

Abstract
Background
Dapoxetine (DPX) is the drug of choice for the specific treatment of premature ejaculation. DPX is characterized by relatively low bioavailability (42%) and short half-life (1.5 h). The aim of this study was to improve DPX bioavailability and delivery across the blood–brain barrier (BBB) using a nanostructured DPX formulation for improved DPX efficacy and patient satisfaction.
Materials and methods
DPX-loaded polymeric micelles (PMs) formulations (F1–F3) were characterized for particle sizes, entrapment efficiencies, and Fourier transform infrared spectroscopic and transmission electron microscopic evaluations. In addition, diffusion profiles of the prepared formulations were investigated. Animal model pharmacokinetic parameters in plasma and brain tissues were investigated and compared with commercial DPX tablets.
Results
Particle size analysis revealed that formulations of DPX PMs showed a narrow range of 62.7±9.3–45.45±9.1 nm for F1–F3. In addition, DPX PMs showed a sustained release pattern with 91.27%±7.64%, 79.43%±7.81%, and 63.78%±5.05% of DPX content permeated after 24 h for F1, F2, and F3, respectively. Plasma pharmacokinetic parameters for DPX PMs showed significant increase (P<0.05) for the area under drug concentration–time curves in plasma and brain tissues compared with commercial DPX tablets.
Conclusion
DPX formulations in the form of PMs improved bioavailability and efficacy across the BBB. This DPX formulation provided improved brain delivery in order to enhance the convenience and compliance of patients.
Keywords: PEG–PLGA, biodegradable copolymer, brain delivery, self-assembled core–shell nanostructures, nanotechnology
Introduction
Premature ejaculation (PE) is a widely distributed disease that affects men at any age. PE is characterized by uncontrolled ejaculation before or immediately after intercourse. Accordingly, anxiety due to partner dissatisfaction could affect the quality of life of PE patients. PE was first classified into types A and B.1 Type A PE develops later in life and is usually associated with erectile dysfunction, whereas type B PE consists of rapid ejaculation (short latency) occurring in <1 min. Four PE subtypes with different pathogeneses have subsequently been proposed.2
Dapoxetine (DPX) is considered the first compound to be developed and the drug of choice for the specific treatment of PE. DPX is one of the several selective serotonin reuptake inhibitors (SSRIs). DPX is a recently approved European drug (European Medicines Agency approval in 2012).3,4 Serotonin reuptake inhibition by DPX increases serotonin’s action at the postsynaptic cleft that promotes ejaculatory delay. DPX is characterized by relatively low bioavailability (42%) and short half-life (1.5 h).
Poly(ethylene glycol) methyl ether-block-poly(lactide-co-glycolide) (PEG–PLGA) copolymers are composed of a hydrophobic PLGA core and a hydrophilic PEG coat. PEG–PLGA amphiphilic copolymers form core–shell polymeric micelles (PMs) that self-assemble in an aqueous solution.5–7 The PEG shell surrounding the core acts as a stabilizer for micelles in the aqueous phase and in vivo acts as protection from protein and cellular adhesion.8,9 The dilution of micelle formulations is unavoidable following their administration, but the PMs resistance to dilution effects is attributed to the extremely low critical micelle concentration (CMC) that ensures micellar integrity and stability for a prolonged period.10 Dilution of other micelle types, especially those with high CMC could split micellar structure and release drug content instantly. PEG–PLGA copolymers have excellent biodegradability and biocompatibility, which have led to PEG–PLGA PMs applications in several studies, including clinical investigations, and both PEG and PLGA have already been approved by the US Food and Drug Administration.11–14
The human brain is protected by a network of capillaries and tissues forming the BBB, which protects the brain from harmful endogenous and exogenous substances or bacteria. Substances that need to enter the brain should be hydrophobic with a molecular size of <400 Da or be able to enter via active efflux transporters.15 Permeability through the BBB relies on drug hydrophobicity. There is a direct relation with lipid solubility and BBB permeability for parameters such as increased logarithm of octanol/water partition coefficient. Another way for passing into the brain is via delivery systems that enhance drug passage through the blood–central nervous system barrier. Recently, amphiphilic block copolymers gained significant importance in this way due to their characteristics as stable, self-assembled, and nano-range-sized structures. The parameters that affect BBB crossing using nanoparticles are the subject of discussions by several reviews.16–18 Studies have shown improved BBB penetration with a reduction in nanoparticle size.17,19,20 In addition, shape, charge (zeta potential), and surface modified ligands and PEGylation (steric stabilization) can have a significant effect in crossing BBB.16,21,22 PMs showed the ability to improve BBB permeability through copolymer interaction with cell membranes that enhance membrane fluidity, inhibit P-glycoprotein and multidrug efflux transporters, deplete cellular ATP, and enhance insulin receptor-mediated transport.23,24
Using a PMs formula, the aim of this study was to improve DPX bioavailability and brain delivery in order to enhance the convenience for patients and reduce the medication dependence of patients. A DPX-loaded PM formulation was prepared and evaluated for particle size measurement, morphological characterization using transmission electron microscopy (TEM), and performance of an ex vivo diffusion study. In addition, determination of pharmacokinetic parameters in plasma and brain tissues in animal models has been compared with commercial DPX tablets (reference formula). This work is the first step in the development of a DPX PM preparation for improving DPX efficacy.
