

Abstract
Background
Bacterial invasive infection and host immune response is fundamental to the understanding of pathogen pathogenesis and the discovery of effective therapeutic drugs. However, there are very few experimental studies on the signaling cross-talks between bacteria and human host to date.
Methods
In this work, taking M. tuberculosis H37Rv (MTB) that is co-evolving with its human host as an example, we propose a general computational framework that exploits the known bacterial pathogen protein interaction networks in STRING database to predict pathogen-host protein interactions and their signaling cross-talks. In this framework, significant interlogs are derived from the known pathogen protein interaction networks to train a predictive l2-regularized logistic regression model.
Results
The computational results show that the proposed method achieves excellent performance of cross validation as well as low predicted positive rates on the less significant interlogs and non-interlogs, indicating a low risk of false discovery. We further conduct gene ontology (GO) and pathway enrichment analyses of the predicted pathogen-host protein interaction networks, which potentially provides insights into the machinery that M. tuberculosis H37Rv targets human genes and signaling pathways. In addition, we analyse the pathogen-host protein interactions related to drug resistance, inhibition of which potentially provides an alternative solution to M. tuberculosis H37Rv drug resistance.
Conclusions
The proposed machine learning framework has been verified effective for predicting bacteria-host protein interactions via known bacterial protein interaction networks. For a vast majority of bacterial pathogens that lacks experimental studies of bacteria-host protein interactions, this framework is supposed to achieve a general-purpose applicability. The predicted protein interaction networks between M. tuberculosis H37Rv and Homo sapiens, provided in the Additional files, promise to gain applications in the two fields: (1) providing an alternative solution to drug resistance; (2) revealing the patterns that M. tuberculosis H37Rv genes target human immune signaling pathways.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4873-9) contains supplementary material, which is available to authorized users.
Keywords: Pathogen-host protein interaction networks, Pathogen-host coevolution, Signaling pathways, Transfer learning, l2-regularized logistic regression
Background
Bacterial invasive infection and host immune response is fundamental to the understanding of pathogen pathogenesis and the discovery of effective therapeutic drugs. As an example, Mycobacterium tuberculosis is the causative agent of tuberculosis, an infectious disease that causes millions of deaths each year [1]. In recent years, M. tuberculosis H37Rv has attracted much attention partly due to its co-infection with HIV [2] and drug resistance [3–6]. From the point of view of interactome, bacterial-host protein interaction networks can be viewed as the interface/cross-talks between pathogen protein-protein interaction (PPI) networks and host PPI protein-protein networks. Bacteria-host signaling cross-talks potentially help us understand the underlying mechanism of M. tuberculosis infection and human defence.
To date, most of the experimental work focuses on detecting protein-protein interactions within bacterial cells. The database STRING [7] (https://string-db.org/) has curated massive PPI networks of 1678 bacterial pathogens such as M. tuberculosis, B. anthracis, F. tularensis, Y. pestis, etc. However, there are very few experimental studies on protein interactions between bacteria and their host. From a computational view of point, M. tuberculosis H37Rv has been extensively studied in recent years in terms of drug resistance analysis [4–6, 8] and PPI networks reconstruction [9, 10]. In [9, 10], interlogs are derived as M. tuberculosis H37Rv PPIs from the known PPIs of other source species. In [9], the known M. tuberculosis H37Rv PPIs are laid aside unused and instead the E. coli PPIs are used as training data to predict M. tuberculosis H37Rv PPIs. In [10], the interlogs derived from lostridium difficile are used to expand the known M. tuberculosis H37Rv PPI networks, and the expanded PPI networks are further used as training data to train a random forest model for the discovery of novel M. tuberculosis H37Rv PPIs.
Bacterial pathogen PPI networks are useful to study the signaling mechanism and drug resistance machinery within bacteria cell. However, we need further reconstruct bacteria-host PPI networks to understand the cross-talk mechanism of bacterial infection and host immunity. In recent years, pathogen-host PPI networks reconstruction and pathogen-host signaling cross-talk modeling have attracted much attention from computational biologists [11–18], most of which focus on virus-host protein interactions. Comparatively, t the experimental studies on bacteria-host protein interactions are much less than that on virus-host protein interactions, partly because of the complex bacterial cell wall, which forms a strong permeability barrier to the mutual access of the bacterial genome and the host genome [19]. The two genomes could come across to physically interact only if bacterial proteins are located at the surface or membrane of bacterial cell, or bacterial proteins could transport or secret into the host cell. To our knowledge, experimental studies on bacteria-host protein interactions have been conducted for a very limited number of species such as Salmonella [20], Bacillus anthracis, Francisella tularensis, and Yersinia pestis [21]. For these bacterial pathogens, the known pathogen-host PPIs can be used as training data of machine learning modeling or be treated as templates to infer interlogs [22]. In [22], the known 62 Salmonella-human PPIs are used to derive interlogs as novel Salmonella-human PPIs.
Nevertheless, no experimental studies on pathogen-host protein interactions have been conducted for the overwhelming majority of bacteria, e.g. M. tuberculosis H37Rv. To study the signaling cross-talks between bacteria and host, two solutions to inferring bacteria-host protein interactions have been proposed, one solution is ortholog knowledge transfer that transfers ortholog knowledge between two different hosts, e.g. knowledge transfer between human and plant to infer Salmonella-plant PPIs from the known Salmonella-human PPIs [23]; and the other solution is interlog knowledge transfer that infers interlogs from known PPIs of different bacteria and different hosts [24, 25]. These two solutions are effective for cross-species knowledge transfer, especially when no experimental data are available to the species to be studied. Nevertheless, these two methods both resort to third-party species that may not physically co-exist with parasitic relationships, e.g. Homo sapiens versus plant [23]. Ortholog or interlog knowledge transfer across widely-variant species are prone to yield a certain level of noise and false interactions due to a large evolutionary divergence.
Actually, the parasitic or co-evolution relationships between bacteria and host indicate that bacterial protein-protein interaction alone is sufficient for us to infer bacteria-host protein interaction without resorting to third-party distant species. Knowledge transfer between two co-evolving species is more credible than that between two evolutionarily distant species. The bacterial pathogen protein interaction networks of 1678 bacteria in STRING [7] provide rich information for us to study bacteria-host protein interactions, because many evidences have demonstrated that bacterial genome is co-evolving with its host genome [19, 26, 27]. In [27], it has been concluded that Mycobacterium tuberculosis complex (MTBC) has been anatomically co-evolving with modern humans for tens of thousands of years on the basis of the evidences of its origin in Africa, the congruence in phylogeography and the dating of major branching events. Moreover, the drug resistance of Mycobacterium tuberculosis (MTB) strains is also evolving with the host genome. In [19], it has been claimed that human genetic factors may play important roles in MTB drug resistance and different MTB lineages (e.g. lineage 1: Indo-Oceanic; lineage 2: East Asian; lineage 3: Central Asian; lineage 4: Euro American; lineage 7: Ethiopia) acquire different levels of drug resistance. Molecular interactions are an effective way to unravel bacteria-host co-evolution relationship and the progression of bacterial drug resistance, which is at present hampered by the limited knowledge of bacteria-host interactions. For instance, the molecular mechanism involved in sensing of extracellular signals for inducing its metabolic adaptation still remains unclear [4].
Furthermore, the interaction between bacteria and host also somewhat contributes to bacterial drug resistance. As suggested in [19], the interaction of MTB with its macrophage microenvironment may play an important role in the risk of progression to drug-resistant TB. Meanwhile from the host side, polymorphisms within genes involved in macrophage activity (SLC11A1, VDR and HLA genes) have been reported to be associated with susceptibility to MTB drug resistance. As reviewed in [28], the amino acid residues from the PPI interfaces are more conserved than those from other parts of the protein surface, and PPI inhibitors can perhaps be more resistant to spontaneous mutations at their binding site versus inhibitors of the active site, thus bacteria-host PPI inhibitors may be of particular interest as antimicrobial drugs that induce less risk of drug resistance. Therefore, the exploration of protein interaction networks between co-evolving bacterial pathogens and host could potentially achieve two goals: (1) deriving more reliable interlogs to study pathogen-host signaling cross-talks; (2) choosing pathogen-host PPI inhibitors to provide an alternative solution to bacterial drug resistance.
In this work, taking M. tuberculosis H37Rv as an example, we propose a general computational framework that transfers the knowledge of bacterial pathogen protein interaction networks to predict pathogen targeted human genes and immune signaling pathways. Due to lack of experimental studies, we take advantage of the co-evolution relationship between M. tuberculosis H37Rv and Homo sapiens [19, 26, 27] to derive interlogs as the training data from M. tuberculosis H37Rv protein interactions alone. We confine the search of M. tuberculosis H37Rv ortholog genes within its human host without resorting to a third-party species. The interlogs derived in this way are presumably more reliable than those derived from remote species. Given two interacting M. tuberculosis H37Rv genes (m1, m2) and their corresponding Homo sapiens ortholog genes (h1, h2), we deem (m1, h2) and (m2, h1) as two interlogs, since the human ortholog gene products are functionally or structurally similar to M. tuberculosis H37Rv gene products. To ensure the quality of data, only the significant interlogs are used as training data, and the less significant interlogs need to be further validated by the trained model. Ortholog and interlog knowledge transfer are prone to introduce a certain level of noise as well as increase the computational complexity. To solve this problem, we adopt theoretically well-established l2-regularized logistic regression as the base machine learning model. Finally, we further conduct gene ontology (GO) and pathway enrichment analyses on the predicted interactions to provide insights into the machinery of M. tuberculosis H37Rv infection and host response. As a major concern in recent years, bacterial drug resistance is also discussed in terms of bacteria-host PPI inhibition to provide a potential alternative solution to M. tuberculosis H37Rv drug resistance.
