

Abstract
The production of cytokines in response to DNA damage events may be an important host defense response to help prevent the escape of pre-cancerous cells. The innate immune pathways involved in these events are known to be regulated by cellular molecules such as STING (stimulator of interferon genes), which controls type I interferon and pro-inflammatory cytokine production in response to the presence of microbial DNA or cytosolic DNA that has escaped from the nucleus. STING signaling has been shown to be defective in a variety of cancers, such as colon cancer and melanoma, actions which may enable damaged cells to escape the immunosurveillance system. Here, we report through examination of databases that STING signaling may be commonly suppressed in a greater variety of tumors due to loss-of-function mutation or epigenetic silencing of the STING/cGAS promoter regions. In comparison, RNA activated innate immune pathways controlled by RIG-I/MDA5 were significantly less affected. Examination of reported missense STING variants confirmed that many exhibited a loss of function phenotype and could not activate cytokine production following exposure to cytosolic DNA or DNA-damage events. Our data implies that the STING signaling pathway may be recurrently suppressed by a number of mechanisms in a considerable variety of malignant disease and be a requirement for cellular transformation.
Introduction
The innate immune system plays a key role in preventing the development of malignant disease, although the signaling pathways that principally initiate antitumor immune responses remain to be fully determined1. Carcinogens can activate DNA damage response (DDR) pathways which include the production of cytokines that alert the immunosurveillance system and facilitate tumor cell elimination2. Recent data has indicated that the production of cytokines in response to DNA damage can be triggered through innate immune cellular receptors, such as STING (stimulator of interferon genes; TMEM173)3, 4. STING can be activated following binding to cyclic dinucleotides (CDN’s) such as cyclic di-AMP which are secreted by intracellular bacteria, to trigger host defense gene transcription5. Alternatively, microbial DNA or self-DNA leaked from the nucleus, in response to DNA damage can associate with a cellular CDN synthase referred to as cGAS (MB21D1, C6orf150) which manufactures STING activating cyclic GMP-AMP (cGAMP)6. STING undergoes a conformational change and associates with TANK-binding kinase 1 (TBK1) to activate transcription factors such as interferon regulatory factor 3 (IRF3) and nuclear factor-κB (NF-κB) that expedite type I interferon (IFN) and pro-inflammatory gene expression and the stimulation of adaptive immunity5.
Studies have shown that STING signaling is essential for efficient antitumor immune responses7, 8. For example, when dying tumor cells are phagocytosed by antigen presenting cells (APC) such as dendritic cells (DC), tumor cell-derived DNA conceivably activates exogenous STING signaling within the APC, resulting in the production of IFN and other cytokines. IFN stimulates DC’s in autocrine manner to promote the cross-presentation of tumor antigens as well as stimulates antitumor CD8+ T cells in paracrine mode5. Intratumoral delivery of STING agonists have been shown to exert considerable therapeutic activity, plausibly through promoting antitumor T cell responses9.
It is unclear how phagocytes are drawn towards potentially tumorigenic cells10. However, it is known that DNA-damage may produce cytokines that can attract APC’s to the damaged cell, and that these actions may involve the STING pathway2, 3. That STING may have an important antitumor function is now indicated by a number of studies. For example, in mice, transient STING signaling is required to initiate wound repair processes in the colon in response to carcinogenic azoxymethane (AOM)/dextran sulfate sodium (DSS) treatment11. Loss of STING leads to augmented colitis-associated cancer (CAC) in these models. The importance of STING signaling in host defense against cancer development may similarly be inferred by observing that cGAS and/or STING expression is commonly suppressed in malignant cells including melanoma and colon cancer2, 4. Recent studies have showed that decreased STING or cGAS expression correlates with poor survival in gastric or lung cancer patients12, 13. Collectively, evidence suggests that intrinsic STING signaling may exert a tumor suppressive effect, as well as trigger cytokine production that attracts the immunosurveillance system, resulting in pre-cancerous cell clearance via recruitment and activation of DC’s and antitumor CD8+ cells7, 8.
In this report, we provide evidence to indicate that STING signaling is commonly suppressed in a wide variety of cancer, predominantly through loss-of-function mutation or by epigenetic silencing of the cGAS or STING promoter regions. Further, we show that a variety of the observed missense mutants failed to function and generate cytokines in response to cytosolic DNA or DNA-adduct forming agents such as DMBA (7,12-dimethylbenz[α]anthracene) or cisplatin. These findings indicate that suppression of STING signaling may be a common, important requirement for the cellular transformation process to proceed and may enable pre-cancerous cells to avoid antitumor immune responses.
