

Abstract

We report a new and robust strategy toward the development of high-performance pressure sensitive adhesives (PSAs) from chemicals directly obtained from raw biomass deconstruction. A particularly unique and translatable aspect of this work was the use of a monomer obtained from real biomass, as opposed to a model compound or lignin-mimic, to generate well-defined and nanostructure-forming polymers. Herein, poplar wood depolymerization followed by minimal purification steps (filtration and extraction) produced two aromatic compounds, 4-propylsyringol and 4-propylguaiacol, with high purity and yield. Efficient functionalization of those aromatic compounds with either acrylate or methacrylate groups generated monomers that could be easily polymerized by a scalable reversible addition–fragmentation chain-transfer (RAFT) process to yield polymeric materials with high glass transition temperatures and robust thermal stabilities, especially relative to other potentially biobased alternatives. These lignin-derived compounds were used as a major component in low-dispersity triblock polymers composed of 4-propylsyringyl acrylate and n-butyl acrylate (also can be biobased). The resulting PSAs exhibited excellent adhesion to stainless steel without the addition of any tackifier or plasticizer. The 180° peel forces were up to 4 N cm–1, and tack forces were up to 2.5 N cm–1, competitive with commercial Fisherbrand labeling tape and Scotch Magic tape, demonstrating the practical significance of our biomass-derived materials.
Short abstract
We demonstrated the successful incorporation of materials obtained directly from raw biomass deconstruction in the development of nanostructured pressure sensitive adhesives for use in tapes.
Introduction
Lignin, a major component in lignocellulosic biomass, is the most abundant source of aromatic building blocks in nature,1 and the depolymerization of lignin is capable of producing value-added small aromatic molecules with great potential as fuel additives and platform chemicals.2−8 Unfortunately, the aromatic moieties in lignin are linked by various kinds of robust C–C and C–O bonds,9 and deconstruction of lignin therefore generates a complex mixture of disparate compounds (monophenols, dimers, and oligomers).4,10−14 Few studies have focused on the direct use of chemicals from raw biomass deconstruction.15,16 Instead, lignin model compounds have been extensively explored toward the formulation of new products for applications, such as thermoplastics, thermoplastic elastomers, coatings, pressure sensitive adhesives (PSAs), composites, and resins.17−26 However, a major unanswered question is “Can these biobased compounds be harvested from raw biomass to produce designer materials in a scalable and cost-effective manner?” Essentially, a huge gap exists between deriving well-defined chemicals from raw biomass and directly utilizing these chemicals for the formulation of specialized consumer products. Robust and scalable depolymerization, purification, functionalization, and polymerization steps, as well as potential recycle and reuse of catalyst and solvents in the process,27 are necessary to reduce the energy and cost associated with “green” materials fabrication and to encourage the sustained use of biomass-derived materials in mainstream applications.
This report demonstrates, for the first time, the harnessing of aromatic chemicals obtained at high purity and yield directly from raw biomass depolymerization for the preparation of PSAs with commercially viable adhesion properties. A distinguishing feature of this work is that real biomass, and not model compounds or lignin-mimics, was used in the generation of self-assembling, nanostructured, polymer materials.
PSAs are a family of adhesives that instantaneously adhere to a substrate after a small external pressure is applied. PSAs are ubiquitous in everyday life with numerous uses, such as packaging, labels, sticky notes, and plastic wraps,28 and the global PSA market is growing rapidly with an expected market of $13 billion by 2023.29 One important class of PSAs is composed of triblock polymers with glassy end blocks and a rubbery midblock, and the viscoelastic properties of the chosen polymers greatly impact the adhesion performance of the resulting PSAs.30−32 When the rubbery block has a low entanglement molecular weight (Me), such as polyisoprene [PI] (Me = 6.1 kg mol–1) or polybutadiene [PB] (Me = 1.7 kg mol–1),33,34 the resulting composites exhibit high storage moduli and relatively poor adhesion to substrates. Hence, rubbery segment-miscible tackifiers or plasticizers often are blended with these PI- or PB-based block polymers to lower the material’s storage modulus and enhance adhesion with the substrate.28 Acrylate-based block polymers, such as those containing glassy poly(methyl methacrylate) (PMMA) end blocks with a rubbery poly(n-butyl acrylate) (PBA) middle block, typically exhibit good adhesion with and without tackifier additives because of the lower effective modulus,35 and much higher Me (Me = 28 kg mol–1) of the PBA block.36 Furthermore, n-butyl acrylate is an attractive choice as it can be derived from biosourced n-butanol.37 Thus, if the PMMA can be replaced by a renewable and sustainable component,38,39 it will be possible to realize fully sustainable PSAs, as will be demonstrated herein.