Methods
Overview flowchart of the proposed framework
As shown in Fig. 1, this work is divided into three phases: (I) data construction; (II) model training and prediction; (III) analyses. The critical phase is to derive interlogs as training data from the known M. tuberculosis H37Rv PPI networks in STRING [9]. This phase takes advantage of the co-evolution relationship between M. tuberculosis H37Rv and human host to construct training data that are experimentally not available. The second phase is to construct feature representation, train an l2-regularized logistic regression model, and then predict interactions from less significant interlogs and non-interlogs. The final phase is to comprehensively analyse the reconstructed MTB-human PPI networks for further understanding of the machinery of bacterial infection and host response. Especially, we analyse the MTB-human PPIs related to M. tuberculosis H37Rv drug resistance. Proper selection or design of PPI inhibitors could potentially provide an alternative solution to bacterial drug resistance.
Fig. 1.
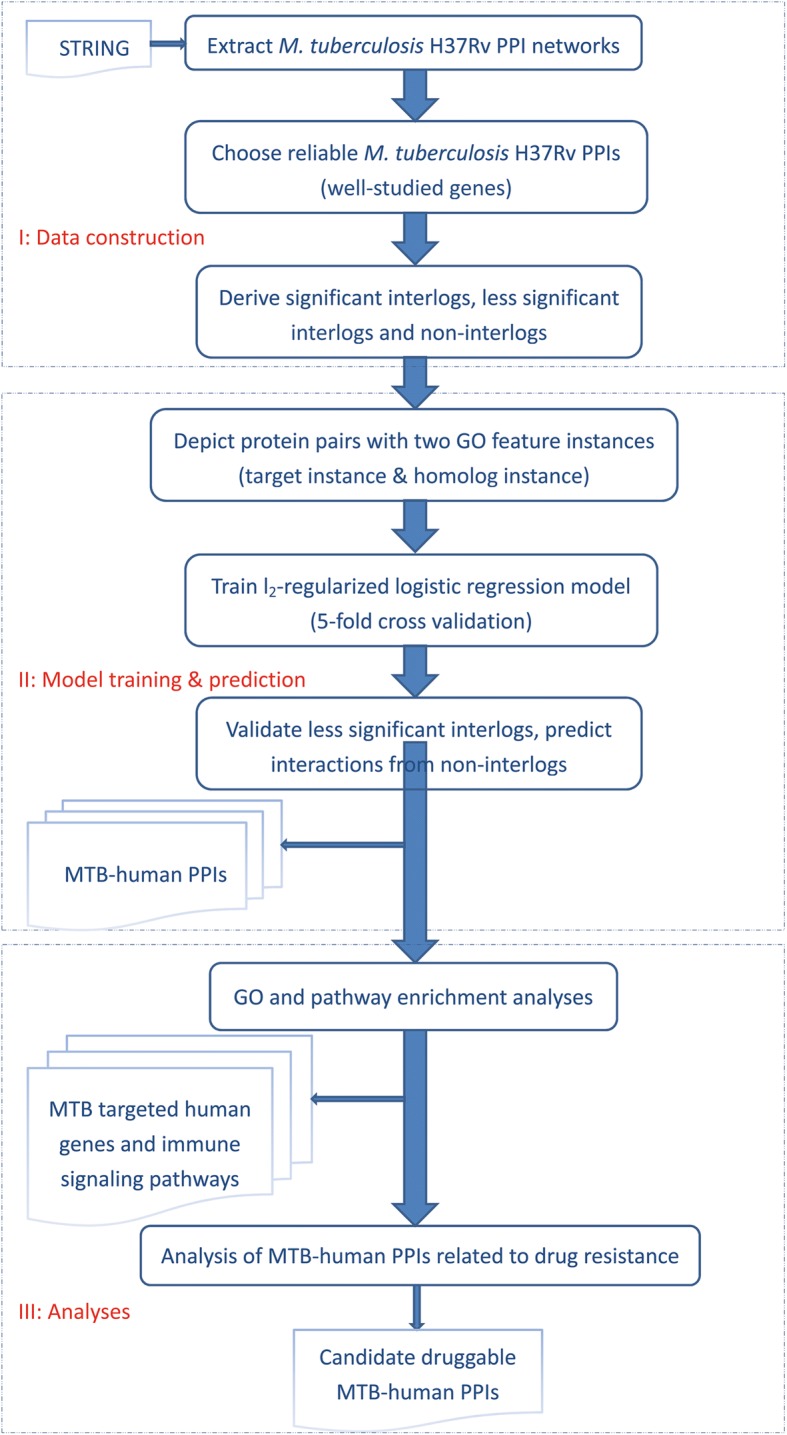
Overview flowchart of the proposed framework
Data construction via interlog knowledge transfer from M. tuberculosis H37Rv PPI networks
The M. tuberculosis H37Rv protein interaction networks in STRING [9] contain 309,715 interactions. Unfortunately, the PPI networks have been estimated to be of low quality [24, 25, 29]. To choose quality M. tuberculosis H37Rv protein interactions, we take the following three measures. Firstly, we only choose the PPIs with experimental evidences. Secondly, we remove the proteins that have no gene names. Lastly, we only choose the well-studied genes that have been annotated with at least one specific gene ontology (GO) term of molecular function or biological process, except the generic root GO terms (GO:0005575, GO:0008150, GO:0003674) in the GO directed acyclic graph (DAG). As results, we obtain 5224 well-studied M. tuberculosis H37Rv genes that correspond to 7835 gene products/proteins and 28,347 M. tuberculosis H37Rv protein-protein interactions. In the same way, we obtain 20,081 well-studied human genes that correspond to 60,126 gene products/proteins.
Now we exploit the knowledge of M. tuberculosis H37Rv PPI networks via orthologous relationships to construct the training data for MTB-human PPI predictions. To formally formulate the method of data construction, we denote the obtained M. tuberculosis H37Rv PPI networks as G, which contains 28,347 interactions and 1469 well-studied M. tuberculosis H37Rv proteins. After removing the M. tuberculosis H37Rv proteins that have no human orthologs, we obtain 1359 well-studied M. tuberculosis H37Rv proteins in total. We search the human ortholog genes in SwissProt [30] simply using PSI-BLAST [31] with default E-value (E-value = 10). Orthologs are defined as homologous genes diverging after a speciation event [32]. Actually, there are some advanced methods to search or predict orthologs, e.g. Reciprocal Best Hits (RBH) [32, 33], which relies on BLAST [34] to identify pairwise orthologs between two species [33], that’s, two genes residing in two different genomes are deemed orthologs if their protein products find each other as the best hit in the opposite genome [32]. For each ortholog pair (A, A’), RBH algorithm needs to run BLAST twice in two directions, one direction is against A query genome, and the opposite direction is against A’ query genome. RBH algorithm yields quality orthologs at the cost of high computational intensity. In this work, we adopt simple homolog-search method instead of RBH algorithm for the two reasons: (1) we need more orthologs including the distant orthologs to derive interlogs as training data, because no experimental data are available to computational modeling; (2) the RBH algorithm would computationally worsen the efficiency of the sophisticated framework as illustrated in Fig. 1. If we choose lower BLAST E-value cutoff, e.g. 1e-50 versus 1e-6 [32], we still could obtain quality orthologs. The noise from distant orthologs or non-orthologs could be counteracted using the regularization technique that is discussed in the next subsection.
From G, we derive the interlogs as follows. Given two interacting M. tuberculosis H37Rv genes (mi, mj), we use Hi, Hj to denote the sets of ortholog genes mi and mj, respectively. In particular, if an ortholog gene yields more than one ortholog protein, only one ortholog protein is randomly chosen as the interacting partner. Given the cutoff of ortholog significance δ, e.g. E-value of PSI-BLAST, we further split Hi, Hj into two subsets , respectively. Here we define as the sets of significant ortholog genes and as the set of less significant ortholog genes. For any two interacting M. tuberculosis H37Rv genes (mi, mj), we create the positive training instances from the set of significant ortholog genes as follows.
| 1 |
where (mi, g) or (mj, g) denotes significant interlog. Formula (1) is based on the assumption that the M. tuberculosis H37Rv gene mi(mj) functionally or structurally co-evolves with its human host ortholog genes (). The interaction of mi with mj to a great extent indicates the interaction of mi with mj ‘s ortholog gene , and the interaction of mj with mi ‘s ortholog genes vice versa. All the positive training instances are then merged to generate the whole positive training set.
| 2 |
From the evolutionary point of view, the less significant interlogs are not so reliable as the significant interlogs, so that they need to be further validated by the predictive model, which is trained on the significant interlogs. The set of less significant interlogs is defined as follows.
| 3 |
where (mi, g) or (mj, g) denotes insignificant interlog. For each M. tuberculosis H37Rv gene mi, let Pi = {g| {mi, g} ∈ Upos} denote the set of its human partner genes, Mi = {g| {mi, g} ∈ G} denote the set of its M. tuberculosis H37Rv partner genes, and denote the set of human ortholog genes of all the genes in Mi. Then we randomly sample the human genes that potentially do not interact with genemi from the set non-ortholog genes Ni = {g| g ∉ All_orthi ^ g ∈ Homowell} to construct the negative training data, where Homowell denotes the well-studied human genes. To obtain well-balanced training data, we impose the constraint ∣Ni ∣ = ∣ Pi∣ on the sampling of negative training data. Then the whole negative training set is defined as follows.
| 4 |
where M denotes the set of M. tuberculosis H37Rv genes in the M. tuberculosis H37Rv PPI networks G.