Materials and methods
Materials
DPX hydrochloride was a kind gift from Spimaco Addwaeih (Riyadh, Saudi Arabia). PEG–PLGA, PLGA Mn 4,500, PEG Mn 2,000, acetone, and other chemicals used were purchased from the Sigma-Aldrich Co. (St Louis, MO, USA).
Formulation of DPX PMs
Formulations of PMs (F1–F3) were prepared. Briefly, PEG–PLGA (100, 150, and 200 mg) was dissolved in acetone (15 mL). DPX (20 mg) was dissolved in the polymeric solution and then added to a 20 mL buffered solution (pH 9). The resulting dispersion was stirred for 4 h to remove the organic solvent. DPX-loaded PMs were dialyzed to remove free DPX using a dialysis membrane tube composed of cellulose (molecular weight cut off =12,000–14,000). The dispersion was then lyophilized using the ALPHA 1-2/LD Plus freeze dryer (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany).
Particle size analysis of the prepared DPX PMs
The prepared DPX formulations were assayed for particle size using the laser diffraction technology by Zetatrac particle size analyzer (Microtrac Inc., Montgomeryville, PA, USA). DPX formulation (2 mL sample) was placed into an analyzer chamber and measured at room temperature. The analysis was performed in triplicate.
Determination of the prepared DPX PMs entrapment efficiency (EE)
DPXs EE% was determined indirectly by measuring the concentration of free drug in the aqueous phase of DPX-loaded PEG–PLGA dispersion. A set volume of the prepared PMs was centrifuged for 60 min at room temperature (Nanosep® centrifugal devices; Pall Corporation, Port Washington, NY, USA). The amount of free drug in the aqueous phase in the recovery chamber was estimated using a standard reported HPLC method (as described in the “Quantification of DPX by high-performance liquid chromatography” section). The analysis was performed in triplicate. DPX PMs EE% was calculated:
| (1) |
Fourier transform infrared (FTIR) analysis of DPX PMs formulation
The lyophilized PM formula (F1) was assessed via FTIR spectroscopy. FTIR spectra of DPX, PEG–PLGA, and the prepared formulations were recorded over the wavelength range from 400 to 4,000/cm using an FTIR spectrophoto meter (Nicolet IZ 10; Thermo Fisher Scientific, Waltham, MA, USA).
Investigations of the prepared DPX PMs using TEM
DPX PMs (F3) were examined using TEM (Model JEM-1230; JOEL, Tokyo, Japan). Briefly, sample drops were mounted on a carbon-coated grid with phosphotungstic acid and then examined by TEM.
Ex vivo permeation of DPX PMs
Permeation studies of the prepared DPX formulations (F1–F3) were accomplished using an equivalent amount of 250 μg of DPX and utilizing an automated Hanson Franz diffusion cell apparatus (Hanson Research Microette Plus, Chatsworth, CA, USA) as previously reported.25 The diffusion was carried out using bovine intestines (collected from slaughter house, Jeddah, Saudi Arabia). A buffered solution (pH 7.0) was used as the diffusion medium. Aliquots of 24 h samples were analyzed for DPX content by HPLC (as described in the “Quantification of DPX by high-performance liquid chromatography” section).
Plasma and brain in vivo pharmacokinetic studies
An in vivo study was carried out to compare the pharmacokinetics of a nanostructured DPX formulation (F3) with DPX commercial tablets. A single oral dose of the prepared DPX formula in addition to the reference product was given to Wistar rats.
Experimental animals
Forty-two Wistar rats, with body masses of 200–220 g, were obtained from the animal house, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia. Every single exploratory convention complied with the approval of the Institutional Ethics Review Board of College of Pharmacy, Umm Al-Qura University, Makkah, Saudi Arabia, and the requirements of the Declaration of Helsinki, the Guiding Principle in the Care and Use of Animals (DHEW production NIH 80-23), and the Standards of Laboratory Animal Care (NIH distribution # 85-23, reconsidered in 1985). The rats were allowed to acclimate to their surroundings for no <14 days in naturally controlled enclosures. Rats were isolated in two groups, each with 21 rats. The commercial DPX tablets (control) group received the DPX once orally (5 mg/kg). The second group (DPX PMs, F3) received DPX equivalent to 5 mg/kg. Three animals were sacrificed to collect plasma and brain samples for each group at specific time intervals (0, 1, 2, 4, 8, 12, and 24 h). The plasma was separated by centrifugation at 5,000 rpm for 5 min, kept in labeled glass tubes with Teflon®-lined caps, and frozen at −20°C until analysis. The brain tissue was homogenized with a buffer at pH 7.5 (10 mL/g tissue) and, then, centrifuged at 30,000 rpm for 15 min at 4°C.