Results
cGAS or STING gene is mutated in a variety of human tumors
We have previously shown that STING signaling is impaired in a variety of human malignancies such as melanoma and colon cancer2, 4. In many cases, the underlining mechanisms in these studies involved loss of STING or cGAS expression through epigenetic silencing2, 4. However, it was also observed that in some cell-lines, STING signaling was defective even though cGAS and STING expression was readily detectable2, 4. This suggested that cGAS and/or STING may be defective through missense mutation, or that as yet unknown accessory molecules that facilitate STING signaling may themselves be defective. This possibility led us to investigate the incidence of cGAS or STING gene alterations in documented human tumors. To start to evaluate this, we principally searched the cBioPortal database14, 15 and confirmed the existence of missense, nonsense, or frame shift mutations in the cGAS or STING gene, albeit at a low percentage (< 1%) (Figure 1a, b and Supplementary Figure 1a, b). Similar results were found following examination of other two databases, COSMIC (catalogue of somatic mutations in cancer) and ICGC (international cancer genome consortium)16 (Supplementary Figure 1c).
Figure 1.

STING and cGAS genes are mutated in a variety of human tumors. (a and b) We searched three databases, eBioPortal, COSMIC, and ICGC that provide cancer genomics data to find mutations in STING and cGAS genes. The missense mutations in red were tested in the following studies. TM; transmembrane, DD; dimerization domain, CTT; C-terminal tail, Mab21; Mab21 domain. (c) The mutations we tested in the following studies are most likely tumor-specific mutations. We confirmed if the missense mutations exist in the general population using ExAC database. Case indicates how many times the mutations were found in human tumors.
Our hypothesis is that mutations could affect STING function and enable cells expressing such variants to escape STING-dependent host defense responses. To confirm if these changes indeed influenced STING activity, six STING variants were generated that contained the observed single amino acids missense substitutions found in the databanks (R169W, R180Q, R191W, P203S, R238W, R253W) (Figure 1a). STING R169W, R180Q, and P203S missense mutations were observed to recurrently occur in different tumor types (Figure 1c and Supplementary Figure 1b). Theses amino acid changes did not appear to be SNP’s that occurred in the general population, as determined by ExAC (Exome Aggregation Consortium) database that provides about 60,000 unrelated individuals in an Exome sequence data set (Figure 1c and Supplementary Figure 1b). R191W and R253W were found in 0.039% and 0.010% of examined alleles following analysis of 120,000 individuals, respectively (Figure 1c and Supplementary Figure 1d). In addition, a number of these mutations appeared to be haploid in nature (Supplementary Figure 1b). When overexpressed in HEK293T cells, wild-type human STING aggregates and potently stimulates IRF3/7, NF-κB transcription factor-dependent activation of the IFNβ promoter (IFNβ-luc). Of the six STING variants, two, R169W and P203S, completely failed to activate the IFNβ promoter (Figure 2a). We complemented this approach by stably reconstituting primary Sting−/− mouse embryonic fibroblast cells (MEFs) independently with the six STING variants, using a retroviral approach, and examined their ability to stimulate IFN production in the presence of STING activating dsDNA agonists. This analysis confirmed that in contrast to cells containing a non-mutated human STING, the R169W and P203S variants exhibited a comparable loss of function phenotype (Figure 2b). However, we also noted that R238W and R253W, which exhibited some function when overexpressed in HEK293T cells, did not appear to function in the MEFs, even though they were efficiently expressed in the MEFs (Figure 2c). Thus, R238W and R253W may also be significantly defective and only exert activity in overexpressed circumstances. STING R180Q and R191W also exhibited defective activity compared to wild-type human STING (Figure 2b). In contrast, none of the STING variants expressed in the reconstituted MEFs affected IFNβ production by synthetic dsRNA, Poly I:C, which activates the RIG-I like helicase (RLH) pathway17 (Figure 2b). Thus, 4 out of 6 mutants exhibited significantly defective activity and two exhibited reduced ability to function. Exactly why these mutations affect STING function is unclear. However, we found that R169W, P203S, R238W, and R253W that appeared inactive also failed to form higher order aggregates, similar to WT STING (Figure 2d). Higher order STING structures have been noticed to occur after activation and precede STING trafficking18. This data suggests that the mutations may affect STING-STING interactions and may affect STING trafficking (Supplementary Figure 2).