Results and Discussion
Poplar wood was deconstructed in methanol with a commercially available Ru/C catalyst (Scheme 1), which is effective and selective to C–O bonds cleavage.27,40 The depolymerization details are provided in the Methods section. The aromatic compound-enriched solution was separated from the solid cellulose residue and the catalyst by vacuum filtration, and two prominent monophenolic compounds, 4-propylsyringol (4pS) and 4-propylguaiacol (4pG), were collected, along with small portions of other dihydroxyl-containing components (Figure 1a). Following a simple extraction with cyclohexane, high-purity monophenolic compounds, 4pS and 4pG, were obtained (Figure 1b) with relative mass fractions of 0.6 and 0.4, respectively, at a total yield of 10 wt % on the basis of weight of dry poplar wood. The removal of the dihydroxyl species is critical to prevent the formation of a cross-linked network during the polymerization process. Then, 4pS and 4pG were efficiently functionalized with either acrylate or methacrylate groups, followed by polymerization via a scalable reversible addition–fragmentation chain-transfer (RAFT) approach (details are located in the Methods section and the Supporting Information).41 All polymerizations proceeded in a controlled manner, generating relatively high-molecular-weight polymers with relatively low dispersities. The characteristics of the resulting polymers, poly(4-propylsyringyl acrylate) (P4pSA), poly(4-propylguaiacyl acrylate) (P4pGA), poly(4-propylsyringyl methacrylate) (P4pSMA), and poly(4-propylguaiacyl methacrylate) (P4pGMA), are summarized in Table 1. The 1H NMR spectra of synthesized monomers and polymers are provided in the Supporting Information (Figures S1–S3). The glass transition temperatures (Tg’s) of P4pSA (98 °C) and P4pSMA (169 °C) were attractive relative to polystyrene (PS, Tg ∼ 100 °C) and PMMA (Tg ∼ 110 °C). The Tg’s of P4pGA (56 °C) and P4pGMA (80 °C) with one methoxy group at the ortho position were lower than those of P4pSA and P4pSMA, as the existence of two ortho methoxy groups constrained rotation of the pendant groups and raised the Tg’s of the dimethoxy-based materials.18 A copolymer of 4pSMA and 4pGMA also was synthesized to demonstrate the feasibility of making polymers from the original biomass mixture, without fractionation into individual components. The monomer ratio of 4pSMA/4pGMA (0.60/0.40) and the segment content in the polymer (0.58/0.42, as determined via 1H NMR spectroscopy) were consistent, suggesting a random incorporation of each substituent in the polymer backbone. The resulting copolymer had a Tg of 135 °C, which was close to that estimated for a random copolymer of 4pSMA and 4pGMA on the basis of the Flory–Fox equation (∼127 °C). The thermal behavior of these lignin-derived polymers, especially the desirable glass transition temperatures, provides an initial indication of their utility in the PSA materials as will be discussed below.
Scheme 1. Process from Raw Biomass to PSAs (4-Propylsyringol, R = OCH3; 4-Propylguaiacol, R = H).

Figure 1.
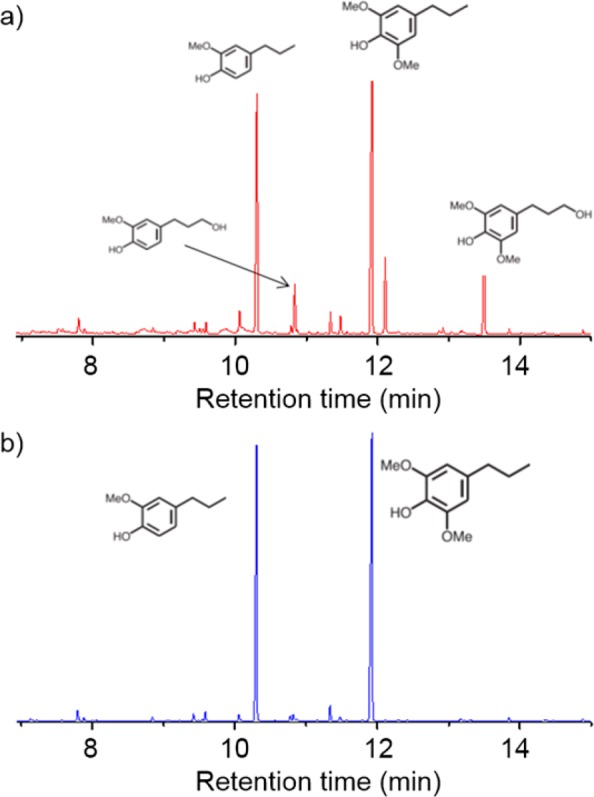
Gas chromatography (GC) trace of raw biomass depolymerization products before (a) and after (b) extraction with cyclohexane.
Table 1. Characteristics of Lignin-Derived Polymers.
| polymer | Mna (kg mol–1) | Đb | Tgc (°C) |
|---|---|---|---|
| P4pSA | 19.1 | 1.44 | 98 |
| P4pGA | 29.6 | 1.29 | 56 |
| P4pSMA | 30.4 | 1.16 | 169 |
| P4pGMA | 12.4 | 1.29 | 80 |
| P(4pSMA-co-4pGMA) (0.58/0.42)d | 26.7 | 1.26 | 135 |
Number-average molecular weight, determined by size-exclusion chromatography (SEC) (see the Methods section).
Dispersity, determined by SEC.
Determined by differential scanning calorimetry (DSC) (see the Methods section).
A mixture of 4pSMA and 4pGMA was fed, and the numbers denote the composition of 4pSMA and 4pGMA (mol/mol) in the resulting copolymer.