Quantification of DPX by high-performance liquid chromatography (HPLC)–tandem mass spectrometry
DPX concentrations in plasma and brain tissue were assayed using the liquid chromatography–tandem mass spectrometry (LC–MS)/MS method following Kim et al’s26 reported procedure with slight modifications. HPLC used the Agilent 1200 system (Agilent Technologies, Santa Clara, CA, USA). The detector was Agilent 6420, a triple quad mass spectrometer controlled by the MassHunter Software. The separation was performed on an Agilent ZORBAX Eclipse Plus C18 column, 5 μm, 4.6×150 mm (Agilent Technologies). The mobile phase was composed of 0.1% formic acid and acetonitrile (20:80, v/v), and the flow rate was 0.5 mL/min. The total run time was 6.0 min. DPX and Sildenafil (internal standard) were detected in the multiple reaction monitoring (MRM) scan mode with positive ion detection. The precursor–product ion pairs used for the MRM detection were m/z 306 → 261 for DPX and m/z 475 → 283 for Sildenafil.
Maximum DPX plasma concentration (Cmax, ng/mL) and time to reach this concentration (Tmax, h), area under the curve (AUC0–∞) (ng h/mL), and mean residence time (MRT0–∞, h) were estimated according to the noncompartmental analysis for DPX plasma concentrations, while a dual compartmental analysis model was used to estimate Cmax, AUC0–∞, and MRT0–∞ for DPX in the brain, using the WinNonlin® software Ver.1.5 (Scientific Consulting, Inc., Cary, NC, USA). The data obtained were expressed as mean values (±SD), except for Tmax, which was presented as the median (range). The statistical significance of the results was checked at a P-value of <0.05 by the analysis of variance followed by Sidak’s multiple comparisons test (GraphPad Prism 6; GraphPad Software, Inc., La Jolla, CA, USA).
Results
Formulation and characterization of DPX PMs
DPX was included in the PMs formula in three different ratios (F1–F3) as indicated in Table 1. Particle size analysis of the prepared DPX PM formulations was 62.7±9.3, 56.7±6.1, and 45.45±9.1 nm for F1, F2, and F3, respectively. The results revealed particle size reductions corresponding to increases in PEG–PLGA polymer content. EE% data in Table 1 show an increase in DPX entrapment within the PMs with the increase in PEG–PLGA ratio in the prepared formulations. EE% showed 39.72±9.36, 51.33±6.82, and 68.38±7.83 for F1, F2, and F3, respectively.
Table 1.
Composition, particle size, and EE% of DPX-loaded PMs’ formulations
| Composition and characterization | F1 | F2 | F3 |
|---|---|---|---|
| Formulations | |||
| DPX (mg) | 20 | 20 | 20 |
| PEG–PLGA (mg) | 100 | 150 | 200 |
| Characterization (mean ± SD) | |||
| Particle size (nm) | 62.7±9.3 | 56.7±6.1 | 45.45±9.1 |
| EE% | 39.72±9.36 | 51.33±6.82 | 68.38±7.83 |
Abbreviations: DPX, dapoxetine; EE, entrapment efficiency; PEG–PLGA, poly (ethylene glycol) methyl ether-block-poly(lactide-co-glycolide); PMs, polymeric micelles.
FTIR analysis of DPX PMs formulation
Interaction studies of drugs and polymers in the prepared formulations can be evaluated using FTIR data. Changes in the characteristic wavenumbers of functional group peaks and excipient peaks can be used to predict chemical incompatibility in formula components. Figure 1 shows the characteristic DPX peaks of 3,057, 2,953, 1,583, 1,456, and 1,394. The IR spectrum of PEG–PLGA showed a broad band at 2,992/cm (peak) indicating the presence of a hydroxyl group. In addition, appearance of a characteristic band at 1,730/cm confirmed the presence of a carbonyl group. The shift of both the hydroxyl and carbonyl groups can be attributed to hydrogen bonding. The absorption bands of the prepared formula did not show interference with the characteristic drugs peaks, indicating the compatibility of formula components.
Figure 1.

FTIR spectra of DPX, DPX PMs, and PEG–PLGA.
Abbreviations: DPX, dapoxetine; FTIR, Fourier transform infrared; PEG–PLGA, poly(ethylene glycol) methyl ether-block-poly(lactide-co-glycolide); PMs, polymeric micelles.