Figure 2.
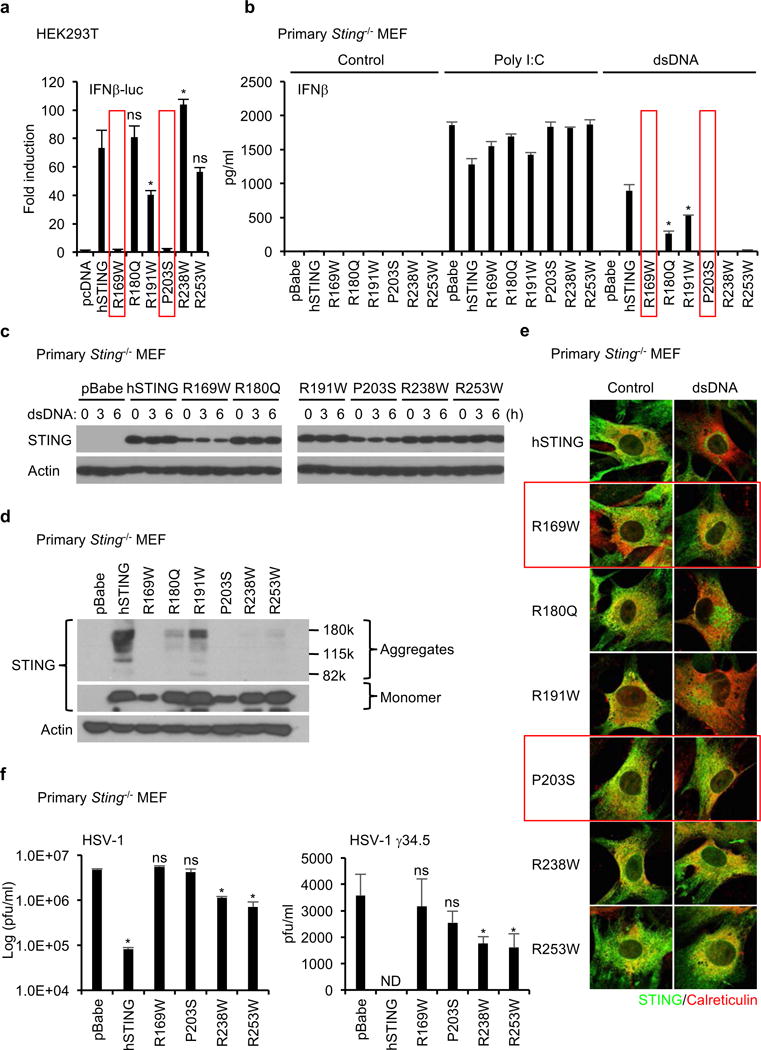
STING mutants found in human tumors fail to activate dsDNA-induced inflammation. (a) The indicated plasmids were transfected into HEK293T cells with reporter plasmids (IFNβ-luc and TK-luc). After 24 hr, the luciferase activity in the cell lysates was measured. (b) Primary Sting−/− MEF cells were reconstituted with the indicated STING mutants using retrovirus. IFNβ in the supernatants was measured by ELISA after treated with Poly I:C (2 μg/ml) or dsDNA (4 μg/ml) for 16 hr. (c and d) The reconstituted Sting−/− MEF cells were treated with dsDNA (4 μg/ml) for the indicated times (c). Wester blots were performed with the indicated antibodies. (e) After treated with dsDNA as described in Figure 2c, the reconstituted Sting−/− MEF cells were stained with antibodies for STING and an ER marker, calreticulin, to observe the localization of STING using a confocal microscope. (f) The reconstituted Sting−/− MEF cells were infected with HSV-1 or HSV-1 γ34.5 for 24 hr and then the viral titer was determined by plaque assay. Data shown here are the averages ± SD (n = 3). Asterisks indicate significant difference (p < 0.05) compared to hSTING (a and b) or pBabe (f) determined by Student’s t-test (ns; not significant, ND; not detected).
Given this data, we thus evaluated if the STING variants were able to traffic to perinuclear regions in response to cytosolic dsDNA, an event which is required for STING activation of the transcription factor IRF319. However, only R180Q and R191W were observed to exhibit translocation function, confirming that the missense mutations R169W, P203S, R238W and R253W were defective in trafficking (Figure 2e). This observation is consistent with our findings that IFNβ could be produced in the reconstituted Sting−/− MEFs with wild-type STING or only the R180Q, or R191W variants (Figure 2b). Thus, the amino acid substitutions may function to impede STING activity after association with CDN’s to affect STING trafficking function.