To demonstrate the ease of producing model consumer products directly from raw biomass depolymerization products, a triblock polymer was synthesized with P4pSA as the glassy end blocks and PBA as the midblock, generating poly(4pSA-b-BA-b-4pSA) (SaBSa) (Scheme 1 step 3, details are located in the Methods section and the SI). The SEC chromatograms of PBA and SaBSa are presented in Figure 2, and the clean shift in molecular weight from before (Mn = 49.7 kg mol–1, Đ = 1.11) vs after addition of the P4pSA end blocks is clearly indicative of the ability to chain extend using the biomass-derived monomers, while retaining substantial control over the polymerization. The final SaBSa polymer had a Mn of 66.4 kg mol–1, a Đ of 1.15, and a P4pSA weight percentage of 22%, as determined via 1H NMR spectroscopy (Figure S4). These macromolecular characteristics were targeted to approximate a commercial poly(MMA-b-BA-b-MMA) (MBM, Kurarity LA2140e, Mn = 66.9 kg mol–1, Đ = 1.12, 23 wt % PMMA) produced by Kuraray Co., Ltd., for use in PSAs.
Figure 2.
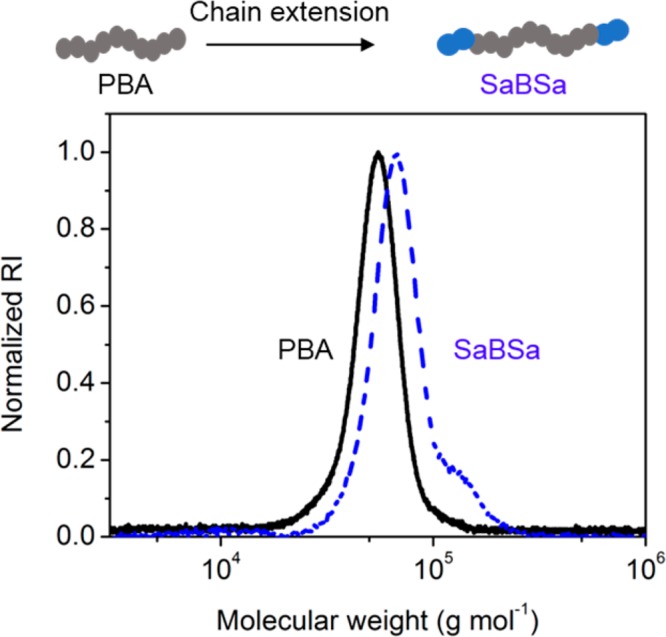
SEC chromatograms of PBA (black solid line, Mn = 49.7 kg mol–1, Đ = 1.11) and SaBSa (blue dashed line, Mn = 66.4 kg mol–1, Đ = 1.15). RI denotes refractive index detector response.
The thermal behavior and phase separation characteristics of SaBSa and MBM were compared by measuring Tg’s and size-scales of microphase separation via DSC and small-angle X-ray scattering (SAXS), respectively. Tg’s corresponding to the PBA-rich domains were detected at −45 °C (SaBSa) and −49 °C (MBM), see also Figure S5. No clear transition of P4pSA (∼98 °C) or PMMA (∼110 °C) was found. Similar results also have been reported in PSAs with a PBA midblock,39 likely due to the low weight fraction of the end block in each polymer (∼11 wt % for each end block); however, the similarity of PBA Tg to that of PBA homopolymer is suggestive of block immiscibility and microphase separation. Nanostructuring in the biobased SaBSa triblock copolymer was confirmed by SAXS (experimental details are given in the SI). A principle scattering peak (q*) at 0.030 Å–1, corresponding to a domain spacing (D* = 2π/q*) of ∼21 nm, was clearly visible in the 1-D azimuthally integrated SAXS data (Figure S6a,b). A similar scattering pattern also was obtained for MBM (Figure S6c,d), which suggested a domain spacing of ∼20 nm.
The overall thermal stability of SaBSa was evaluated by thermogravimetric analysis (TGA), and the bioderived triblock exhibited excellent thermal stability. SaBSa was thermally stable up to 337 °C under air (the temperature at which the weight of the polymer was reduced by 5%), see also Figure S7. The thermal degradation of MBM also was characterized in air flow, and the thermal stability was much lower, with a 5% degradation temperature of 285 °C (Figure S7). Furthermore, when comparing SaBSa to other model bioinspired PSAs reported in the literature, the lignin-derived materials herein have thermal degradation temperatures in air that are at least 30 °C higher than acrylate-functionalized glucose, acrylate-functionalized isosorbide, and polyester-based systems,38,39,42 providing a much larger temperature window for processing.