Investigations of the prepared DPX PMs using TEM
According to the data obtained from the characterization of DPX PM formulations, F3 was selected for the TEM investigation. TEM offers detailed images, including internal structures, for the investigated sample. Additionally, TEM is considered as an important tool in validating the reliability of the particle size measurement by laser diffraction technology. Figure 2A shows the particle sizes of the prepared formula, revealing that the majority (85.4%) of the particles’ population was <50 nm and some aggregates of PMs were <15%, according to the peaks summary data gained from the instrument. TEM images of this formulation illustrated in Figure 2B indicate the presence of nanoparticles with spherical structures and a size range in accordance with the data obtained from the particle size analyzer.
Figure 2.
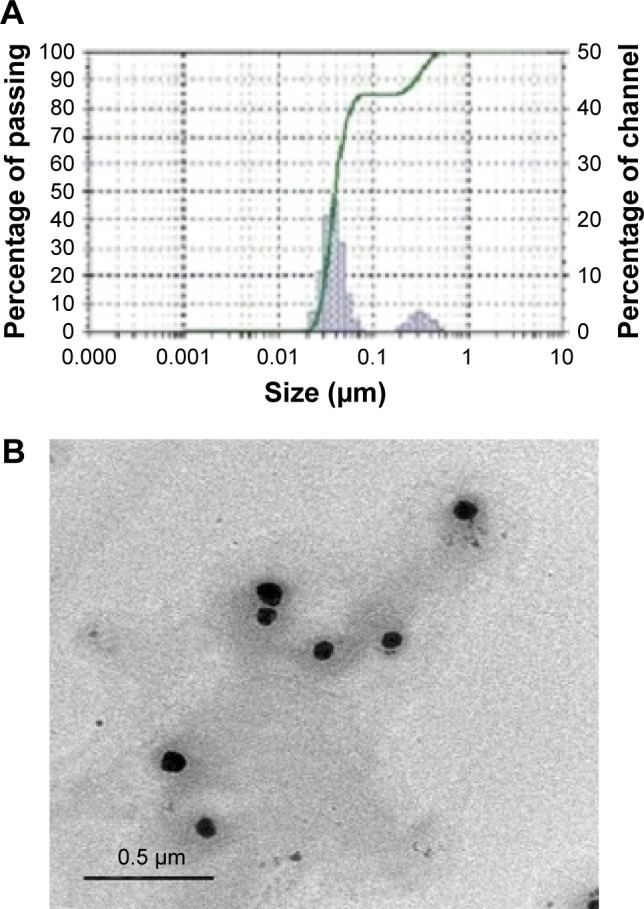
Particle size distribution of DPX PMs obtained from the particle size analyzer.
Notes: (A) Particle sizes in the prepared formula. The majority of the population (85.4%) showed a size of <50 nm with the rest of the population (14.6%) showing the growth of particle size to 320 nm. (B) The TEM image of the prepared DPX PMs’ formula. The image shows a dark internal structure and a size range in accordance with the data obtained from the particle size analyzer.
Abbreviations: DPX, dapoxetine; PMs, polymeric micelles; TEM, transmission electron microscopy.
Ex vivo permeation of DPX PMs
The prepared DPX PMs’ formulations (F1–F3) were assessed for permeation characteristics through bovine intestine as indicated in Figure 3. The results revealed a significant difference in the release pattern (P<0.05) for the F3 compared with F1 and F2 DPX PM formulations. The data in Figure 3 reveal that DPX PM formulations showed a sustained permeation pattern. Permeation data for DPX PMs showed 91.27%±7.64%, 79.43%±7.81%, and 63.78%±5.05% of DPX content diffused after 24 h for F1, F2, and F3, respectively (Figure 3). In contrast, DPX PM formulation (F1) revealed a high initial burst after 1 h with ~40.87%±6.37% of permeated DPX content compared with 23.8%±3.15% of permeated DPX content from the F3 formulation as indicated in Figure 3.
Figure 3.
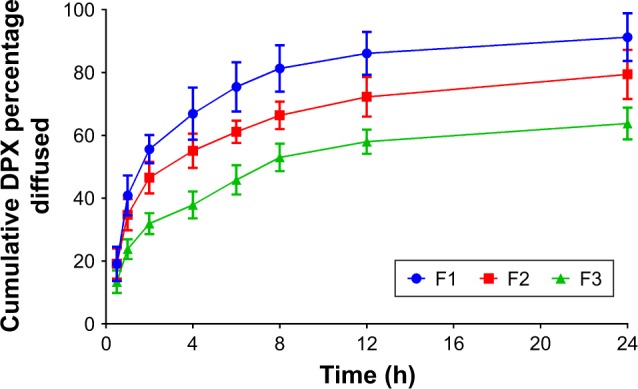
Ex vivo diffusion profiles of DPX PMs’ formulations F1–F3 through bovine intestine.