Finally, we have previously shown that STING is essential for the inhibition of DNA virus infection, such as herpes simplex virus-1 (HSV-1). For example, Sting−/− mice are extremely susceptible to lethal HSV-1 infection19. Attenuated versions of HSV-1 (T-Vec) have been evaluated in clinical trials as a therapeutic oncolytic agent for the treatment of cancer20. We compared two HSV-1 strains for their ability to replicate in the reconstituted MEFs. Wild-type HSV-1 retains the ability to suppress type I IFN production which affects its replication. In contrast, an HSV-1 variant that lacks the γ34.5 gene cannot prevent viral-mediated type I IFN induction for reasons that remain to be fully determined and is significantly attenuated21.
While wild-type STING could suppress HSV-1 replication in reconstituted Sting−/− MEFs, the STING variants exhibited defective function. R169W, P203S, R238W, and R253W, did not exert anti-viral activity (Figure 2f). More strikingly, HSV-1 γ34.5 completely failed to replicate in the wild-type STING reconstituted Sting−/− MEFs. However, HSV-1 γ34.5 could replicate in cells expressing the STING defective variants, R169W, P203S, R238W, or R253W (Figure 2f). Thus, it is plausible that cancer cells expressing defective STING proteins render cells vulnerable to HSV-1 infection. Therefore, analysis of STING signaling function in cancer cells may help predict outcome to select oncolytic virotherapy.
Analysis of cGAS variants expressed in cancer
To complement this study, we performed a similar examination of cGAS, which in the presence of cytosolic dsDNA generates STING-activating CDN’s (cGAMP)22. We evaluated 11 missense cGAS variants that we noted were recurrently expressed in different human tumors as indicated in the databases (Figure 1b and Supplementary Figure 1b). Principally, we generated a cell-based assay and tested if the cancer-related cGAS variants were functional, or not. Since, neither cGAS nor STING are expressed in HEK293T cells, we transfected cGAS with STING into these cells (Figure 3a). Transfection of cGAS generated STING activating CDN’s (the transfected plasmid DNA serves as a ligand for cGAS activation) and in the presence of wild-type STING resulted in IFNβ-luc activation (Figure 3a). This study indicated that the majority of the 11 examined cGAS variants were able to bind dsDNA and generate CDN’s to activate the IFNβ promoter (Figure 3a). However, two of the 11 cGAS variants, R376Q and E383K, did not exhibit any ability to stimulate IFNβ promoter activity through STING (Figure 3a). To extend this analysis, the non-functioning cGAS variants (R376Q and E383K) were stably expressed in primary cGas−/− MEFs using retroviruses. This assay confirmed that in the presence of cytosolic dsDNA, R376Q and E383K, did not exhibit any significant ability to stimulate STING and subsequent TBK1/IRF3 activity or IFNβ production (Figure 3b, c). It is plausible that the reasons for R376Q and E383K lack of function could arise though their inability to bind dsDNA and generate CDN’s. However, the R376Q and E383K mutations were not in the known DNA binding domain of cGAS (Figure 1b). To confirm this, cell lysates from cGAS variant transfected HEK293T cells were incubated with biotin-labeled dsDNA and dsDNA-protein complexes were precipitated by neutravidin-conjugated beads. This data confirmed that all the cGAS variants analyzed were able to bind to dsDNA (Figure 3d). Next, we evaluated if R376Q and E383K exhibited enzymatic activity and were able to generate CDN’s. After the cGAS variants were transfected into HEK293T cells, CDN’s (cGAMP) were purified from the cell lysates and quantitated by HPLC-mass spectrometry (LC/MS). We found that cGAMP was not detected in the lysates from R376Q or E383K expressing HEK293T cells in contrast to cells expressing the wild-type cGAS (Figure 3e). Taken together, our data indicates that cGAS missense mutants, R376Q and E383K, have lost the ability to generate cGAMP following binding to dsDNA. Thus, R376 and E383 are probably critically involved in the catalytic enzymatic activity of cGAS. Our data therefore indicates that not only STING but also cGAS gene can undergo genetic loss-of-function mutation to inhibit STING signaling.
Figure 3.