Model evaluation is a hard problem in the case of lack of experimental data. Nevertheless, random sampling of a tiny fraction of data in the huge space of non-interlogs is convincingly to capture non-interactions with a large probability. Hence we obtain the negative data to estimate the model as follows. Let denote the set of human genes, from which the negative data to be validated are defined as follows.
| 5 |
For each M. tuberculosis H37Rv gene mi, we sample the human genes from the set to obtain the prediction set as follows.
| 6 |
The prediction set is further reduced for the sake of computational complexity by imposing a constraint on the space as follows.
| 7 |
Formula (7) means that no more than 300 human proteins are randomly sampled for each M. tuberculosis H37Rv protein. The data and analyses are referred to the section Results.
Multi-instance GO feature construction via homolog knowledge transfer
State-of-art feature construction is a critical step of machine learning modeling in solving specific problems. Gene ontology (GO) has been widely used as features to predict protein-protein interactions [14–18, 35–40]. In [35], GO has been claimed to be the most discriminative feature for PPI prediction [35]. Gene ontology is a hierarchically organized and controlled vocabulary that characterizes gene products [41], and the annotations of genes or gene products are provided in terms of GO terms in GOA [42]. Despite its powerful predictive capability, GO feature representation could encounter a serious problem for those less-studied or novel genes, because the sparsity of GO terms potentially yields null feature vectors. In this work, homolog knowledge transfer is conducted via independent homolog instances to solve this problem, that is, each gene/protein is depicted with two instances, namely target instance and homolog instance. The target instance depicts the GO knowledge of the gene/protein itself, and the homolog instance depicts the GO knowledge of the homologs. The homologs are extracted from SwissProt [30] using PSI-BLAST [31] (E-value = 10) against all species. We treat all types of evidence codes equally including ISS (Inferred from Sequence or structural Similarity), IEA (Inferred from Electronic Annotation), etc. The reason that we choose the default E-value is that we need to capture distant homologs. Similarly, the reason that we do not filter out those indirectly-derived or uncurated annotations is to overcome the sparsity and enlarge the coverage of GO terms. Undoubtedly, a certain level of noise would be introduced into the computational framework, which will be discussed in the next subsection. The GO terms are extracted from GOA [42]. For each protein i in the training set U, we obtain two sets of GO terms, one set denoted as contains the GO terms of the homologs, and the other set denoted as contains the GO terms of the protein itself. Then the whole set of GO terms of the training set is defined as follows:
| 8 |
For each protein pair (i1, i2), the target instance and the homolog instance are formally defined as follows:
| 9 |
For each GO term g ∈ S, denotes the component g of the target instance and denotes the component g of the homolog instance . Those GO terms g ∉ S are discarded. Formula (9) means that if the protein pair (i1, i2) shares the same GO term g, then the corresponding component in the feature vector or is set 2; if neither protein in the protein pair possesses the GO term g, then the value is set 0; otherwise the value is set 1. The GO terms of the protein pair (i1, i2) that do not belong to the whole set of GO terms of the training set, formally defined as , are ignored in the feature construction.
L2-regularized logistic regression for large data training and noise tolerance
The existing interlog modeling methods [22–25] have demonstrated two major disadvantages. First, they do not discriminate less significant interlogs from significant interlogs, less significant interlogs need to be further validated; second, they cannot detect those interactions that exist inexplicitly in the form of interlogs. To solve the two problems, we combine machine learning approach with interlog modeling via two-level knowledge transfer, namely interaction-level interlog knowledge transfer and protein−/gene-level homolog knowledge transfer. The former level of knowledge transfer is to derive the significant interlogs as the training data, and the second level of knowledge transfer is to make up for the sparsity of GO terms. The two-level knowledge transfer demands that the machine learning methods we choose should be resistant to noise. SVM support vector machine (SVM) [43] is a theoretically established machine learning method known for its regularization technique resistant to noise/outliers. Unfortunately, SVM is not applicable to large training data due to its time complexity O(n2). Here we adopt the l2-regularized logistic regression method [44], implemented in the toolbox LIBLINEAR [45], to counteract the ortholog/homolog noise and fit the large training data in linear time.
Given a set of instance-label pairs (xi, yi), i = 1, 2, …, l; xi ∈ Rn; yi ∈ {−1, +1}, linear regression attempts to derive a decision function f(xi) = ωTxi + b, which is further converted to probability via the logistic function p(y = ± 1| ω, xi) = 1/1 + exp(−yi(ωTxi + b)). The weight vector and bias (ω, b) could be estimated by minimizing the negative log-likelihood . L2-regularized logistic regression imposes a constraint on the l2-norm of the weight vector ω to solve the following unconstrained optimization problem [44].
| 10 |
where C denotes the penalty parameter/regularizer that balances the two terms in Formula (10) to achieve good generalization ability. The second term could penalize potential noise/outlier fitting. The optimization of objective function (10) is solved via its dual form.
| 11 |
where αi denotes Lagrangian operator and .
In the test and prediction phase, the decision function f(x) yields two outputs for each protein-protein pair (i1, i2), which are further combined into one final decision as follows.
| 12 |
where ∣Δ∣ denotes the absolute value of Δ. The final label for protein pair (i1, i2) is defined as below.
| 13 |
The threshold ζ is used to filter out those weak positive predictions.
Experimental setting and model evaluation
As described above, each protein pair (i1, i2) is represented with two instances, the target instance and the homolog instance , so that the proposed framework yields three outputs for decision, i.e., , , respectively. Accordingly, we design three experimental settings, namely combined-instance (), homolog-instance () and target-instance (), to validate the effectiveness of homolog knowledge transfer. The combined-instance setting combines the outputs of the target instance and the homolog instance, the homolog-instance setting uses the homolog instance alone to evaluate the model robustness to GO term sparsity, and the target-instance setting uses the target instance alone to yield the baseline performance, equivalence to or excellence over which indicates that homolog knowledge transfer is effective.
Five performance metrics, i.e. ROC-AUC (Receiver Operating Characteristic AUC), SE (sensitivity), SP (specificity), MCC (Matthews correlation coefficient) and F1 score, are used to evaluate the proposed model via 5-fold cross validation. The dataset is randomly split into five disjoint parts. For five folds, each fold treats one part as test set and the other four parts are merged as training set. For each test example, the true label and the predicted label are recorded into the confusion matrix M. When the five folds complete, we use M to calculate the performance metrics. Except ROC-AUC, all the other metrics are derived from the confusion matrix M. From M, we define several intermediate variables as formula (14). Based on these intermediate variables, we further define SPl, SEl and MCCl for each label as formula (15) and the overall MCC as formula (16).
| 14 |
| 15 |
| 16 |
where the element of confusion matrix Mi, j records the counts that class i are classified to class j, and L denotes the number of labels. AUC is calculated based on the decision values defined in formula (12). F1 score is defined as follows:
| 17 |
Results
Quality validation on the constructed data via GO enrichment analysis
The E-value cut-off for significant interlogs is set δ = 1e ‐ 50. To date, there is no commonly-accepted standard to choose PSI-Blast E-value cut-off. We are inclined to choose a small E-value cut-off to obtain quality interlogs. As results, we obtain 15,287 significant interlogs as the positive examples (see Additional file 1), 15,287 randomly sampled non-interlogs as the negative examples (see Additional file 2), a set containing 98,187 less significant interlogs, a set containing 98,187 non-interlogs that are potentially negative examples, and a prediction set containing 1359,00 protein pairs. The sampling ratio of negative examples to positive examples is set 1:1 for the two reasons: (1) skewed distributions between the positive class and the negative class (e.g. ratio 1:10, 1:100, etc.) could increase the risk of model bias; (2) there is actually no direct mapping from the biological problem space to the computational space, so it is improper to simulate the huge negative space by sampling a much larger negative data set to train a predictive model from a computational point of view.
The significant interlogs are directly viewed as MTB-human protein interactions, so we need to assess the quality of the derived interlogs and their potential applications. As there is no independent benchmark measure, we analyse the quality of interlogs only from the aspects of similar GO terms. Of course, it is the feature vector of GO terms as Formula (9) defines that determines the predictive output. In addition, we analyse the drug resistance related interlogs to reveal the role of the host factors in the progression of bacterial antibiotic resistance. The less significant interlogs need to be further validated by the proposed framework and will be analysed in the following subsection. Antimicrobial peptides (AMPs) represent a potential alternative to available antibiotics. Raman et al. [46] exploit the M. tuberculosis H37R PPI networks to find so-called co-target genes whose co-inhibition with the resistance genes could effectively blockade M. tuberculosis H37RV signaling pathways. Nevertheless, inhibitors of pathogen-host PPI interface could be more therapeutically specific to bacterial infection with less risk of drug resistance and drug side-effects. Figure 2a illustrates the significant interlogs related to M. tuberculosis H37Rv drug-resistant genes that get involved in cytochromes and other target-modifying enzymes [46], which could cause potential chemical modification of drug molecules (see Additional file 3 for detailed GO enrichment analysis). Figure 2b illustrates the significant interlogs related to M. tuberculosis H37Rv drug-resistant genes that get involved in SOS-response and DNA replication [46], which could lead to mutations in the gene or its regulatory region (see Additional file 4 for detailed GO enrichment analysis).
Fig. 2.