Abbreviations: DPX, dapoxetine; PMs, polymeric micelles.
Plasma and brain in vivo pharmacokinetic studies
Plasma pharmacokinetic DPX PM parameters showed a significant increase (P<0.05) for area under the curve from 0 to infinity observed (AUC0–inf_obs) and MRT0–∞ compared with commercial DPX tablets as shown in Table 2. DPX PMs’ formula AUC0–inf_obs increased by 2.7-fold compared with commercial tablets. Moreover, Tmax was dramatically delayed for the DPX PM formulation groups (6.4±0.2 h) compared to 1.2±0.2 h for the control group (Table 2). In contrast, commercial tablets showed a significant difference in Cmax compared with the DPX PMs formula.
Table 2.
Pharmacokinetic parameters for DPX PMs and DPX commercial tablets in the plasma and brain
| Parameter | Tmax (h) | Cmax (ng/mL) | AUC0–inf_obs (ng h/mL) | MRT (h) |
|---|---|---|---|---|
| Plasma | ||||
| DPX PMs formula (F3) | 6.4±0.5* | 458.84±45.3* | 6,061.36±212.3* | 9.46±1.1 |
| Commercial DPX tablets | 1.2±0.2 | 624.8±50.1 | 2,214.2±132.1 | 7.4±1.2 |
| Brain tissue | ||||
| DPX PMs formula (F3) | 1.5±0.3* | 242.96±21.1 | 2,571.35±118.6* | 10.86±2.1 |
| Commercial DPX tablets | 0.97±0.2 | 216.9±11.2 | 1,337.63±99.21 | 9.21±1.5 |
Note:
Significant at P<0.05 using Student’s t-test.
Abbreviations: AUC, area under the curve; DPX, dapoxetine; MRT, mean residence time; PMs, polymeric micelles; Cmax, maximum DPX plasma concentration; Tmax, time to reach this concentration.
In the case of brain tissue, no DPX was detected at 24 h and pharmacokinetic parameters showed a significant increase in the AUC0–inf_obs for DPX PMs when compared with the commercial tablets’ group. DPX PMs showed an ~1.65-fold increase in AUC0–inf_obs compared to the marketed tablets’ group. Pharmacokinetics data are presented in more detail in Figure 4A and B.
Figure 4.

DPX plasma and DPX brain concentrations (A and B, respectively) from DPX PMs (F3) and a DPX commercial tablet.
Notes: The data showed DPX PMs’ sustained release pattern with increased AUC0–inf_obs and delayed Tmax values in both plasma and brain compared with commercial DPX tablets. *Significant difference at P<0.05, two-way analysis of variance followed by Sidak’s multiple comparisons test. Tmax, time to reach maximum DPX plasma concentration.
Abbreviations: AUC, area under the curve; DPX, dapoxetine; PMs, polymeric micelles.
Discussion
Oral drug delivery is the first choice for chronic therapy. The main obstacle for oral drug delivery is controlled release and protection from gastrointestinal environmental variations. PMs are a promising approach as a drug delivery system for drugs as they have low aqueous solubility and poor bioavailability characters. The core–shell structure of PMs with an inner hydrophobic core and hydrophilic coat allows the encapsulation and delays the release of drugs.27 PMs have the advantage of improved thermodynamic stability upon a series of dilutions because of low CMC values compared with micelles prepared from low molecular weight surfactants.28 In addition, PMs show high kinetic stability below the CMC that allows the slow PM disassembly and PM integrity and drug content retention before reaching the target sites relative to regular surfactants. These characteristics of PMs enhance the oral bioavailability of encapsulated drugs.28
DPX shows a pKa value of 8.6, which indicates that DPX is mainly charged at physiological pH. DPX protonation of DPX is attributed to its water solubility (68 mg/mL). The improved solubility represents a challenge in DPX design used in the PM formulations. Accordingly, the preparation of the PM formulations was carried out in an aqueous solution with a pH of 9.5 in order to reduce the solubility of DPX HCl in water via reduction in the degree of the ionized form of DPX, which then allows more drug entrapment in the PM hydrophobic core. Results of EE% revealed that as the polymer content in the formulation increased (F3 > F2 > F1), DPX entrapment is improved and could be attributed to the ability of the copolymer molecules to efficiently encapsulate the drug in the core of the formed micelles. The prepared F3 DPX PMs showed sizes <50 nm (Figure 2A). The results concur with resulting TEM images for the prepared DPX PMs (Figure 2B) and with the previous reports that utilized different hydrophobic (PLGA)-to-hydrophilic (PEG) ratios in the structures of the copolymer used for the preparation of PMs.27,29,30 FTIR results revealed no possible chemical interactions between DPX and PEG–PLGA.