While the thermal parameters are important, adhesion performance is most critical in evaluating the potential of materials as PSAs. Herein, three tests were performed to assess adhesive properties: a 180° peel test (ASTM D3330, width 12.7 mm), a loop tack force test (ASTM D6195, width 12.7 mm), and a shear strength test (ASTM D3654, contact area of 12.7 mm × 12.7 mm). The 180° peel test determines the force needed to tear a strip off the adherend at a constant speed; the loop tack force test measures the bonding strength that forms instantly between adhesive and adherend when they are brought into contact; and the shear strength test probes the resistance of the adhesive to creep under an applied load. In addition to the above test, the mode of PSA failure is qualitatively evaluated as either adhesive (interfacial failure with no residue left on the substrate) or cohesive (failure within the adhesive layer itself, leaving residue on the substrate), noting that adhesive failure is more desirable for removable PSAs, while cohesive failure is more favorable for permanent PSAs.28,39
The neat SaBSa polymers exhibited excellent adhesion properties, without the addition of any tackifiers or other additives. The 180° peel force and loop tack force data (stainless steel as the adherend) of SaBSa are presented in Figure 3, and Figures S8 and S9 (data for MBM, Scotch Magic tape, and Fisherbrand labeling tape also are included for comparison). All samples had adhesive failure, leaving no residue on the adherend. The 180° peel force for SaBSa was in the range 2–4 N cm–1, with an average of 3.1 N cm–1, which was comparable to commercial Fisherbrand labeling tape (3.5–5 N cm–1) and Scotch Magic tape (1.7–2.0 N cm–1). By contrast, the 180° peel force for commercial MBM was very low (0.2–0.5 N cm–1). The average loop tack force of SaBSa was 2.2 N cm–1, comparable to Scotch Magic tape (1.4 N cm–1) and Fisherbrand labeling tape (3.9 N cm–1), while the MBM samples were not able to bind with the adherend during the loop tack test. In the shear test, a 500 g weight was suspended from a SaBSa polymer strip adhered to a stainless-steel plate, and the time for the SaBSa to detach from the adherend was recorded. Three samples of SaBSa were tested, with a range of failure time 570–810 min. The shear resistance of SaBSa was better than those of commercial duct tape (width 25 mm, failure time ∼200 min), electrical tape (width 20 mm, failure time ∼500 min), and Post-it notes (width 16 mm, failure time <0.5 min), but not as good as those of Scotch tape (width 20 mm, failure time >10 000 min) and paper tape (width 25 mm, failure time ∼1400 min).43 The peel and loop tack forces of biomass-derived SaBSa also were as good as or better than those reported in potentially biobased polyesters-based PSAs (with the addition of tackifier) and acrylic PSAs with glucose or isosorbide components (no addition of tackifier).38,39,42−44 These comparisons reveal that biomass-derived SaBSa polymers are extremely promising for PSA applications, without the addition of tackifiers or any other additives.
Figure 3.

Illustration of 180° peel test (a) and loop tack test (d). Photos showing the setup of 180° peel test (b) and loop tack test (e). Comparison of 180° peel force (c) and loop tack force (f) of SaBSa triblock polymer with commercial MBM, Scotch Magic tape, and Fisherbrand labeling tape. In panel f, the red star associated with MBM indicates that MBM did not form a bond with the adherend under the testing conditions.
The tensile strength of PSAs is an inverse indicator of adhesion performance. Typically, when PSAs have high tensile strength (>2 MPa), tackifying additives are needed to afford good adhesion, and when PSAs have relatively low tensile strength (<1 MPa), no tackifiers are required.38,39,42−44 The stress–strain curves of SaBSa and MBM from dog-bone-shaped specimens (ASTM D638) are shown in Figure S10. The average tensile strength and elongation at break were 0.73 ± 0.15 MPa and 450 ± 60% for SaBSa, respectively, and 8.1 ± 1.9 MPa and 580 ± 64% for MBM. The relatively low tensile strength of the SaBSa (<1 MPa), when combined with the previously mentioned adhesion properties, demonstrates the potential of neat SaBSa for PSA applications.
Conclusion
Triblock polymers containing an aromatic monomer from real raw biomass, as opposed to lignin model compounds or lignin-mimics, exhibit excellent thermal and adhesion properties, suitable for applications as PSAs. A major component of this biobased material was obtained from the depolymerization of poplar wood, which led to two aromatic compounds, 4-propylsyringol and 4-propylguaiacol, with high purity and yield after simple purification. Efficient functionalization of the aromatic compounds with either acrylate or methacrylate groups generated monomers that were polymerized by RAFT method to produce homopolymers and random copolymers with high glass transition temperatures. Low-dispersity triblock polymers (SaBSa) with lignin-based and n-butyl acrylate monomers also were prepared by RAFT. The polymers exhibited superior thermal stability up to 337 °C in air (more stable than commercial MBM, polyester PSAs, and acrylic PSAs with glucose or isosorbide components). Most importantly, this lignin-based polymer had 180° peel forces up to 4 N cm–1 and tack forces up to 2.5 N cm–1, which are comparable to commercial Fisherbrand labeling tape and Scotch Magic tape. Thus, this study demonstrates a robust strategy toward the development of high-performance PSAs from chemicals derived directly from raw biomass.
Methods
See the Supporting Information for additional descriptions of all methods, characterization data, and experiments; major details are provided below.
General Procedure of Lignocellulosic Biomass Depolymerization and Purification
Poplar wood powder (1 g, particle size <0.5 mm), methanol (20 mL), and catalyst (5 wt % Ru/C, Sigma-Aldrich, 100 mg) were added to a 50 mL high-pressure Parr reactor. The reactor was purged with H2 three times and then pressurized with H2 to a pressure of 40 bar. The reactor was heated to 250 °C and held for 15 h while stirring. After the reaction was completed, the reactor was cooled to room temperature with an external flow of compressed air. The solution containing aromatic monomers was separated from the resultant slurry by filtration. The solid (cellulose, catalyst) was washed with methanol (10 mL, 3 times), and the solution was combined with the previous methanol solution. Methanol was removed using a rotary evaporator at 60 °C, and the residue was extracted with cyclohexane (10 mL, 3 times) to obtain pure 4-propylsyringol (4pS) and 4-propylguaiacol (4pG) mixture in the cyclohexane phase (light brown color). The aromatic monomers from the poplar wood feedstock were analyzed before and after the cyclohexane extraction on an Agilent 7890B series gas chromatograph (GC) equipped with a HP5 capillary column and an Agilent 5977A series mass spectroscopy detector (Figure 1). The following operating conditions were used: injection temperature of 250 °C, a column temperature program of 50 °C (held for 1 min), heating ramp to 300 °C at 15 °C min–1, and then 300 °C (held for 7 min). The detector temperature was 290 °C.