The derived interlogs between M. tuberculosis H37Rv drug-resistant genes and human genes. a illustrates the interactions of M. tuberculosis H37Rv drug-resistant genes involved in cytochromes and other target-modifying enzymes that could cause potential chemical modification of drug molecules [46]. b illustrates the interactions of M. tuberculosis H37Rv drug-resistant genes involved in SOS-response and DNA replication that lead to mutations in the gene or its regulatory region. The light blue circles denote M. tuberculosis H37Rv genes and the red diamonds denote human genes [46]
Interlog {Rv2193, MT-CO1}
The drug-resistant gene Rv2193 is involved in cytochromes and other target-modifying enzymes that could cause potential chemical modification of drug molecules [46]. The interlog {Rv2193|I6Y8N5, MT-CO1|P003951} is inferred from the known M. tuberculosis H37Rv protein interaction {Rv2193|I6Y8N5, Rv3043c|I6YAZ7} [7], where the human protein P003951 (MT-CO1) is orthologous to the M. tuberculosis H37Rv protein I6YAZ7 (Rv3043c) with E-value equal to 4e-094. From GO enrichment analysis as partially provided in Table 1, the two genes {Rv2193, MT-CO1} both get involved in the common biological processes of oxidation-reduction process (GO:0055114) and hydrogen ion transmembrane transport (GO:1902600). In addition, the two genes are also both involved in aerobic cellular respiration, e.g. Rv2193 respiratory electron transport chain (GO:0022904) and MT-CO1 aerobic respiration (GO:0009060). Besides, the two orthologous proteins {Rv3043c|I6YAZ7, MT-CO1|P003951} are also involved in other highly similar biological processes (see Additional file 3).
Table 1.
GO enrichment analysis of the significant interlog {Rv2193, MT-CO1} LH57_11955 (Rv2193) is classified into the drug resistance type of cytochromes and other target-modifying enzymes that could cause potential chemical modification of drug molecules [46]
| GO term ID | GO aspect | GO term name | |
|---|---|---|---|
| Common GO terms | GO:0016020 | C | membrane |
| GO:0016021 | C | integral to membrane | |
| GO:0016491 | F | oxidoreductase activity | |
| GO:0055114 | P | oxidation-reduction process | |
| GO:1902600 | P | hydrogen ion transmembrane transport | |
| H57_11955|Rv2193 only | GO:0022904 | P | respiratory electron transport chain |
| GO:0019646 | P | aerobic electron transport chain | |
| GO:0015002 | F | heme-copper terminal oxidase activity | |
| MT-CO1 only | GO:0070469 | C | respiratory chain |
| GO:0045277 | C | respiratory chain complex IV | |
| GO:0005751 | C | mitochondrial respiratory chain complex IV | |
| GO:0006979 | P | response to oxidative stress | |
| GO:0009060 | P | aerobic respiration | |
| GO:0046688 | P | response to copper ion | |
| GO:0051602 | P | response to electrical stimulus | |
| GO:0020037 | F | heme binding |
C denotes cellular component, F denotes molecular function, and P denotes biological process
Interlog {Rv2737c, ERCC6}
The drug-resistant gene Rv2737c is involved in SOS-response and DNA replication that lead to mutations in the gene or its regulatory region. The interlog {Rv2737c|I6YE90, ERCC6|Q03468} is inferred from the known M. tuberculosis H37Rv protein interaction {Rv2737c|I6YE90, helZ|Rv2101|I6YCF3} [7], where the human protein Q03468 (ERCC6) is orthologous to the M. tuberculosis H37Rv protein I6YCF3 (Rv2101, gene name helZ) with E-value equal to 5e-058. The GO enrichment analysis of the two genes {Rv2737c, ERCC6} is partially provided in Table 2. We can see that these two genes get involved in the common biological processes of DNA repair (GO:0006281) and response to DNA damage stimulus (GO:0006974). Besides, the gene Rv2737c protects microbial DNA from antibiotics (GO:0046677, response to antibiotic), DNA damage (GO:0009432, SOS response), ultraviolet radiation (GO:0009650, UV protection), ionizing radiation (GO:0010212, response to ionizing radiation), etc. Accordingly, the human gene ERCC6 gets involved in response to gamma radiation (GO:0010332), response to UV (GO:0009411), response to oxidative stress (GO:0006979), DNA damage response, signal transduction resulting in induction of apoptosis (GO:0008630), base-excision repair (GO:0006284), etc. The similar biological processes suggest that the two genes {Rv2737c, ERCC6} potentially interact.
Table 2.
GO enrichment analysis of the derived interlog {Rv2737c, ERCC6}. recA (Rv2737c) is classified into the drug resistance type of SOS-response and DNA replication that leads to mutations in the gene or its regulatory region [46]
| GO term ID | GO aspect | GO term name | |
|---|---|---|---|
| Common GO terms | GO:0005515 | F | protein binding |
| GO:0006281 | P | DNA repair | |
| GO:0006974 | P | response to DNA damage stimulus | |
| GO:0016787 | F | hydrolase activity | |
| GO:0008094 | F | DNA-dependent ATPase activity | |
| GO:0003677 | F | DNA binding | |
| recA|Rv2737c only | GO:0046677 | P | response to antibiotic |
| GO:0009432 | P | SOS response | |
| GO:0009650 | P | UV protection | |
| GO:0010212 | P | response to ionizing radiation | |
| GO:0006310 | P | DNA recombination | |
| GO:0006259 | P | DNA metabolic process | |
| GO:0000725 | P | recombinational repair | |
| ERCC6 only | GO:0000303 | P | response to superoxide |
| GO:0006283 | P | transcription-coupled nucleotide-excision repair | |
| GO:0006284 | P | base-excision repair | |
| GO:0006979 | P | response to oxidative stress | |
| GO:0007256 | P | activation of JNKK activity | |
| GO:0008630 | P | DNA damage response, signal transduction resulting in induction of apoptosis | |
| GO:0010332 | P | response to gamma radiation | |
| GO:0009411 | P | response to UV |
C denotes cellular component, F denotes molecular function, and P denotes biological process
Performance of 5-fold cross validation
The ROC curves of 5-fold cross validation are illustrated in Fig. 3 and the detailed performance metrics are provided in Table 3. The proposed method achieves fairly good performance in terms of all the performance measures, and the performance varies very little between the three experimental settings, which indicate that the homolog instances are effective to substitute the target instances when the GO knowledge of the genes concerned is not available.
Fig. 3.
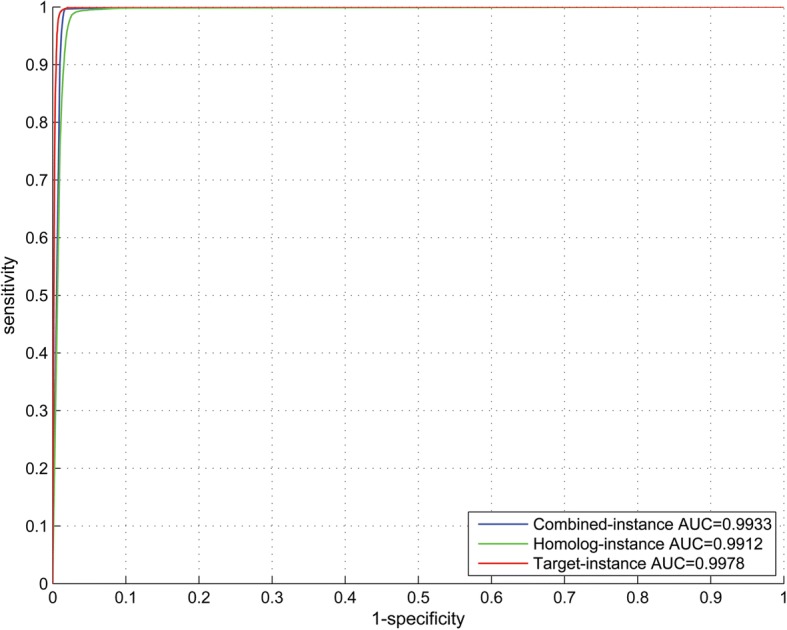
ROC curves for 5-fold cross validation performance evaluation on the artificially created significant interlogs between M. tuberculosis H37Rv and H. sapiens
Table 3.
Performance estimation of 5-fold cross validation and performance comparison with the existing methods
| Size | Combined-instance | Homolog-instance | Target-instance | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SP | SE | MCC | SP | SE | MCC | SP | SE | MCC | ||
| Positive | 15,287 | 0.9823 | 0.9966 | 0.9790 | 0.9684 | 0.9915 | 0.9601 | 0.9820 | 0.9976 | 0.9796 |
| Negative | 15,287 | 0.9965 | 0.9821 | 0.9790 | 0.9912 | 0.9676 | 0.9601 | 0.9975 | 0.9817 | 0.9796 |
| [Acc; MCC] | [98.93%; 0.9789] | [97.95%; 0.9599] | [97.95%; 0.9599] | |||||||
| [ROC-AUC] | [0.9933] | [0.9912] | [0.9978] | |||||||
| F1 Score | 0.9894 | 0.9798 | 0.9897 | |||||||
| KMM-SVM [23] | SP | SE | F1 score | |||||||
| ① human- > mouse | 0.517 | 0.937 | 0.667 | |||||||
| ② E.coli- > human | 0.257 | 0.161 | 0.199 | |||||||
Furthermore, the proposed method achieves quite well-balanced performance on the two classes, indicating that the positive class of significant interlogs and the negative class of non-interlogs are well separated. In the section Analysis of the constructed data, we have analysed the quality of the derived significant interlogs via GO enrichment analysis. To well interpret the good two-class separability, we conduct further GO enrichment analysis on the positive and the negative training data.
