stefanie Czub and John Gray for helpful discussions regarding the project; Anita Mora for graphic art assistance; and Mike Miller, Terry Kreeger, Jean Jewell, and Lynn Creekmore for CWD agent-positive and -negative cervid tissues.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH.
REFERENCES
- 1.Saunders SE, Bartelt-Hunt SL, Bartz JC. 2012. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg Infect Dis 18:369–376. doi: 10.3201/eid1803.110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haley NJ, Hoover EA. 2015. Chronic wasting disease of cervids: current knowledge and future perspectives. Annu Rev Anim Biosci 3:305–325. doi: 10.1146/annurev-animal-022114-111001. [DOI] [PubMed] [Google Scholar]
- 3.Zabel M, Ortega A. 2017. The ecology of prions. Microbiol Mol Biol Rev 81:e00001-. doi: 10.1128/MMBR.00001-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uehlinger FD, Johnston AC, Bollinger TK, Waldner CL. 2016. Systematic review of management strategies to control chronic wasting disease in wild deer populations in North America. BMC Vet Res 12:173. doi: 10.1186/s12917-016-0804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. 1987. The epidemiology of Creutzfeldt-Jakob disease: conclusion of a 15-year investigation in France and review of the world literature. Neurology 37:895–904. doi: 10.1212/WNL.37.6.895. [DOI] [PubMed] [Google Scholar]
- 6.van Duijn CM, Delasnerie-Laupretre N, Masullo C, Zerr I, de Silva R, Wientjens DP, Brandel JP, Weber T, Bonavita V, Zeidler M, Alperovitch A, Poser S, Granieri E, Hofman A, Will RG. 1998. Case-control study of risk factors of Creutzfeldt-Jakob disease in Europe during 1993-95. European Union (EU) Collaborative Study Group of Creutzfeldt-Jakob disease (CJD). Lancet 351:1081–1085. doi: 10.1016/S0140-6736(97)09468-3. [DOI] [PubMed] [Google Scholar]
- 7.Race B, Phillips K, Meade-White K, Striebel J, Chesebro B. 2015. Increased infectivity of anchorless mouse scrapie prions in transgenic mice overexpressing human prion protein. J Virol 89:6022–6032. doi: 10.1128/JVI.00362-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassard H, Torres JM, Lacroux C, Douet JY, Benestad SL, Lantier F, Lugan S, Lantier I, Costes P, Aron N, Reine F, Herzog L, Espinosa JC, Beringue V, Andreoletti O. 2014. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun 5:5821. doi: 10.1038/ncomms6821. [DOI] [PubMed] [Google Scholar]
- 9.Lasmezas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, Robain O, Ironside J, Hauw JJ. 1996. BSE transmission to macaques. Nature 381:743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 10.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 11.Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. 1997. The same prion strain causes vCJD and BSE. Nature 389:448–550. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 12.Waddell L, Greig J, Mascarenhas M, Otten A, Corrin T, Hierlihy K. 2018. Current evidence on the transmissibility of chronic wasting disease prions to humans—a systematic review. Transbound Emerg Dis 65:37–49. doi: 10.1111/tbed.12612. [DOI] [PubMed] [Google Scholar]
- 13.Hannaoui S, Schatzl HM, Gilch S. 2017. Chronic wasting disease: emerging prions and their potential risk. PLoS Pathog 13:e1006619. doi: 10.1371/journal.ppat.1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurt TD, Sigurdson CJ. 2016. Cross-species transmission of CWD prions. Prion 10:83–91. doi: 10.1080/19336896.2015.1118603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurt TD, Jiang L, Fernandez-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, Sigurdson CJ. 2015. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest 125:1485–1496. doi: 10.1172/JCI79408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, Suardi S, Ruggerone M, Moda F, Graziano S, Sbriccoli M, Cardone F, Pocchiari M, Ingrosso L, Baron T, Richt J, Andreoletti O, Simmons M, Lockey R, Manson JC, Barron RM. 2012. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J Gen Virol 93:1624–1629. doi: 10.1099/vir.0.042507-0. [DOI] [PubMed] [Google Scholar]
- 17.Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O'Malley C, Powell C, Asante EA, Linehan JM, Brandner S, Wadsworth JD, Collinge J. 2010. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol 91:2651–2657. doi: 10.1099/vir.0.024380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamguney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. 2006. Transmission of elk and deer prions to transgenic mice. J Virol 80:9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, Chen K, Jenny AL, O'Rourke K, Belay ED, Schonberger LB, Petersen RB, Sy MS, Chen SG, Gambetti P. 2005. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh RF, Kincaid AE, Bessen RA, Bartz JC. 2005. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol 79:13794–13796. doi: 10.1128/JVI.79.21.13794-13796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, Parnell M, Striebel J, Priola SA, Ward A, Williams ES, Race R, Chesebro B. 2009. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis 15:1366–1376. doi: 10.3201/eid1509.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Race B, Meade-White KD, Phillips K, Striebel J, Race R, Chesebro B. 2014. Chronic wasting disease agents in nonhuman primates. Emerg Infect Dis 20:833–837. doi: 10.3201/eid2005.130778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams ES, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 24.Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. 2010. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6:e1001217. doi: 10.1371/journal.ppat.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orru CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, Ghetti B, Gambetti P, Caughey B. 2015. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog 11:e1004983. doi: 10.1371/journal.ppat.1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lasmezas CI, Fournier JG, Nouvel V, Boe H, Marce D, Lamoury F, Kopp N, Hauw JJ, Ironside J, Bruce M, Dormont D, Deslys JP. 2001. Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt-Jakob disease: implications for human health. Proc Natl Acad Sci U S A 98:4142–4147. doi: 10.1073/pnas.041490898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marce D, Auvre F, Ruchoux MM, Ferrari S, Monaco S, Sales N, Caramelli M, Leboulch P, Brown P, Lasmezas CI, Deslys JP. 2008. Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS One 3:e3017. doi: 10.1371/journal.pone.0003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comoy EE, Mikol J, Luccantoni-Freire S, Correia E, Lescoutra-Etchegaray N, Durand V, Dehen C, Andreoletti O, Casalone C, Richt JA, Greenlee JJ, Baron T, Benestad SL, Brown P, Deslys JP. 2015. Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep 5:11573. doi: 10.1038/srep11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasmezas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, Auvre F, Correia E, Lescoutra-Etchegaray N, Sales N, Wells G, Brown P, Deslys JP. 2005. Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783. doi: 10.1016/S0140-6736(05)17985-9. [DOI] [PubMed] [Google Scholar]
- 30.Herzog C, Riviere J, Lescoutra-Etchegaray N, Charbonnier A, Leblanc V, Sales N, Deslys JP, Lasmezas CI. 2005. PrPTSE distribution in a primate model of variant, sporadic, and iatrogenic Creutzfeldt-Jakob disease. J Virol 79:14339–14345. doi: 10.1128/JVI.79.22.14339-14345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Czub S, Schulz-Schaeffer W, Stahl-Hennig C, Beekes M, Schaetzl H, Motzkus D. 25 May 2017, posting date First evidence of intracranial and peroral transmission of chronic wasting disease (CWD) into cynomolgus macaques: a work in progress. YouTube video, 1:32:00, posted by Laurent Bernard https://www.youtube.com/embed/Vtt1kAVDhDQ.
- 32.Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. 1994. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 33.Piccardo P, Cervenak J, Yakovleva O, Gregori L, Pomeroy K, Cook A, Muhammad FS, Seuberlich T, Cervenakova L, Asher DM. 2012. Squirrel monkeys (Saimiri sciureus) infected with the agent of bovine spongiform encephalopathy develop tau pathology. J Comp Pathol 147:84–93. doi: 10.1016/j.jcpa.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gajdusek DC. 1977. Unconventional viruses and the origin and disappearance of kuru. Science 197:943–960. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 35.Gibbs CJ Jr, Gajdusek DC. 1973. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 182:67–68. doi: 10.1126/science.182.4107.67. [DOI] [PubMed] [Google Scholar]
- 36.Gibbs CJ Jr, Gajdusek DC. 1972. Transmission of scrapie to the cynomolgus monkey (Macaca fascicularis). Nature 236:73–74. doi: 10.1038/236073a0. [DOI] [PubMed] [Google Scholar]
- 37.Hayasaka K, Gojobori T, Horai S. 1988. Molecular phylogeny and evolution of primate mitochondrial DNA. Mol Biol Evol 5:626–644. [DOI] [PubMed] [Google Scholar]
- 38.Aristide L, Rosenberger AL, Tejedor MF, Perez SI. 2015. Modeling lineage and phenotypic diversification in the New World monkey (Platyrrhini, Primates) radiation. Mol Phylogenet Evol 82(Part B):375–385. doi: 10.1016/j.ympev.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Schatzl HM, Da Costa M, Taylor L, Cohen FE, Prusiner SB. 1995. Prion protein gene variation among primates. J Mol Biol 245:362–374. doi: 10.1006/jmbi.1994.0030. [DOI] [PubMed] [Google Scholar]
- 40.Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J, Will R, Manson JC. 2001. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J 20:5070–5078. doi: 10.1093/emboj/20.18.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlson GA, Westaway D, DeArmond SJ, Peterson-Torchia M, Prusiner SB. 1989. Primary structure of prion protein may modify scrapie isolate properties. Proc Natl Acad Sci U S A 86:7475–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 43.Meade-White K, Race B, Trifilo M, Bossers A, Favara C, Lacasse R, Miller M, Williams E, Oldstone M, Race R, Chesebro B. 2007. Resistance to chronic wasting disease in transgenic mice expressing a naturally occurring allelic variant of deer prion protein. J Virol 81:4533–4539. doi: 10.1128/JVI.02762-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C, Miller MW, Williams ES, Hoover E, Telling GC. 2004. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol 78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond GJ, Raymond LD, Meade-White KD, Hughson AG, Favara C, Gardner D, Williams ES, Miller MW, Race RE, Caughey B. 2007. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. J Virol 81:4305–4314. doi: 10.1128/JVI.02474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angers RC, Kang HE, Napier D, Browning S, Seward T, Mathiason C, Balachandran A, McKenzie D, Castilla J, Soto C, Jewell J, Graham C, Hoover EA, Telling GC. 2010. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 328:1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrott MR, Sigurdson CJ, Mason GL, Hoover EA. 2012. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J Gen Virol 93:212–221. doi: 10.1099/vir.0.035006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duque Velasquez C, Kim C, Herbst A, Daude N, Garza MC, Wille H, Aiken J, McKenzie D. 2015. Deer prion proteins modulate the emergence and adaptation of chronic wasting disease strains. J Virol 89:12362–12373. doi: 10.1128/JVI.02010-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 50.McDowell KL, Nag N, Franco Z, Bu M, Piccardo P, Cervenak J, Deslys JP, Comoy E, Asher DM, Gregori L. 2015. Blood reference materials from macaques infected with variant Creutzfeldt-Jakob disease agent. Transfusion 55:405–412. doi: 10.1111/trf.12841. [DOI] [PubMed] [Google Scholar]
- 51.Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. 1996. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]