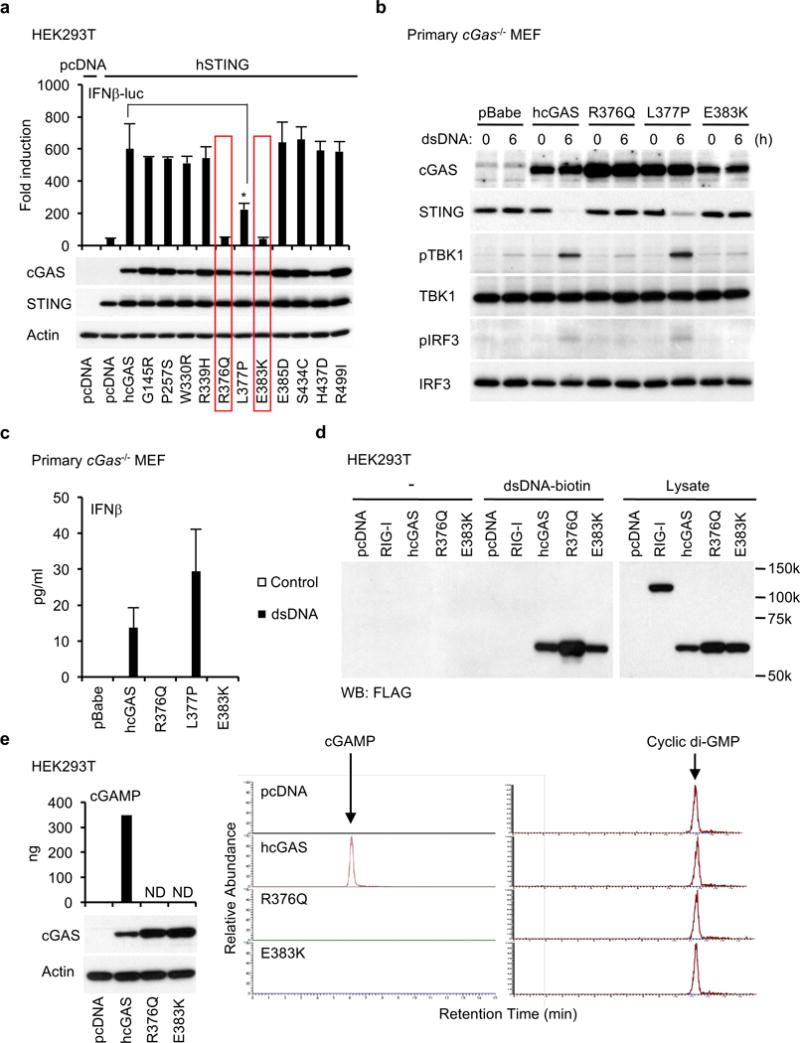
cGAS mutants, R376Q and E383K, found in human tumors cannot synthesize cGAMP. (a) cGAS or its mutants were expressed into HEK293T cells with human STING. As described in Figure 2a, the luciferase activity was measured. The expression of STING and cGAS was confirmed by western blots. (b and c) Primary cGas−/− MEF cells were reconstituted with the indicated cGAS mutants as described in Figure 2b and then treated with dsDNA (4 μg/ml) for 6 hr (b) or 24 hr (c). Wester blots were performed with the indicated antibodies (b) or IFNβ was measured by ELISA (c). (d) The cell lysates from HEK293T cells transfected with the indicated plasmids were incubated with biotin-labeled dsDNA and then subjected to precipitation with neutravidin-conjugated beads. After washing, precipitated proteins were eluted by boiling in SDS-sample buffer and then western blots were performed with the anti-FLAG antibody. (e) cGAS, E376Q, or E383K was expressed in HEK293T cells, which was verified by wester blots. cGAMP was purified from the cell lysates and quantified by LC/MS with calibration samples that were prepared by spiking known amount of cGAMP into the cell lysates from HEK293T cells. Cyclic di-GMP was added as an internal control to the cell lysates before purification of cGAMP to validate the purification process. Data shown here are the averages ± SD (n = 3). Asterisks indicate significant difference (p < 0.05) determined by Student’s t-test (ND; not detected).