The permeation of DPX from bovine intestine is shown in Figure 3. The permeation data revealed a sustained pattern of DPX formed from the PMs’ formula. This could be attributed to the gradual release of DPX from the hydrophobic PLGA core and, to a lower extent, the hydrolysis of the biodegradable polymers of the PMs structure. The hydrophobic PLGA core acts as a reservoir for DPX molecules. The core is coated by a hydrophilic PEG that is exposed to the aqueous environment. The PEG coat confers aqueous solubility and steric stability to the PMs in the biological fluids.31 Additionally, the results revealed variation in the release rates for F1, F2, and F3 that are attributed to the variation in copolymer ratio and particle size of the investigated formulations.
For a long time, PMs have been extensively focused on for oral drug delivery at the preclinical level. After oral administration, PMs face the GI tract’s aggressive physicochemical environment. Gastric fluids affect particles’ properties even before coming into contact with the intestinal cells.32 PMs survive a variety of pH levels, digesting enzymes, and bile salts. In vitro tests in gastric and intestinal simulated liquids demonstrated enhanced PM permeability as represented in previous studies.6,9,12,33 Studies have demonstrated that polymeric nanoparticles improved biopharmaceutical bioavailability in preclinical models.34–36 PMs’ properties could achieve valuable bioavailability enhancement for our study. In addition, PMs’ mucoadhesion properties, as a result of direct retention by both covalent and ionic bonds and vander Waals’ forces, increase the PMs’ residency time and delay the drug release from the micelles.37,38 PMs’ small particle size and surface properties favor the enhancement of the extent and rate of absorption.39 A previous study showed that PMs’ major route of cell line internalization is via fluid-phase pinocytosis.40 PMs’ surface properties affect distribution through body fluids. Some reports have confirmed that part of PMs is involved in the prolongation of the drug’s elimination.4,41 The smallest particle size enhances its ability to pass the intestinal barrier of the intestinal epithelium after absorption, but there are still unique properties for PMs as a result of the PEG coating, which protects it from uptake by the reticulo-endothelial system (RES).42,43 The resulting prolonged circulation half-life allows diffusion in the tissues or micelles taken up by absorptive enterocytes that will be predominantly transported via the bloodstream.44,45 Both M cells and enterocytes are assimilated into the blood and lymph vessels in addition to participating in mucoadhesion, which delays the drugs’ residence time.
In the brain, DPX elimination can be decreased by the micelles due to a delay in residency time. At 1.5 h after administration, DPX concentration from the PM formulas reached a maximum concentration (242.96±21.1 ng/g), which was 1.2-fold (216.9±11.2 ng/g) more than that of commercial DPX tablets (Table 2). Moreover, AUC0–inf_obs increased by 1.92-fold for the DPX PMs’ formula compared with commercial DPX. These results indicate that DPX PMs could enhance the distribution of DPX into the brain tissues. PMs’ particle diameter (,50 nm) has the advantage of a small diameter in addition to distinct core–shell architecture, which contributes substantially to reduced uptake into the RES in the liver and spleen.46 The BBB might be overcome by particles with a size of ~<50 nm.47 PMs’ surface when modified by PEG is a good nonionic surfactant and can act as an emulsifier and absorption enhancer for oral drug formulations with a prolonged circulation time in the blood and low toxicity; this prolonged time promotes brain DPX accumulation and increases its cerebral concentration.48
Conclusion
The study showed that the development of DPX PM formulation improved both bioavailability and delivery across the BBB. Brain DPX level is a major parameter for DPX efficacy. This leads to patient satisfaction and compliance and reduces a patient’s medication dependence. The development of DPX formulations specifically for PE with improved efficiency in the administered dose eliminates reliance on other unapproved treatments. This achievement means that effective and safe treatment, representing a major advance in sexual medicine, has been introduced to relieve a major health problem in men.