Synthesis and Characterization of 4-Propylsyringyl Acrylate (4pSA), 4-Propylguaiacyl Acrylate (4pGA), 4-Propylsyringyl Methacrylate (4pSMA), and 4-Propylguaiacyl Methacrylate (4pGMA)
The aromatic monomer mixture was acrylated following a procedure adapted from the literature,18 with acryloyl chloride (1.2 molar equiv, Sigma-Aldrich, 97%) used for the synthesis of the acrylates and methacryloyl chloride (1.2 molar equiv, Alfa Aesar, 97%) used for the synthesis of the methacrylates. Additional details and 1H NMR characterization data are provided in the Supporting Information text and Figures S1 and S2.
Synthesis of Poly(4pSA), Poly(4pGA), Poly(4pSMA), Poly(4pGMA), and Poly(4pSMA-co-4pGMA)
Poly(4pSA) and poly(4pGA) (see Figure S3 for 1H NMR spectra) were synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization, using a procedure described in the literature.18 The initiator, 2,2′-azobis(isobutyronitrile) (AIBN, Sigma-Aldrich, 98%), was recrystallized twice from methanol prior to use. The chain-transfer agent (CTA), 3,5-bis(2-dodecyl-thiocarbonothioylthio-1-oxopropoxy)benzoic acid (BTCBA, Sigma-Aldrich, 98%), was used as received. The polymerization solvent, anisole (Sigma-Aldrich, ≥99.7%) with 5 wt % N,N-dimethylformamide (DMF, Sigma-Aldrich, ≥99.9%), was prepared and stored on molecular sieves to minimize water content. The same procedure was employed in the synthesis of poly(4pSMA) and poly(4pGMA), except that 2-cyano-2-propyl benzodithioate (CPB, STREM Chemicals, 97%) was utilized as the CTA. A copolymer of 4pSMA and 4pGMA also was made following the procedure mentioned above, but a mixture of 4pSMA and 4pGMA at a ratio of 0.6/0.4 was fed instead of the single monomers.
Synthesis of Poly(4-propylsyringyl acrylate-b-butyl acrylate-b-4-propylsyringyl acrylate) (SaBSa) Triblock Polymer
SaBSa triblock polymer was synthesized in a two-step RAFT polymerization, using BTCBA as the CTA. Poly(n-butyl acrylate) (PBA) was synthesized first, following the general procedure described in the previous section, except that the polymer was precipitated into cold methanol. Then, PBA was chain-extended to make the SaBSa triblock polymer. SaBSa was isolated by precipitation in cold methanol three times to remove unreacted 4pSA and dried in vacuum for 2 days at 40 °C. 1H NMR data and weight fraction calculations are provided in Figure S4.
Characterization of Polymers
The number-average molecular weight (Mn) and dispersity (Đ) of the synthesized polymers and MBM were obtained using a Viscotek VE2001 size-exclusion chromatography (SEC) instrument with THF (Optima) as the eluent (1.0 mL min–1) and polystyrene standards (1.78–205 kg mol–1) as the reference. Glass transition temperatures (Tg’s) of all polymers were determined using a differential scanning calorimeter (DSC, Discovery Series, TA Instruments). The thermal degradation behavior of SaBSa was characterized using thermogravimetric analysis (TGA, Discovery Series, TA Instruments). Further details of thermal analysis are located in the Supporting Information. Tensile testing on lignin-based polymers was performed using dog-bone-shaped testing bars (following ASTM D638, bar type 5, 5.3 mm gauge width, 0.8 mm thickness) that were prepared by compression molding into a Teflon PTFE sheet (McMaster Carr) on a PHI hot press. Tensile testing was performed with a RSA-G2 solids analyzer (TA Instruments) in tension mode. Tensile testing on MBM also was performed for comparison, except that the testing specimens were prepared with an aluminum mold. Detailed protocols are provided in the Supporting Information.
Adhesion Testing
Polymer films were prepared by casting polymer solution onto a 50 μm thick sheet of PET (McMaster Carr). A 30 wt % SaBSa or MBM solution was made by dissolving 100 mg of SaBSa or MBM into 230 mg of o-xylene (Sigma-Aldrich, 97%). Polymer films with ∼20 μm thickness were prepared by casting the solution onto a PET sheet using a home-built flow coater45 at a speed of 10 mm s–1 with a blade width of 12.7 mm and a gap height of 100 μm. The films were dried under ambient conditions for 24 h before adhesion testing. A mirrorlike stainless-steel plate (McMaster Carr) was used as the adherend.
180° Peel Test
A SaBSa film strip was adhered to the stainless-steel plate using a 4.5 lb hand roller. The strips were mounted to a RSA-G2 solids analyzer and tested at a peel rate of 5 mm s–1. The peel force was averaged across the plateau in force for four samples.