A variety of studies indicates that STING can exist as a dimer in the endoplasmic reticulum (ER), to associate with CDN’s5. Similarly, cGAS has also been reported to form a dimer in association with dsDNA ligand23. Since it was noted that the majority of the observed STING/cGAS variants exhibited haploid mutation, we examined whether such variants could exhibit dominant-negative function. To evaluate this, STING variants R169W and P203S or cGAS variants R376Q and E383K were co-transfected into HEK293T cells with wild-type versions of their respective genes and an IFNβ-luc reporter. This data showed that the STING variants could indeed exert a dominant-negative function in this assay (Figure 4a and Supplementary Figure 3a). To further confirm this, the STING variants R169W, P203S, and R253W were co-expressed in Sting−/− MEFs with wild-type STING. This study confirmed that IFNβ production, TBK1/IRF3 activation, and STING trafficking were inhibited by the STING variants (Figure 4b, c and Supplementary Figure 3b). Thus, the STING variants inhibit wild-type STING function. However, less of a dominant-negative effect was observed upon examination of the cGAS variants for reasons that remain unclear (Figure 4d). While two STING variants, R169W and P203S, were not found in the ExAC database, cGAS R376Q and E383K alleles (haploid) were observed in 0.004% and 0.016% of 120,000 alleles examined, respectively (Figure 1c and Supplementary Figure 1b, 1d). Thus, heterologous STING variants may be sufficient to affect STING signaling in tumor cells.
Figure 4.
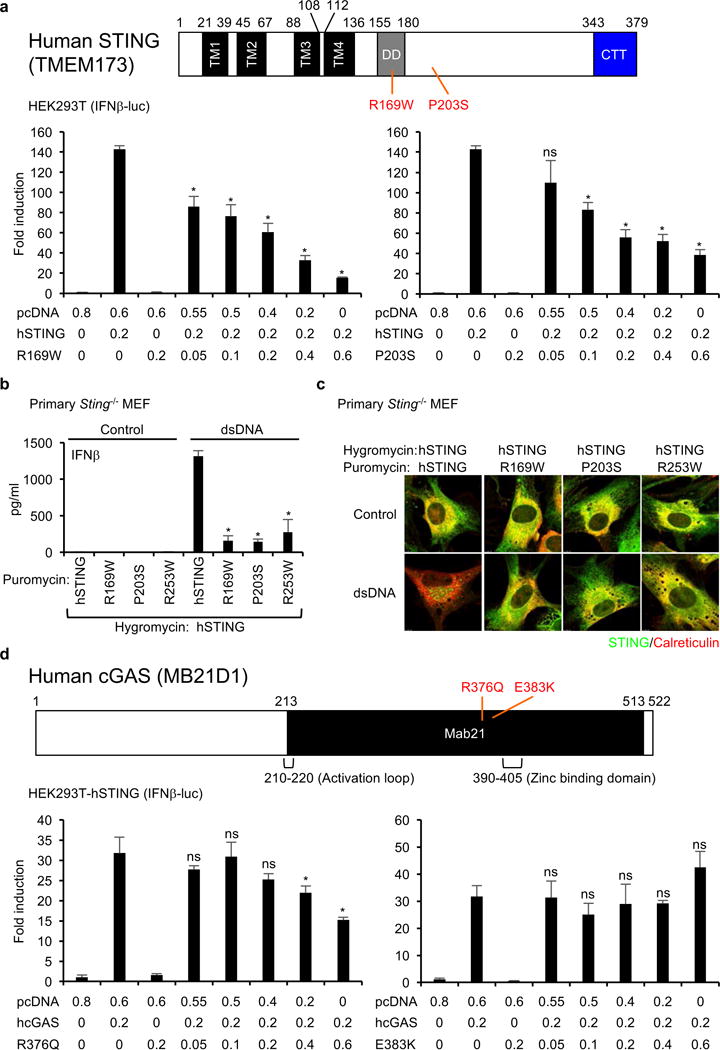
STING mutants are dominant-negative mutants. (a) The luciferase activity in the cell lysates was measured as described in Figure 2a after the transfection of the indicated plasmids into HEK293T cells. The numbers under the graph indicate the amount of transfected plasmids (μg). (b and c) Primary Sting−/− MEF cells were reconstituted using retrovirus vectors encoding STING mutants (puromycin resistant) and wild-type human STING (hygromycin resistant). After drug selection with puromycin and hygromycin, the reconstituted MEF cells were treated with dsDNA (4 μg/ml) for 16 hr. IFNβ in the supernatants was measured by ELISA (b) and the cells were stained with the indicated antibodies to observer the localization of STING as described in Figure 2d (c). (d) HEK293T-hSTING, which stably expresses wild-type human STING, were transfected with the indicated plasmids and the luciferase activity in the cell lysates was measured. The numbers under the graph indicate the amount of transfected plasmids (μg). Data shown here are the averages ± SD (n = 3). Asterisks indicate significant difference (p < 0.05) compared to hSTING (A and B) or cGAS (B) transfected samples determined by Student’s t-test (ns; not significant).