As illustrated in Fig. 4a, the protein pairs in the positive training data (i.e. significant interlogs) show more significant common patterns of subcellular localization, molecular functionality and biological processes than those in the negative training data (i.e. non-interlogs). Such a wide difference of common patterns between the positive data and the negative data presumably contribute much to the two-class separability, which then results in the good performance of 5-fold cross validation. The results as illustrated in Fig. 4a are consistent with the observations that two proteins that interact are more likely to reside in the same cellular compartments, fulfil similar molecular functions and participate in similar biological processes. As mentioned in the section Background, the complex bacterial cell wall that forms a strong permeability barrier to the mutual access of the bacterial genome and the host genome [19], the two partners of significant interlogs may merely functionally interact if no transport or secretion helps the two partners physically contact.
Fig. 4.

Percentage of pathogen-host protein pairs in the training data whose partners share common GO terms
Quality validation on the predicted interactions from less significant interlogs and non-interlogs
At present there is no experimental data available as validation set to evaluate the proposed model. Nevertheless, the less significant interlogs would be more likely to be interacting partners than non-interlogs. For the reasons, we explicitly study the potential interactions from the less significant interlogs and the non-interlogs, partly to evaluate the proposed model as well. The predicted interactions from the less significant interlogs and the non-interlogs are provided in Additional files 5 and 6, respectively. As shown in Table 4, the predicted positive rates on the less significant interlogs and non-interlogs are 18.78 and 1.41%, respectively. We can see that the less significant interlogs are more likely to interact than the non-interlogs. The result can be well interpreted by the wide difference of patterns of common GO terms between the less significant interlogs and the non-interlogs as illustrated in Fig. 4b. Comparing Fig. 4a with Fig. 4b, we see that the interlogs show much are similar distributions of GO terms than non-interlogs. The low positive rate 18.78% indicates that the less significant interlogs should not be equally treated as the significant interlogs as the existing work does [22–25], and need to be further validated by a machine learning framework. The low positive rate 1.41% shows that the sampling method of negative data as described in Formula (4) is rational.
Table 4.
Predicted positive rates on less significant interlogs, non-interlogs and the prediction set
| Less significant interlogs | Non-interlogs | Prediction set | |
|---|---|---|---|
| Size | 98,187 | 98,187 | 407,700 |
| Predicted positive rate | 18.78% | 1.41% | 1.96% |
As reviewed in [19], host factors play important roles in the progression of bacterial drug resistance. Hence the interactions between M. tuberculosis H37Rv drug-resistant genes and human host genes are of special interest to us. Moreover, inhibitors of pathogen-host PPI interface would be more therapeutically with less side-effect on other human genes and pathways. The predicted interactions from less significant interlogs related to drug resistance are illustrated in Fig. 5. Figure 5a illustrates the interlogs that get involved in antibiotic efflux pumps [46] (see Additional file 7 for detailed GO enrichment analysis), and Fig. 5b illustrates the interlogs that get involved in target-modifying enzymes [46] (see Additional file 8 for detailed GO enrichment analysis).
Fig. 5.

Validated less significant interlogs. a illustrates the interactions of M. tuberculosis H37Rv drug-resistant genes that are involved in antibiotic efflux pumps [46]. b illustrates the interactions of M. tuberculosis H37Rv drug-resistant genes that are involved in target-modifying enzymes [46]
Interlog {Rv0849, ABCB1}
The interlog {Rv0849|I6X9Y5, ABCB1|P08183} is derived from the known interaction {Rv0849|I6X9Y5, Rv1348|I6YAB3} [7], wherein the human protein P08183is orthologous to the M. tuberculosis H37Rv protein I6X9Y5 with E-value equal to 6e-043. The interlog {Rv0849|I6X9Y5, ABCB1|P08183} is predicted to be a pathogen-host protein interaction with probability 0.9987 (see Additional file 7). GO enrichment analysis shows that the two genes {Rv0849, ABCB1} both are located at membrane (GO:0005886, plasma membrane; GO:0016021, integral to membrane) and participate the biological process of transport (GO:0055085, transmembrane transport; GO:0006810, transport) (see Table 5). In addition, the human gene ABCB1 also gets involved in the biological processes of drug transmembrane transport (GO:0006855) and xenobiotic transport (GO:0042908). The GO terms (GO:0009986, cell surface; GO:0070062, extracellular vesicular exosome) indicate that the human protein P08183 could have physical contact with the M. tuberculosis H37Rv membrane protein I6X9Y5 to induce immune response (GO:0002485, antigen processing and presentation of endogenous peptide antigen via MHC class I via ER pathway, TAP-dependent).
Table 5.
Gene ontology analysis of the predicted interactions {Rv0849, ABCB1} and {Rv1988, PNP}. Rv0849 is classified into the drug resistance type of antibiotic efflux pumps [46]. Rv1988 is classified into the drug resistance type of target-modifying enzymes [46]
| {Rv0849, ABCB1} | GO term ID | GO aspect | GO term name |
|---|---|---|---|
| Common GO terms | GO:0055085 | P | transmembrane transport |
| GO:0006810 | P | transport | |
| GO:0005886 | C | plasma membrane | |
| GO:0016021 | C | integral to membrane | |
| ABCB1 only | GO:0005215 | F | transporter activity |
| GO:0006855 | P | drug transmembrane transport | |
| GO:0042908 | P | xenobiotic transport | |
| GO:0009986 | C | cell surface | |
| GO:0070062 | C | extracellular vesicular exosome | |
| GO:0002485 | P | antigen processing and presentation of endogenous peptide antigen via MHC class I via ER pathway, TAP-dependent | |
| {Rv1988, PNP} | GO term ID | GO aspect | GO term name |
| Common GO terms | GO:0005737 | C | cytoplasm |
| GO:0016740 | F | transferase activity | |
| Rv1988 only | GO:0000154 | P | rRNA modification |
| GO:0008649 | F | rRNA methyltransferase activity | |
| GO:0031167 | P | rRNA methylation | |
| GO:0046677 | P | response to antibiotic | |
| PNP only | GO:0006139 | P | nucleobase-containing compound metabolic process |
| GO:0006148 | P | inosine catabolic process | |
| GO:0006195 | P | purine nucleotide catabolic process | |
| O:0006738 | P | nicotinamide riboside catabolic process | |
| GO:0006955 | P | immune response | |
| GO:0042493 | P | response to drug | |
| GO:0070970 | P | interleukin-2 secretion | |
| GO:0034356 | P | NAD biosynthesis via nicotinamide riboside salvage pathway |
C denotes cellular component, F denotes molecular function, and P denotes biological process
Interlog {Rv1988, PNP}
The interlog {Rv1988|Q10838, PNP|P00491} is derived from the known interaction {Rv1988|Q10838, Rv0535|I6Y409} [7]. The human protein P00491 is orthologous to the M. tuberculosis H37Rv protein I6Y409 with E-value equal to 6e-034. The M. tuberculosis H37Rv gene Rv1988 is classified into the drug resistance type of target-modifying enzymes [46]. As shown in Table 5, the M. tuberculosis H37Rv Rv1988 gets involved in the biological processes of rRNA modification (GO:0000154), rRNA methylation (GO:0031167) and response to antibiotic (GO:0046677), while the human gene PNP also gets involved in the biological processes of protein modifications, e.g. the catabolic processes of nucleobase-containing compound (GO:0006139), inosine (GO:0006148), purine nucleotide (GO:0006195), etc. In addition, PNP is involved in the biological processes of immune response (GO:0006955, GO:0070970) and response to drug (GO:0042493).
Predicted interactions on the prediction set
The prediction set that contains 407,700 MTB-human protein pairs is derived from the huge space of non-interlogs according to Formula (7). As shown in Tables 1, 4.96% of protein pairs are predicted to be pathogen-host PPIs. Such a low positive rate is presumably rational with a low risk of false positive predictions. The predicted interactions on the prediction set are provided in Additional file 9. For the convenience of analysis, we merge the significant interlogs together with the predicted interactions from the less significant interlogs, non-interlogs and the prediction set into Additional file 10. We totally obtain 43,116 predicted protein interactions between M. tuberculosis H37Rv and Homo sapiens, which is still incomplete since the prediction set is only a small part of the prediction space.
Taking advantage of the predicted MTB-human PPI networks, we need to address two concerns (1) how many human genes a M. tuberculosis H37Rv gene is likely to target; (2) what roles the targeted human genes play in the human PPI networks. The two concerns are actually about two kinds of degree distributions (1) the degree distribution of the M. tuberculosis H37Rv genes in the MTB-human PPI networks (see Fig. 6 (left)); (2) the degree distribution of the human genes in human PPI networks (see Fig. 6 (right)). We can see that the two degrees show a tendency of power-law distribution. As shown in Fig. 6 (left), only a small portion of M. tuberculosis H37Rv genes are densely connected by human genes, indicating that only a small number of M. tuberculosis H37Rv genes intensively target dozens to several hundred of human genes. For instance, M. tuberculosis H37Rv gene Rv0440 (groEL) and Rv1436 (LH57_07850) interact with 209 and 206 human genes, respectively. As shown in Fig. 6 (right), only a small of targeted human genes are highly-connected hub genes and the long tail indicate that many targeted human genes are orphan genes in human PPI networks. It could be concluded that only a small number of M. tuberculosis H37Rv genes target a small number of human hub genes. The human PPI networks are constructed from HPRD [47] and BioGRID [48]. To further reveal the signaling cross-talks between M. tuberculosis H37Rv and Homo sapiens, we will discuss the patterns of M. tuberculosis H37Rv genes manipulating the human immune signaling pathways in the section Discussions.