Loop Tack Test
A SaBSa film strip was formed into a teardrop-shaped loop and mounted to the upper grip of the RSA-G2 solids analyzer. The loop then was lowered onto the stainless-steel plate mounted to the lower grip. The contact length of the strip was ∼30 mm. The upper grip was raised at a speed of 5 mm s–1 until the strip detached from the adherend. The maximum force was recorded as the tack force, and the average of three samples was reported.
Shear Test
The film strip was adhered to a stainless-steel plate with a contact area of 12.7 mm × 12.7 mm, using a 4.5 lb hand roller. A 500 g weight then was suspended from the strip, and the average time to failure of three samples was reported.
For the comparison of 180° peel force and tack force to commercial materials, Fisherbrand labeling tape (19 mm in width) and Scotch Magic tape (19 mm in width) were tested using the procedures described above.
Acknowledgments
The polymer synthesis and characterization work was performed by S.W. and T.H.E. and was supported financially by NSF Grant CHE-1507010 to T.H.E. The biomass deconstruction and monomer production work was conducted by L.S., B.S., and D.G.V. and was supported financially by the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001004. The University of Delaware (UD) NMR facility and RSA-G2 instrument were supported by the Delaware COBRE program with a grant from NIH NIGMS (1 P30 GM110758-01). The authors thank the UD Advanced Materials Characterization Laboratory for the use of DSC and TGA instruments. The authors are grateful for the generous donation of Kurarity LA2140e polymer from Kuraray Co. Ltd. The authors thank Kevin Liedel from UD Delaware Energy Institute for help with generating figures. The authors also acknowledge Dr. Shuang Liu for assistance with the adhesion and tensile testing experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00140.
Additional experimental details; 1H NMR spectra; DSC traces of the second heating; mass remaining and the first derivative of mass change vs temperature; 2-D SAXS pattern and azimuthally integrated 1-D SAXS data; 180° peel force; loop tack force; stress–strain curves; and photo taken during tensile testing (PDF)
Author Present Address
∥ S.W.: Bridgestone Americas, 10 East Firestone Boulevard, Akron, Ohio 44317, United States.
Author Present Address
⊥ L.S.: Department of Sustainable Biomaterials, Virginia Tech, 230B Cheatham Hall, Blacksburg, Virginia 24060, United States.
The authors declare no competing financial interest.
Notes
No unexpected or unusually high safety hazards were encountered.
Supplementary Material
References
- Laurichesse S.; Avérous L. Chemical modification of lignins: towards biobased polymers. Prog. Polym. Sci. 2014, 39 (7), 1266–1290. 10.1016/j.progpolymsci.2013.11.004. [DOI] [Google Scholar]
- Sun Z.; Fridrich B.; de Santi A.; Elangovan S.; Barta K. Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 2018, 118 (2), 614–678. 10.1021/acs.chemrev.7b00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.; Washburn N. R. Chemistry of lignin-based materials. Green Mater. 2013, 1 (3), 137–160. 10.1680/gmat.12.00009. [DOI] [Google Scholar]
- Shuai L.; Amiri M. T.; Questell-Santiago Y. M.; Héroguel F.; Li Y.; Kim H.; Meilan R.; Chapple C.; Ralph J.; Luterbacher J. S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 2016, 354 (6310), 329–333. 10.1126/science.aaf7810. [DOI] [PubMed] [Google Scholar]
- Shuai L.; Saha B. Towards high-yield lignin monomer production. Green Chem. 2017, 19 (16), 3752–3758. 10.1039/C7GC01676J. [DOI] [Google Scholar]
- Liu W.; Jiang H.; Yu H. Thermochemical conversion of lignin to functional materials: a review and future directions. Green Chem. 2015, 17 (11), 4888–4907. 10.1039/C5GC01054C. [DOI] [Google Scholar]
- Wu W.; Dutta T.; Varman A. M.; Eudes A.; Manalansan B.; Loqué D.; Singh S. Lignin valorization: two hybrid biochemical routes for the conversion of polymeric lignin into value-added chemicals. Sci. Rep. 2017, 7 (1), 8420. 10.1038/s41598-017-07895-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall D. L.; Ralph J.; Donohue T. J.; Noguera D. R. Biochemical transformation of lignin for deriving valued commodities from lignocellulose. Curr. Opin. Biotechnol. 2017, 45, 120–126. 10.1016/j.copbio.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Deuss P. J.; Barta K. From models to lignin: transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. 10.1016/j.ccr.2015.02.004. [DOI] [Google Scholar]
- Ferrini P.; Rinaldi R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem., Int. Ed. 2014, 53 (33), 8634–8639. 10.1002/anie.201403747. [DOI] [PubMed] [Google Scholar]