STING mutants (R169W and P203S) cannot facilitate carcinogen-induced pro-Inflammatory cytokine production
The DNA damage response pathway can be triggered by DNA-adduct forming agents such as DMBA and even chemotherapeutic agents such as cisplatin. These events can lead to the activation of the STING pathway and to the production of type I IFN and pro-inflammatory cytokines which can attract phagocytes and promote antitumor T cell activity2, 3. Thus, the inhibition of STING signaling may enable DNA-damaged cells to escape the immunosurveillance system and eventual eradication. To confirm whether the STING variants (R169W and P203S) impeded carcinogen-mediated pro-inflammatory gene induction, we treated reconstituted primary Sting−/− MEFs with DMBA or cisplatin. We noted that MEFs containing wild-type human STING expressed low levels of a variety of chemokines (Cxcl10, Ccl5) as well as interferon-stimulated genes (Stat1, Irf7), compared to empty vector control, even when unstimulated (Figure 5). However, DMBA treatment significantly enhanced cytokine production in cells reconstituted with wild-type STING but not in those cells expressing the R169W or P203S variants (Figure 5). Cisplatin also induces in interferon-stimulated genes only in the reconstituted cells with wild-type STING (Figure 5). These results clearly show that inflammatory responses induced by DNA-adduct forming agents can be blocked by the cancer-related mutations in STING gene. Thus, certain mutations or loss of STING expression can enable DNA damaged cells to avoid the production of pro-inflammatory cytokines that could alert the immunosurveillance system.
Figure 5.
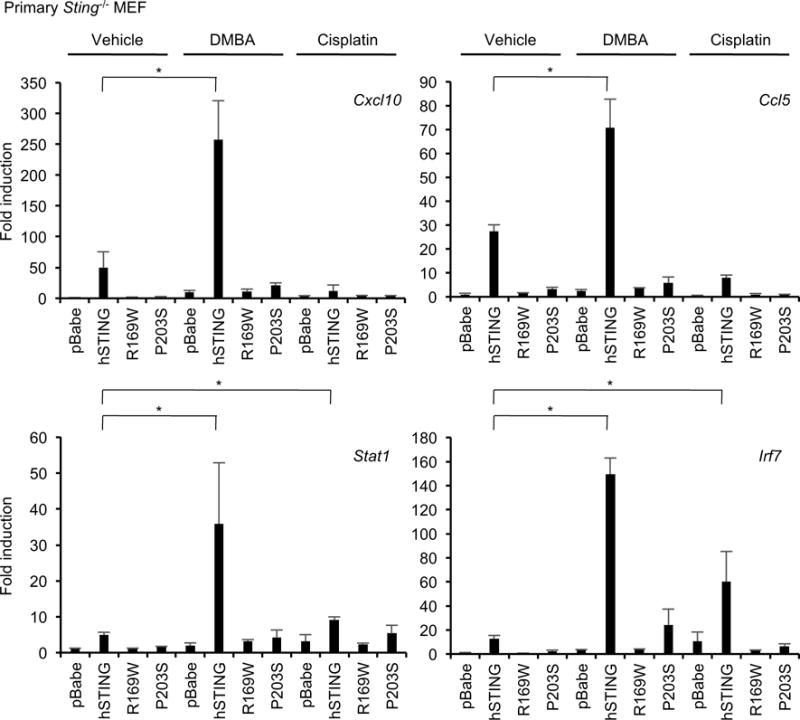
STING mutants fail to respond to DNA-adduct forming agents. The reconstituted primary Sting−/− MEF cells with empty vector (pBabe), hSTING, R169W, or P203S were treated DMBA (20 μg/ml) or cisplatin (10 μM) for 48 hr. Total RNA was extracted and realtime PCR was performed with the indicated probes after cDNA synthesis. Data shown here are the averages ± SD (n = 3). Asterisks indicate significant difference (p < 0.05) determined by Student’s t-test.
cGAS or STING promoter regions exhibit hypermethylation in a variety of human tumors
We have previously observed that cGAS or STING expression can be prevented in colon cancer and melanoma cells through epigenetic silencing of the promoter region2, 4. To extend these studies beyond these tumor types, we analyzed the methylation level for cGAS and STING gene in pan-tumor based on 32 different TCGA (The Cancer Genome Atlas) tumor types including primary solid tumors (n=8426) and solid normal tissue (n=747) samples. The TCGA tumor methylation was assayed on the Illumina Infinium Human Methylation 450K chip (HM-450K). The methylation level for the 16 individual cGAS- and 11 STING-probes on the HM-450K are shown (Figure 6a).