Fig. 6.

The degree distribution of the M. tuberculosis H37Rv genes in the derived MTB-human PPI networks (left); the degree distribution of M. tuberculosis H37Rv targeted human genes in human physical PPI networks (right)
Discussions
To date, there are very few experimental studies on protein interactions between bacterial pathogens and their hosts. The latest database STRING has curated 1678 bacterial pathogen protein-protein interaction networks, but no work has been reported to exploit these networks to predict pathogen-host protein interactions thus far. Pathogen-host protein interactions play a critical role of signaling cross-talks between pathogen PPI networks and host PPI networks, which is of significance to understand the underlying mechanism of bacterial invasive infection and host immune response.
Mycobacterium tuberculosis is an obligate pathogenic bacterial species in the family of Mycobacteriaceae and the causative agent of tuberculosis. The physiology of M. tuberculosis is highly aerobic and requires high levels of oxygen. As primarily a pathogen of the mammalian respiratory system, M. tuberculosis mainly infects the lungs as well as other tissues. M. tuberculosis H37Rv has received much attention in recent years partly due to its co-infection with HIV and increasingly serious drug resistance. To date, the cross-talks or interactions between M. tuberculosis and H. sapiens proteins are much less understood than the individual genome of M. tuberculosis. To the best of our knowledge, there is no experimental study on protein interactions between M. tuberculosis H37Rv and Homo sapiens.
Methodology comparison with the related computational methods
The related computational methods generally infer interlogs via one or more third-party species. For instances, Kshirsagar et al. [23] use the known Salmonella-human PPIs as templates to infer Salmonella-plant PPIs via plant-human ortholog mapping (see Fig. 7a). Zhou et al. [24, 25] use the known prokaryote-eukaryote PPIs as templates to infer M. tuberculosis-H. sapiens PPIs via the ortholog mappings of prokaryote-M. tuberculosis and eukaryote-H. sapiens (see Fig. 7b). However, the large gap between the source species (e.g. plant) and the target species (e.g. human) is to a large extent prone to yield false pathogen-host protein interactions. In addition, the interlog-only methods [24, 25] neither validate the less significant interlogs nor train a predictive model to predict the non-interlogs that also potentially interact. Similar to the methods that combine interlog with machine learning approach [9, 23], we also use the derived interlogs as training data since there are no experimental data available, but differently we confine the search of M. tuberculosis H37Rv ortholog genes within the human host without resorting to a third-party species, meanwhile we do not need prior pathogen-host PPIs of other species as templates (see Fig. 7c). As illustrated in Fig. 7c, the ortholog genes of the two interacting M. tuberculosis H37Rv genes (A, B) are searched within the human genome space, presumably A’ and B′, respectively, then it is assumed that A interacts with B′ and B interacts with A’. The assumption is based on the accumulated evidences that the different strains of the obligate human pathogen M. tuberculosis have co-evolved, migrated, and expanded with their human hosts [27]. Knowledge transfer between co-evolving species is more credible than that between evolutionarily distant species.
Fig. 7.

Illustration of the way of deriving interlogs. The ellipses in red and blue denote the target species and the ellipses in other colors denote the source species. The circles denote genes. The red full line denotes the experimental protein-protein interactions. The red dotted line denotes ortholog mapping. The blue full line with arrows at two ends denotes the derived interlogs. The methods illustrated in (a) and (b) exploited the pathogen-host PPIs of other species to derive the interlogs of the target species, while the method illustrated in (c) transferred the knowledge of intra-species M. tuberculosis H37Rv PPI networks to predict protein interactions between M. tuberculosis H37Rv and H. sapiens
It is noted that the quality of M. tuberculosis H37Rv protein interaction networks in STRING [9] directly affects the quality of inferred interlogs between M. tuberculosis H37Rv and Homo sapiens. Among the experimental data in STRING [9], only 32 MTB PPIs are actually derived by experiments. Obviously, such a small data size cannot satisfy our needs, so we resort to the other experimental data in STRING [9] that are actually interlogs inferred from other experimentally-verified PPIs. Yu et al. [49] have testified the feasibility of interolog mapping, i.e. the transfer of interaction annotation from one organism to another using comparative genomics.
Performance comparison with the related computational methods
The interlog-only methods [24, 25] do not provide baseline performance for comparison. The method that combines interlog with machine learning approach [23] derives Salmonella-plant interlogs from the known plant-human PPIs as the training data to train a KMM-SVM model for novel Salmonella-plant PPI predictions. As shown in Table 3, the knowledge of Salmonella-human PPI networks is transferred in the first experiment to predict Salmonella-mouse protein interactions, achieving SE 0.937 and SP 0.517; and the knowledge of Salmonella-Ecoli PPI networks is transferred in the second experiment to predict Salmonella-human protein interactions, achieving SE 0.257 and SP 0.161. The results in the second experiment are obviously much poorer in that the gap of species between E.coli and human is much larger than that between mice and human. Even in the first experiment, the method [23] neither achieves satisfactory performance partly due to the other two reasons: (1) the less significant interlogs are not explicitly excluded out of the positive training data; (2) the two-class skew distribution also contributes to the low performance (SP = 0.517). In this work, the knowledge is only transferred across co-evolving pathogen and host, so that the proposed method achieves much better performance (see Table 3). Nevertheless, the proposed method also yields a certain level of bias and performance overestimation for the two reasons (1) similar interlogs in the training data could overestimate the performance of 5-fold cross validation performance, though they do not affect the final trained model and predictions; (2) the positive training data do not contain non-interlogs that also potentially interact because there are no such experimental data, so that the two classes of training data are easily separated. How to choose the representative interlogs to more objectively evaluate the proposed model is worth further consideration in the future work.
GO enrichment analysis of the targeted human genes
Figure 8 illustrates the top 20 GO terms of human genes that are predicted to be targeted by M. tuberculosis H37Rv genes. As shown in Fig. 8 (left), M. tuberculosis H37Rv genes are inclined to target those human genes located in the cellular compartments of membrane (GO:0016020), integral to membrane (GO:0016021), cytoplasm (GO:0005737), nucleus (GO:0005634), mitochondrion (GO:0005739), etc. As shown in Fig. 8 (middle), the targeted human genes fulfil the molecular functions of protein binding (GO:0005515), hydrolase activity (GO:0016787), ATP binding (GO:0005524), metal ion binding (GO:0046872), oxidoreductase activity (GO:0016491), etc. As shown in Fig. 8 (right), the targeted human genes get involved in the biological processes of transport (GO:0006810), oxidation-reduction process (GO:0055114), metabolic process (GO:0008152), ion transport (GO:0006811), proteolysis (GO:0006508), regulation of transcription, DNA-dependent (GO:0006355), etc.
Fig. 8.

GO enrichment analysis of the human genes that are predicted to be targeted by M. tuberculosis H37Rv drug-resistant genes
Pathway enrichment analysis of the targeted human genes
Bacterial invasion could induce host inflammatory response, for instances, TNF-α is thought to play a role in the activation of resting macrophages and inhibition of bacterial dissemination, and IL-10 might play a role in controlling the trade-off between the anti-microbial activity and host-derived tissue caseation [50, 51]. We map the targeted human genes onto the known human immune signaling pathways curated in NetPath [52] to study how M. tuberculosis genes interact with human defence system. For simplicity, the pathways IL1~IL11 in NetPath are merged into one IL signaling pathway, thus we totally obtain 27 human immune signaling pathways. The predicted MTB-human PPIs related to human immune signaling pathways are provided in Additional file 11. As shown in Fig. 9, M. tuberculosis H37Rv genes are inclined to target the human immune signaling pathways of AR (Androgen receptor), TNF-alpha (Tumor necrosis factor alpha), TGF-beta (Transforming growth factor beta receptor), IL (Interleukin), BCR (B cell receptor), TSH (Thymic stromal lymphopoietin),, etc. In most cases, many M. tuberculosis H37Rv genes are predicted to target more than one human immune signaling pathways (see Additional file 12), for instances, M. tuberculosis H37Rv gene recA (Rv2737c) is predicted to target five signaling pathways (TNF-alpha;IL2;BCR;AR;TSH). Partial GO enrichment analysis of the targeted human genes on TNF-alpha and IL signaling pathways are given in Table 6.
Fig. 9.

Statistics of MTB-human PPIs and MTB genes that are predicted to target specific human immune signaling pathways
Table 6.