Acknowledgments
The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for the continuous support. This work was supported financially by the Deanship of Scientific Research at Umm Al-Qura University (grant code: 15-MED-3-1-0044).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Patrick DL, Althof SE, Pryor JL. Premature ejaculation: an observational study of men and their partners. J Sex Med. 2005;2(3):358–367. doi: 10.1111/j.1743-6109.2005.20353.x. [DOI] [PubMed] [Google Scholar]
- 2.Giuliano F, Hellstrom WJ. The pharmacological treatment of premature ejaculation. BJU Int. 2008;102:668–675. doi: 10.1111/j.1464-410X.2008.07719.x. [DOI] [PubMed] [Google Scholar]
- 3.McMahon CG. Dapoxetine: a new option in the medical management of premature ejaculation. Ther Adv Urol. 2012;4(5):233–251. doi: 10.1177/1756287212453866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim JO, Sahay G, Kabanov AV, Bronich TK. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules. 2010;11(4):919–926. doi: 10.1021/bm9013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu CH, Kuo SW, Chen JK, Ko FH, Liao CS, Chang FC. Self-assembly behavior of AB diblock and CD random copolymer mixtures in the solution state through mediated hydrogen bonding. Langmuir. 2008;24:7727–7734. doi: 10.1021/la703960g. [DOI] [PubMed] [Google Scholar]
- 6.Deng C, Jiang Y, Cheng R, Meng F, Zhong Z. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: promises, progress and prospects. Nano Today. 2012;7:467–480. [Google Scholar]
- 7.Discher DE, Ahmed F. Polymersomes. Annu Rev Biomed Eng. 2006;8:323–341. doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Tang X, Zhang J, et al. PEG–PLGA copolymers: their structure and structure-influenced drug delivery applications. J Control Release. 2014;183:77–86. doi: 10.1016/j.jconrel.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 9.Jones MC, Leroux JC. Polymeric micelles–a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48(2):101–111. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 10.Gao WP, Bai Y, Chen EQ, et al. Controlling vesicle formation via interpolymer hydrogen-bonding complexation between poly (ethylene oxide)-block-polybutadiene and poly (acrylic acid) in solution. Macromolecules. 2006;39:4894–4898. [Google Scholar]
- 11.Matsumura Y, Hamaguchi T, Ura T, et al. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br J Cancer. 2004;91(10):1775–1781. doi: 10.1038/sj.bjc.6602204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 2010;7(2):145–158. doi: 10.1517/17425240903436479. [DOI] [PubMed] [Google Scholar]
- 13.Swierczewska M, Lee KC, Lee S. What is the future of PEGylated therapies? Expert Opin Emerg Drugs. 2015;20(4):531–536. doi: 10.1517/14728214.2015.1113254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Wen Q, Choi S. FDA’s Regulatory Science Program for generic PLA/PLGA-based drug products. Am Pharm Rev. 2016;19(4):5–9. [Google Scholar]
- 15.Banks WA. Characteristics of compounds that cross the blood–brain barrier. BMC Neurol. 2009;9(suppl 1):S3. doi: 10.1186/1471-2377-9-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Liu L. Modern methods for delivery of drugs across the blood–brain barrier. Adv Drug Deliv Rev. 2012;64(7):640–665. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: overcoming blood–brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34–47. doi: 10.1016/j.jconrel.2016.05.044. [DOI] [PubMed] [Google Scholar]
- 18.Kabanov AV, Batrakova EV, Miller DW. Pluronic block copolymers as modulators of drug efflux transporter activity in the blood-brain barrier. Adv Drug Deliv Rev. 2003;55(1):151–164. doi: 10.1016/s0169-409x(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 19.Etame AB, Smith CA, Chan WCW, Rutka JT. Design and potential application of PEGylated gold nanoparticles with size-dependent permeation through brain microvasculature. Nanomedicine. 2011;7(6):992–1000. doi: 10.1016/j.nano.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Hanada S, Fujioka K, Inoue Y, Kanaya F, Manome Y, Yamamoto K. Cell-based in vitro blood–brain barrier model can rapidly evaluate nanoparticles’ brain permeability in association with particle size and surface modification. Int J Mol Sci. 2014;15(2):1812–1825. doi: 10.3390/ijms15021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778(3):729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Watson PM, Anderson JM, Vanltallie CM, Doctrow SR. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci Lett. 1991;129(1):6–10. doi: 10.1016/0304-3940(91)90708-2. [DOI] [PubMed] [Google Scholar]