- Deepa A. K.; Dhepe P. L. Lignin depolymerization into aromatic monomers over solid acid catalysts. ACS Catal. 2015, 5 (1), 365–379. 10.1021/cs501371q. [DOI] [Google Scholar]
- Song Q.; Wang F.; Xu J. Hydrogenolysis of lignosulfonate into phenols over heterogeneous nickel catalysts. Chem. Commun. 2012, 48 (56), 7019–7021. 10.1039/c2cc31414b. [DOI] [PubMed] [Google Scholar]
- Choi H. S.; Meier D. Fast pyrolysis of Kraft lignin-vapor cracking over various fixed-bed catalysts. J. Anal. Appl. Pyrolysis 2013, 100, 207–212. 10.1016/j.jaap.2012.12.025. [DOI] [Google Scholar]
- Calvaruso G.; Clough M. T.; Rinaldi R. Biphasic extraction of mechanocatalytically-depolymerized lignin from water-soluble wood and its catalytic downstream processing. Green Chem. 2017, 19 (12), 2803–2811. 10.1039/C6GC03191A. [DOI] [Google Scholar]
- van de Pas D. J.; Torr K. M. Biobased epoxy resins from deconstructed native softwood lignin. Biomacromolecules 2017, 18 (8), 2640–2648. 10.1021/acs.biomac.7b00767. [DOI] [PubMed] [Google Scholar]
- Rouméas L.; Billerach G.; Aouf C.; Dubreucq É.; Fulcrand H. Furylated flavonoids: fully biobased building blocks produced by condensed tannins depolymerization. ACS Sustainable Chem. Eng. 2018, 6 (1), 1112–1120. 10.1021/acssuschemeng.7b03409. [DOI] [Google Scholar]
- Holmberg A. L.; Stanzione J. F.; Wool R. P.; Epps T. H. III. A facile method for generating designer block copolymers from functionalized lignin model compounds. ACS Sustainable Chem. Eng. 2014, 2 (4), 569–573. 10.1021/sc400497a. [DOI] [Google Scholar]
- Wang S.; Bassett A. W.; Wieber G. V.; Stanzione J. F. III; Epps T. H. III. Effect of methoxy substituent position on thermal properties and solvent resistance of lignin-inspired poly(dimethoxyphenyl methacrylate)s. ACS Macro Lett. 2017, 6 (8), 802–807. 10.1021/acsmacrolett.7b00381. [DOI] [Google Scholar]
- Holmberg A. L.; Nguyen N. A.; Karavolias M. G.; Reno K. H.; Wool R. P.; Epps T. H. III. Softwood lignin-based methacrylate polymers with tunable thermal and viscoelastic properties. Macromolecules 2016, 49 (4), 1286–1295. 10.1021/acs.macromol.5b02316. [DOI] [Google Scholar]
- Holmberg A. L.; Reno K. H.; Nguyen N. A.; Wool R. P.; Epps T. H. III. Syringyl methacrylate, a hardwood lignin-based monomer for high-Tg polymeric materials. ACS Macro Lett. 2016, 5 (5), 574–578. 10.1021/acsmacrolett.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzione J. F. III; Giangiulio P. A.; Sadler J. M.; La Scala J. J.; Wool R. P. Lignin-based bio-oil mimic as biobased resin for composite applications. ACS Sustainable Chem. Eng. 2013, 1 (4), 419–426. 10.1021/sc3001492. [DOI] [Google Scholar]
- Stanzione J. F. III; Sadler J. M.; La Scala J. J.; Reno K. H.; Wool R. P. Vanillin-based resin for use in composite applications. Green Chem. 2012, 14 (8), 2346–2352. 10.1039/c2gc35672d. [DOI] [Google Scholar]
- Holmberg A. L.; Reno K. H.; Wool R. P.; Epps T. H. III. Biobased building blocks for the rational design of renewable block polymers. Soft Matter 2014, 10 (38), 7405–7424. 10.1039/C4SM01220H. [DOI] [PubMed] [Google Scholar]
- Saito T.; Brown R. H.; Hunt M. A.; Pickel D. L.; Pickel J. M.; Messman J. M.; Baker F. S.; Keller M.; Naskar A. K. Turning renewable resources into value-added polymer: development of lignin-based thermoplastic. Green Chem. 2012, 14 (12), 3295–3303. 10.1039/c2gc35933b. [DOI] [Google Scholar]
- Zhao S.; Abu-Omar M. M. Renewable thermoplastics based on lignin-derived polyphenols. Macromolecules 2017, 50 (9), 3573–3581. 10.1021/acs.macromol.7b00064. [DOI] [Google Scholar]
- Gioia C.; Lo Re G.; Lawoko M.; Berglund L. Tunable thermosetting epoxies based on fractionated and well-characterized lignins. J. Am. Chem. Soc. 2018, 140 (11), 4054–4061. 10.1021/jacs.7b13620. [DOI] [PubMed] [Google Scholar]
- Van den Bosch S.; Schutyser W.; Vanholme R.; Driessen T.; Koelewijn S. F.; Renders T.; De Meester B.; Huijgen W. J. J.; Dehaen W.; Courtin C. M.; Lagrain B.; Boerjan W.; Sels B. F. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8 (6), 1748–1763. 10.1039/C5EE00204D. [DOI] [Google Scholar]
- Creton C. Pressure-sensitive adhesives: an introductory course. MRS Bull. 2003, 28 (6), 434–439. 10.1557/mrs2003.124. [DOI] [Google Scholar]
- Sinha B.Pressure sensitive adhesives market by chemical composition (acrylic, rubber, ethylene vinyl acetate (EVA), silicone, polyurethane, and others), type (water based, hot melts, solvent based, and radiation based), application (labels, medical, graphics, tapes, and others) and end-use industry (automotive, packaging, building & construction, electronics, medical, consumer goods, and others) - global opportunity analysis and industry forecast, 2017–2023. https://www.alliedmarketresearch.com/pressure-sensitive-adhesives-market (accessed March 5, 2018).