GO and pathway enrichment analysis of the M. tuberculosis H37Rv genes that are predicted to target human TNF-alpha and IL-1~IL11 signaling pathways
| TNF-Alpha | GO term ID | GO aspect | GO term name | % | Shared |
| MTB genes that target human gene PSMC2 | GO:0005524 | F | ATP binding | 26.92 | Yes |
| GO:0016020 | C | membrane | 20.51 | Yes | |
| GO:0055114 | P | oxidation-reduction process | 16.67 | No | |
| GO:0016310 | P | phosphorylation | 5.98 | No | |
| GO:0006412 | P | translation | 5.13 | No | |
| GO:0055085 | P | transmembrane transport | 2.14 | Yes | |
| GO:0046933 | F | hydrogen ion transporting ATP synthase activity, rotational mechanism | 1.71 | No | |
| Human gene PSMC2 | GO:0002479 | P | antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent | – | – |
| GO:0043687 | P | post-translational protein modification | – | – | |
| GO:0045899 | P | positive regulation of RNA polymerase II transcriptional preinitiation complex assembly | – | – | |
| Targeted human genes | CASP10,BID,RUVBL2,DDX21,RPL4,BRINP1,PSMC2,PSMD1,FANCD2,MTIF2,PSMD2,PSMB5,RPS11,PSMC3,GLUL,PDCD2,KTN1 | ||||
| IL-1 ~ IL-11 | GO term ID | GO aspect | GO term name | % | Shared |
| M.TB genes that target the human gene VCP | GO:0003824 | F | catalytic activity | 32.17 | No |
| GO:0016021 | C | integral to membrane | 16.78 | No | |
| GO:0005524 | F | ATP binding | 27.27 | Yes | |
| GO:0055114 | P | oxidation-reduction process | 9.79 | No | |
| GO:0006810 | P | transport | 3.50 | Yes | |
| GO:0006281 | P | DNA repair | 2.10 | No | |
| Human gene VCP | GO:0005515 | F | protein binding | – | – |
| GO:0006914 | P | autophagy | – | – | |
| GO:0016236 | P | macroautophagy | – | – | |
| GO:0010918 | P | positive regulation of mitochondrial membrane potential | – | – | |
| GO:0006974 | P | cellular response to DNA damage stimulus | – | – | |
Targeted human genes BCL2L11,UNC119,IRS1,IL2,DOK2,PTPN6,BAD,IL11,VCP,IRS2
C denotes cellular component, F denotes molecular function, and P denotes biological process
TNF-alpha signaling pathway
The tumor necrosis factor alpha (TNF-alpha) is a pro-inflammatory cytokine that belongs to the TNF superfamily [48]. M. tuberculosis H37Rv is predicted to invade human TNF-alpha signaling pathway through 443 MTB-human PPIs and the targeted human genes {CASP10, BID, RUVBL2, DDX21, RPL4, BRINP1, PSMC2, PSMD1, FANCD2, MTIF2, PSMD2, PSMB5, RPS11, PSMC3, GLUL, PDCD2, KTN1} (see Additional file 11). Taking the targeted human gene PSMC2 for example (see Table 6), among the M. tuberculosis H37Rv genes that target the human gene PSMC2, 20.51% of proteins are located at membrane (GO:0016020); 16.67% of genes are involved in the biological process of oxidation-reduction process (GO:0055114); 2.14% of genes are involved in the biological process of transmembrane transport (GO:0055085). Besides the GO terms marked with “shared” in Table 6, the human gene PSMC2 also gets involved in translation (GO:0046933;hydrogen ion transporting ATP synthase activity, rotational mechanism) and post-translational protein modification (GO:0043687). Especially, the targeted human gene PSMC2 plays an important role in the host immune response (GO:0002479; antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent).
IL1~IL11 signaling pathway
Interleukins are a group of cytokines (secreted proteins and signal molecules) that were first seen to be expressed by white blood cells (leukocytes). The function of immune system depends in a large part on interleukins that promote the development and differentiation of T and B lymphocytes, and hematopoietic cells [53]. M. tuberculosis H37Rv is predicted to interact with human IL-1~ 11 signaling pathways through 157 MTB-human PPIs and the targeted human genes {BCL2L11, UNC119, IRS1, IL2, DOK2, PTPN6, BAD, IL11, VCP, IRS2} (see Additional file 11). Taking the targeted human gene VCP for example (see Table 6), among the M. tuberculosis H37Rv genes that target human gene VCP, 16.78% of genes are located at integral to membrane (GO:0016021); 27.27% of genes fulfil the molecular function of ATP binding (GO:0005524); 9.79% of genes are involved in oxidation-reduction process (GO:0055114); 3.50% of genes are involved in transport (GO:0006810); 9.79% of genes are involved in DNA repair (GO:0006281), etc. Besides ATP binding (GO:0005524) and transport (GO:0006810), the targeted human gene VCP gets involved in autophagy (GO:0006914), macroautophagy (GO:0016236) and cellular response to DNA damage stimulus (GO:0006974), etc.
Conclusions
In this work, we provide a general computational framework to exploit the knowledge of the pathogen protein interaction networks in the database STRING for the rapid reconstruction of pathogen-host protein interaction networks. We take full advantage of the co-evolution relationship between M. tuberculosis H37Rv and H. sapiens to derive significant interlogs, which are used as the training data to build a predictive model. The knowledge transfer model effectively solves the problem that no experimental bacteria-host protein interactions are available as training data. The predicted protein interactions provided in the Additional files promise to gain applications in the two fields (1) providing an alternative solution to drug resistance; (2) revealing the patterns that M. tuberculosis H37Rv genes target human immune signaling pathways.
Additional files
Text file contains the positive training data consisting of the derived significant interlogs. (TXT 1201 kb)
Text file contains the negative training data consisting of randomly sampled non-interlogs. (TXT 769 kb)
Text file contains the gene ontology analysis of the derived interlogs that get involved in drug resistance of cytochromes and other target-modifying enzymes. (TXT 1703 kb)
Text file contains the gene ontology analysis of the derived interlogs that get involved in drug resistance of SOS-response and DNA replication. (TXT 1157 kb)
Text file contains the predicted results on the positive independent test set consisting of less significant interlogs. (TXT 1055 kb)
Text file contains the predicted results on the negative independent test set consisting of randomly sampled non-interlogs. (TXT 80 kb)
Text file contains the gene ontology analysis of the validated less significant interlogs that get involved in drug resistance of antibiotic efflux pumps. (TXT 234 kb)
Text file contains the gene ontology analysis of the validated less significant interlogs that get involved in drug resistance of target-modifying enzymes. (TXT 21 kb)
Text file contains the predicted results on the prediction set consisting of randomly sampled non-interlogs. (TXT 552 kb)
Text file contains the summary of the derived or predicted M.TB-human PPIs. (TXT 2888 kb)
Text file contains the human cancer/immune signaling pathways that M. tuberculosis H37Rv genes are predicted to target. (TXT 106 kb)
Text file contains the M. tuberculosis H37Rv genes that target human cancer/immune signaling pathways. (TXT 21 kb)
Acknowledgments
Availability of supporting data
All data generated or analysed during this study are included in this published article [and its Additional files].
Funding
This work is partly supported by the funding from the NIH grants 5G12MD007595, P01CA214091 and 5P2OGM103424-15, DOD ARO grant W911NF-15-1-0510 and the Louisiana Cancer Research Consortium (LCRC). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, DOD or LCRC.
Availability of data and materials
The source codes and tools for this proposed framework are publicly available at https://github.com/suyumei/MTBH37RvHomo.git.
Abbreviations
- AR
Androgen receptor
- B. anthracis
Bacillus anthracis
- BCR
B cell receptor
- Ecoli
Escherichia coli
- F. tularensis
Francisella tularensis
- GO
Gene ontology
- H. sapiens
Homo sapiens
- IL
Interleukins are a group of cytokines
- M. tuberculosis H37Rv
Mycobacterium tuberculosis
- MTB
M. tuberculosis H37Rv
- MTBC
Mycobacterium tuberculosis complex
- RBH
Reciprocal best hits
- TNF-alpha
The tumor necrosis factor alpha
- TSH
Thyroid stimulating hormone
- Y. pestis
Yersinia pestis
Authors’ contributions
MS conducted the study and wrote the paper. FEK and ZK revised the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4873-9) contains supplementary material, which is available to authorized users.
Contributor Information
Suyu Mei, Email: meisygle@gmail.com.
Erik K. Flemington, Email: erik@tulane.edu
Kun Zhang, Email: kzhang@xula.edu.