- 23.Shao K, Huang R, Li J, et al. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release. 2010;147(1):118–126. doi: 10.1016/j.jconrel.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 24.Sharma AK, Zhang L, Li S, et al. Prevention of MDR development in leukemia cells by micelle-forming polymeric surfactant. J Control Release. 2008;131(3):220–227. doi: 10.1016/j.jconrel.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashem FM, Al-Sawahli MM, Nasr M, Ahmed OA. Optimized zein nanospheres for improved oral bioavailability of atorvastatin. Int J Nanomedicine. 2015;10:4059–4069. doi: 10.2147/IJN.S83906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TK, Kim IS, Hong SH, Choi YK, Kim H, Yoo HH. Determination of dapoxetine in rat plasma by ultra-performance liquid chromatography–tandem mass spectrometry. J Chromatogr B. 2013;926:42–46. doi: 10.1016/j.jchromb.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Ling P, Zhang T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J Drug Deliv. 2013;2013:340315. doi: 10.1155/2013/340315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–1355. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 29.Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA–PEG block copolymer. J Control Release. 2001;70(1–2):63–70. doi: 10.1016/s0168-3659(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 30.Jin C, Qian N, Zhao W, et al. Improved therapeutic effect of DOX–PLGA–PEG micelles decorated with bivalent fragment HAb18 F (ab′) 2 for hepatocellular carcinoma. Biomacromolecules. 2010;11(9):2422–2431. doi: 10.1021/bm1005992. [DOI] [PubMed] [Google Scholar]
- 31.Gaucher G, Satturwar P, Jones MC, Furtos A, Leroux JC. Polymeric micelles for oral drug delivery. Eur J Pharm Biopharm. 2010;76(2):147–158. doi: 10.1016/j.ejpb.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Song Z, Feng R, Sun M, et al. Curcumin-loaded PLGA–PEG–PLGA triblock copolymeric micelles: preparation, pharmacokinetics and distribution in vivo. J Colloid Interface Sci. 2011;354(1):116–123. doi: 10.1016/j.jcis.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 33.Wang G, Wang JJ, Li F, To SS. Development and evaluation of a novel drug delivery: pluronics/SDS mixed micelle loaded with myricetin in vitro and in vivo. J Pharm Sci. 2016;105(4):1535–1543. doi: 10.1016/j.xphs.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Xu R, Zhang Y, Ye X, et al. Inhibition effects and induction of apoptosis of flavonoids on the prostate cancer cell line PC-3 in vitro. Food Chem. 2013;138(1):48–53. doi: 10.1016/j.foodchem.2012.09.102. [DOI] [PubMed] [Google Scholar]
- 35.Maronpot RR, Koyanagi M, Davis J, et al. Safety assessment and single-dose toxicokinetics of the flavouring agent myricitrin in Sprague–Dawley rats. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2015;32(11):1799–1809. doi: 10.1080/19440049.2015.1084653. [DOI] [PubMed] [Google Scholar]
- 36.Ghahremankhani AA, Dorkoosh F, Dinarvand R. PLGA–PEG–PLGA tri-block copolymers as an in-situ gel forming system for calcitonin delivery. Polym Bull. 2007;59:637–646. [Google Scholar]
- 37.Wang Y, Wang C, Gong C, et al. Polysorbate 80 coated poly (ɛ-caprolactone)–poly (ethylene glycol)–poly (ɛ-caprolactone) micelles for paclitaxel delivery. Int J Pharm. 2012;434(1–2):1–8. doi: 10.1016/j.ijpharm.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Kim BS, Park SW, Hammond PT. Hydrogen-bonding layer-by-layer-assembled biodegradable polymeric micelles as drug delivery vehicles from surfaces. ACS Nano. 2008;2(2):386–392. doi: 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- 39.Jung T, Kamm W, Breitenbach A, Kaiserling E, Xiao JX, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50(1):147–160. doi: 10.1016/s0939-6411(00)00084-9. [DOI] [PubMed] [Google Scholar]
- 40.Luo L, Tam J, Maysinger D, Eisenberg A. Cellular internalization of poly (ethylene oxide)-b-poly (ε-caprolactone) diblock copolymer micelles. Bioconjug Chem. 2002;13(6):1259–1265. doi: 10.1021/bc025524y. [DOI] [PubMed] [Google Scholar]
- 41.Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 42.Behzadi S, Serpooshan V, Tao W, et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46:4218–4244. doi: 10.1039/c6cs00636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Idan IJ, Abdullah LC, Mahdi DS, Obaid MK, Jamil SN. Adsorption of anionic dye using cationic surfactant-modified kenaf core fibers. Open Access Libr J. 2017;4:1–18. [Google Scholar]
- 44.Yang Q, Jones SW, Parker CL, Zamboni WC, Bear JE, Lai SK. Evading immune cell uptake and clearance requires PEG grafting at densities substantially exceeding the minimum for brush conformation. Mol Pharm. 2014;11(4):1250–1258. doi: 10.1021/mp400703d. [DOI] [PubMed] [Google Scholar]
- 45.Draffehn S, Eichhorst J, Wiesner B, Kumke MU. Insight into the modification of polymeric micellar and liposomal nanocarriers by fluorescein-labeled lipids and uptake-mediating lipopeptides. Langmuir. 2016;32(27):6928–6939. doi: 10.1021/acs.langmuir.6b01487. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Yang G, Guo X, Tang Z, Zhong Z, Zhou S. Redox-responsive polyanhydride micelles for cancer therapy. Biomaterials. 2014;35(9):3080–3090. doi: 10.1016/j.biomaterials.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 47.Nance E, Timbie K, Miller GW, et al. Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood−brain barrier using MRI-guided focused ultrasound. J Control Release. 2014;189:123–132. doi: 10.1016/j.jconrel.2014.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni SA, Feng SS. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. 2013;30(10):2512–2522. doi: 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]