- Peykova Y.; Lebedeva O. V.; Diethert A.; Müller-Buschbaum P.; Willenbacher N. Adhesive properties of acrylate copolymers: Effect of the nature of the substrate and copolymer functionality. Int. J. Adhes. Adhes. 2012, 34, 107–116. 10.1016/j.ijadhadh.2011.12.001. [DOI] [Google Scholar]
- O’Connor A. E.; Willenbacher N. The effect of molecular weight and temperature on tack properties of model polyisobutylenes. Int. J. Adhes. Adhes. 2004, 24 (4), 335–346. 10.1016/j.ijadhadh.2003.11.005. [DOI] [Google Scholar]
- Tobing S. D.; Klein A. Molecular parameters and their relation to the adhesive performance of acrylic pressure-sensitive adhesives. J. Appl. Polym. Sci. 2001, 79 (12), 2230–2244. . [DOI] [Google Scholar]
- Gotro J. T.; Graessley W. W. Model hydrocarbon polymers: rheological properties of linear polyisoprenes and hydrogenated polyisoprenes. Macromolecules 1984, 17 (12), 2767–2775. 10.1021/ma00142a058. [DOI] [Google Scholar]
- Roovers J.; Toporowski P. M. Characteristic ratio and plateau modulus of 1,2-polybutadiene. A comparison with other rubbers. Rubber Chem. Technol. 1990, 63 (5), 734–746. 10.5254/1.3538286. [DOI] [Google Scholar]
- Nakamura Y.; Adachi M.; Tachibana Y.; Sakai Y.; Nakano S.; Fujii S.; Sasaki M.; Urahama Y. Tack and viscoelastic properties of an acrylic block copolymer/tackifier system. Int. J. Adhes. Adhes. 2009, 29 (8), 806–811. 10.1016/j.ijadhadh.2009.06.004. [DOI] [Google Scholar]
- Tong J. D.; Jerôme R. Synthesis of poly(methyl methacrylate)-b-poly(n-butyl acrylate)-b-poly(methyl methacrylate) triblocks and their potential as thermoplastic elastomers. Polymer 2000, 41 (7), 2499–2510. 10.1016/S0032-3861(99)00412-7. [DOI] [Google Scholar]
- Vendamme R.; Schüwer N.; Eevers W. Recent synthetic approaches and emerging bio-inspired strategies for the development of sustainable pressure-sensitive adhesives derived from renewable building blocks. J. Appl. Polym. Sci. 2014, 131 (17), 40669. 10.1002/app.40669. [DOI] [Google Scholar]
- Nasiri M.; Reineke T. M. Sustainable glucose-based block copolymers exhibit elastomeric and adhesive behavior. Polym. Chem. 2016, 7 (33), 5233–5240. 10.1039/C6PY00700G. [DOI] [Google Scholar]
- Gallagher J. J.; Hillmyer M. A.; Reineke T. M. Acrylic triblock copolymers incorporating isosorbide for pressure sensitive adhesives. ACS Sustainable Chem. Eng. 2016, 4 (6), 3379–3387. 10.1021/acssuschemeng.6b00455. [DOI] [Google Scholar]
- Ye Y.; Zhang Y.; Fan J.; Chang J. Selective production of 4-ethylphenolics from lignin via mild hydrogenolysis. Bioresour. Technol. 2012, 118, 648–651. 10.1016/j.biortech.2012.05.127. [DOI] [PubMed] [Google Scholar]
- Barner-Kowollik C.; Perrier S. The future of reversible addition fragmentation chain transfer polymerization. J. Polym. Sci., Part A: Polym. Chem. 2008, 46 (17), 5715–5723. 10.1002/pola.22866. [DOI] [Google Scholar]
- Lee S.; Lee K.; Kim Y.; Shin J. Preparation and characterization of a renewable pressure-sensitive adhesive system derived from ε-decalactone, l-lactide, epoxidized soybean oil, and rosin ester. ACS Sustainable Chem. Eng. 2015, 3 (9), 2309–2320. 10.1021/acssuschemeng.5b00580. [DOI] [Google Scholar]
- Shin J.; Martello M. T.; Shrestha M.; Wissinger J. E.; Tolman W. B.; Hillmyer M. A. Pressure-sensitive adhesives from renewable triblock copolymers. Macromolecules 2011, 44 (1), 87–94. 10.1021/ma102216d. [DOI] [Google Scholar]
- Ding K.; John A.; Shin J.; Lee Y.; Quinn T.; Tolman W. B.; Hillmyer M. A. High-performance pressure-sensitive adhesives from renewable triblock copolymers. Biomacromolecules 2015, 16 (8), 2537–2539. 10.1021/acs.biomac.5b00754. [DOI] [PubMed] [Google Scholar]
- Emerson J. A.; Toolan D. T. W.; Howse J. R.; Furst E. M.; Epps T. H. III. Determination of solvent-polymer and polymer-polymer Flory-Huggins interaction parameters for poly(3-hexylthiophene) via solvent vapor swelling. Macromolecules 2013, 46 (16), 6533–6540. 10.1021/ma400597j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.