References
- 1.Koul A, Herget T, Klebl B, Ullrich A. Interplay between mycobacteria and host signalling pathways. Nat Rev Microbiol. 2004;2:189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 2.Nunn P, Williams B, Floyd K, Dye C, Elzinga G, et al. Tuberculosis control in the era of HIV. Nat Rev Immunol. 2005;5:819–826. doi: 10.1038/nri1704. [DOI] [PubMed] [Google Scholar]
- 3.Silva ON, de la Fuente-Núñez C, Haney EF, Fensterseifer IC, Ribeiro SM, et al. An anti-infective synthetic peptide with dual antimicrobial and immunomodulatory activities. Sci Rep. 2016;6:35465. doi: 10.1038/srep35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui T, Zhang L, Wang X, He ZG. Uncovering new signaling proteins and potential drug targets through the interactome analysis of Mycobacterium tuberculosis. BMC Genomics. 2009;10:118. doi: 10.1186/1471-2164-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melak T, Gakkhar S. Maximum flow approach to prioritize potential drug targets of Mycobacterium tuberculosis H37Rv from protein-protein interaction network. Clin Transl Med. 2015;4(61) [DOI] [PMC free article] [PubMed]
- 6.Cui ZJ, Yang QY, Zhang HY, Zhu Q, Zhang QY. Bioinformatics identification of drug resistance-associated gene pairs in Mycobacterium tuberculosis. Int J Mol Sci. 2016;17(9). pii: E1417. [DOI] [PMC free article] [PubMed]
- 7.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung BK, Dick T, Lee DY. In silico analyses for the discovery of tuberculosis drug targets. J Antimicrob Chemother. 2013;68:2701–2709. doi: 10.1093/jac/dkt273. [DOI] [PubMed] [Google Scholar]
- 9.Liu ZP, Wang J, Qiu YQ, Leung RK, Zhang XS, et al. Inferring a protein interaction map of Mycobacterium tuberculosis based on sequences and interologs. BMC Bioinformatics. 2012;13(Suppl 7):S6. doi: 10.1186/1471-2105-13-S7-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ananthasubramanian S, Metri R, Khetan A, Gupta A, et al. Mycobacterium tuberculosis and Clostridium difficille interactomes: demonstration of rapid development of computational system for bacterial interactome prediction. Microb Inform Exp. 2012;2:4. doi: 10.1186/2042-5783-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durmuş Tekir SD, Ülgen KÖ. Systems biology of pathogen-host interaction: networks of protein-protein interaction within pathogens and pathogen-human interactions in the post-genomic era. Biotechnol J. 2013;8:85–96. doi: 10.1002/biot.201200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukherjee S, Sambarey A, Prashanthi K, Chandra N. Current trends in modeling host-pathogen interactions. WIREs Data Mining Knowl Discov. 2013;3:109–128. doi: 10.1002/widm.1085. [DOI] [Google Scholar]
- 13.Durmuş S, Çakır T, Özgür A, Guthke R. A review on computational systems biology of pathogen-host interactions. Front Microbiol. 2015;6:235. doi: 10.3389/fmicb.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi Y, Tastan O, Carbonell JG, Klein-Seetharaman J, Weston J. Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics. 2010;26:i645–i652. doi: 10.1093/bioinformatics/btq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei S. Probability weighted ensemble transfer learning for predicting interactions between HIV-1 and human proteins. PLoS One. 2013;8:e79. doi: 10.1371/journal.pone.0079606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei S, Zhu H. A novel one-class SVM based negative data sampling method for reconstructing proteome-wide HTLV-human protein interaction networks. Sci Rep. 2015;5(8034) [DOI] [PMC free article] [PubMed]
- 17.Mei S, Zhang K. Computational discovery of Epstein-Barr virus targeted human genes and signalling pathways. Sci Rep. 2016;6(30612) [DOI] [PMC free article] [PubMed]
- 18.Eid FE, ElHefnawi M, Heath LS. DeNovo: virus-host sequence-based protein-protein interaction prediction. Bioinformatics. 2016; 32:1144-50. [DOI] [PubMed]
- 19.Ben-Kahla I, Al-Hajoj S. Drug-resistant tuberculosis viewed from bacterial and host genomes. Int J Antimicrob Agents. 2016;48:353–360. doi: 10.1016/j.ijantimicag.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Mei S, Zhu H. AdaBoost based multi-instance transfer learning for predicting interactions between Salmonella and human proteins. PLoS One. 2014;9:e110488. doi: 10.1371/journal.pone.0110488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyer MD, Neff C, Dufford M, Rivera CG, Shattuck D, et al. The human-bacterial pathogen protein interaction networks of bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One. 2010;5:e12089. doi: 10.1371/journal.pone.0012089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schleker S, Garcia-Garcia J, Klein-Seetharaman J, Oliva B. Prediction and comparison of Salmonella-human and Salmonella-Arabidopsis interactomes. Chem Biodivers. 2012;9:991–1018. doi: 10.1002/cbdv.201100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kshirsagar M, Schleker S, Carbonell J, Klein-Seetharaman J. Techniques for transferring host-pathogen protein interactions knowledge to new tasks. Front Microbiol. 2015;6(36) [DOI] [PMC free article] [PubMed]
- 24.Zhou H, Rezaei J, Hugo W, Gao S, Jin J, et al. Stringent DDI-based prediction of H. Sapiens-M. Tuberculosis H37Rv protein-protein interactions. BMC Syst Biol. 2013;7(Suppl 6):S6. doi: 10.1186/1752-0509-7-S6-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Gao S, Nguyen NN, Fan M, Jin J, et al. Stringent homology-based prediction of H. Sapiens-M. Tuberculosis H37Rv protein-protein interactions. Biol Direct. 2014;9:5. doi: 10.1186/1745-6150-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palomino JC, Martin A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics (Basel) 2014;3:317–340. doi: 10.3390/antibiotics3030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comas I, Coscolla M, Luo T, Borrell S, Holt K, et al. Out-of-Africa migration and Neolithic co-expansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veselovsky, A.V., Zharkova, M.S., Poroikov, V.V., Nicklaus, M.C. Computer-aided design and discovery of protein-protein interaction inhibitors as agents for anti-HIV therapy. SAR QSAR Environ Res 25, 457–71 (2014). [DOI] [PubMed]
- 29.Zhou H, Wong L. Comparative analysis and assessment of M. Tuberculosis H37Rv protein-protein interaction datasets. BMC Genomics. 2011;12(Suppl 3):S20. doi: 10.1186/1471-2164-12-S3-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward N, Moreno-Hagelsieb G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: how much do we miss? PLoS One. 2014;9:e101850. doi: 10.1371/journal.pone.0101850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salichos L, Rokas A. Evaluating ortholog prediction algorithms in a yeast model clade. PLoS One. 2014;6:e18755. doi: 10.1371/journal.pone.0018755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 35.Maetschke S, Simonsen M, Davis M, Ragan MA. Gene ontology-driven inference of protein–protein interactions using inducers. Bioinformatics. 2012;28:69–75. doi: 10.1093/bioinformatics/btr610. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Zhu L, Guo J, Zhang D, Lin K. Prediction of yeast protein-protein interaction network: insights from the gene ontology and annotations. Nucleic Acids Res. 2006;34:2137–2150. doi: 10.1093/nar/gkl219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeBodt S, Proost S, Vandepoele K, Rouze´ P, Peer Y, et al. Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics. 2009;10:288. doi: 10.1186/1471-2164-10-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller J, Lo RS, Ben-Hur A, Desmarais C, Stagljar I, et al. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A. 2005;102:12123–12128. doi: 10.1073/pnas.0505482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin N, Wu B, Jansen R, Gerstein M, Zhao H, et al. Information assessment on predicting protein-protein interactions. BMC Bioinformatics. 2004;5:154. doi: 10.1186/1471-2105-5-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandyopadhyay S, Ray S, Mukhopadhyay A, Maulik U. A review of in silico approaches for analysis and prediction of HIV-1-human protein-protein interactions. Brief Bioinform. 2015;16:830–851. doi: 10.1093/bib/bbu041. [DOI] [PubMed] [Google Scholar]
- 41.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, et al. The GOA database in 2009--an integrated gene ontology annotation resource. Nucleic Acids Res. 2009;37(Database):D396–D403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang, C.C., & Lin, C.J. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2, 1–27 (2011) [Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm].
- 44.Yu F, Huang F, Lin C. Dual coordinate descent methods for logistic regression and maximum entropy models. Mach Learn. 2011;85:41–75. doi: 10.1007/s10994-010-5221-8. [DOI] [Google Scholar]
- 45.Fan R, Chang K, Hsieh C, Wang X, Lin C. LIBLINEAR: a library for large linear classification. Mach Learn Res. 2008;9:1871–1874. [Google Scholar]
- 46.Raman K, Chandra N. Mycobacterium tuberculosis interactome analysis unravels potential pathways to drug resistance. BMC Microbiol. 2008;8:234. doi: 10.1186/1471-2180-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keshava Prasad, T.S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., et al.. Human protein reference database--2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–D772. [DOI] [PMC free article] [PubMed]
- 48.Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43(Database issue):D470–D478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu H, Luscombe NM, Lu HX, Zhu X, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med. 2016;22:531–538. doi: 10.1038/nm.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Opp MR, Smith EM, Hughes TK Jr. Interleukin-10 (cytokine synthesis inhibitory factor) acts in the central nervous system of rats to reduce sleep. J Neuroimmunol. 1995;60(1-2):165-8. [DOI] [PubMed]
- 52.Kandasamy K, Mohan SS, Raju R, Keerthikumar S, Kumar GS, et al. NetPath: a public resource of curated signal transduction pathways. Genome Biol. 2010;11:R3. doi: 10.1186/gb-2010-11-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics. 2010;5:30–55. doi: 10.1186/1479-7364-5-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text file contains the positive training data consisting of the derived significant interlogs. (TXT 1201 kb)
Text file contains the negative training data consisting of randomly sampled non-interlogs. (TXT 769 kb)
Text file contains the gene ontology analysis of the derived interlogs that get involved in drug resistance of cytochromes and other target-modifying enzymes. (TXT 1703 kb)
Text file contains the gene ontology analysis of the derived interlogs that get involved in drug resistance of SOS-response and DNA replication. (TXT 1157 kb)
Text file contains the predicted results on the positive independent test set consisting of less significant interlogs. (TXT 1055 kb)
Text file contains the predicted results on the negative independent test set consisting of randomly sampled non-interlogs. (TXT 80 kb)
Text file contains the gene ontology analysis of the validated less significant interlogs that get involved in drug resistance of antibiotic efflux pumps. (TXT 234 kb)
Text file contains the gene ontology analysis of the validated less significant interlogs that get involved in drug resistance of target-modifying enzymes. (TXT 21 kb)
Text file contains the predicted results on the prediction set consisting of randomly sampled non-interlogs. (TXT 552 kb)
Text file contains the summary of the derived or predicted M.TB-human PPIs. (TXT 2888 kb)
Text file contains the human cancer/immune signaling pathways that M. tuberculosis H37Rv genes are predicted to target. (TXT 106 kb)
Text file contains the M. tuberculosis H37Rv genes that target human cancer/immune signaling pathways. (TXT 21 kb)
Data Availability Statement
The source codes and tools for this proposed framework are publicly available at https://github.com/suyumei/MTBH37RvHomo.git.