Figure 6.
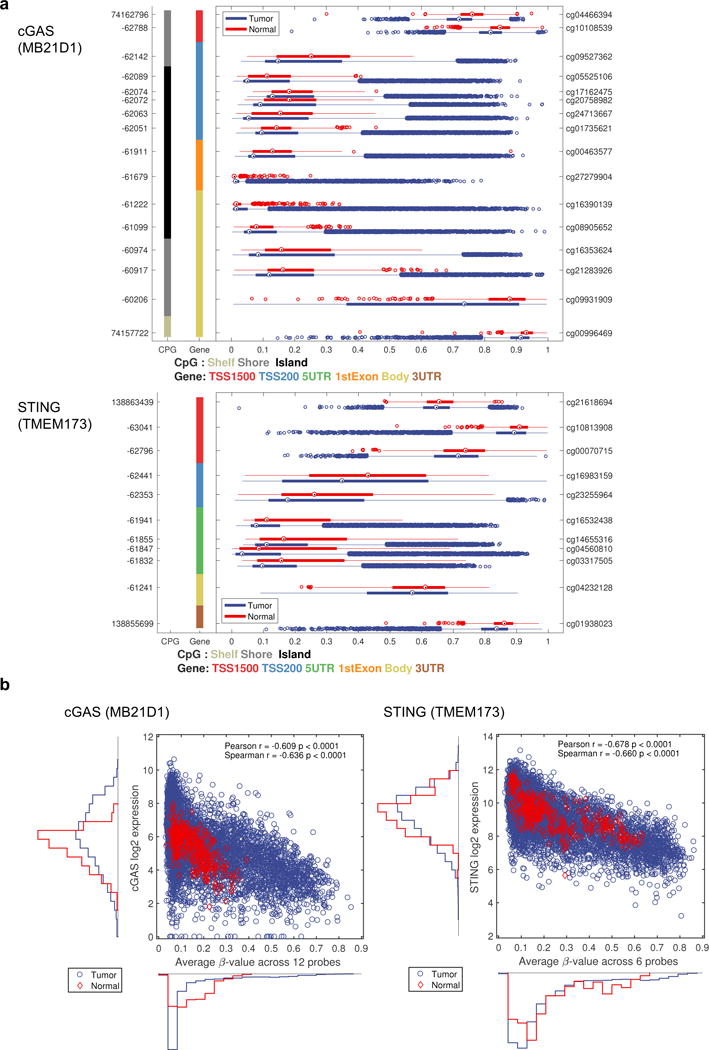
cGAS and STING expression is suppressed by methylation in a pan-tumor analysis. (a) The methylation data 32 different tumors types were obtained from TCGA. The methylation level for the 16 probes available for cGAS and the 11 probes available for STING on Illumina 450K chip are shown as box plots for normal samples (red) and tumor samples (blue). The x-axis indicates methylation level by β-value, ranging from 0 to 1 where 0 means no methylation and 1 means 100% methylated. The y-axis shows the genomic position (left axis) and probe ID (right axis) for all the probes. The colored columns indicate CpG island and gene body type as described in the figure. (b) The gene expression for cGAS or STING versus the average β-value across selected 12 probes for cGAS or 6 probes for STING.
In case of cGAS, a similar methylation pattern was observed for each probe across multiple gene regions including TSS200, 5′UTR, 1st Exon, and gene body (Figure 6a). This was further confirmed by pairwise correlation between all the 16 probes. The 12 middle probes all showed a high correlation between each other (r>0.45), indicating that alternate regions of the cGAS gene are similarly methylated (Supplementary Figure 4a, b). The general methylation pattern shows a low degree of methylation in most samples but there are many tumor samples with a high degree of methylation (Figure 6a). A similar pattern was also observed for STING (Figure 6a). STING was represented by 11 probes on the HM-450K chip and the probes located in the TSS200 and 5′UTR region highly correlated (r>0.63) (Supplementary Figure 4a, b).
The overall methylation level for cGAS or STING was also compared using selected probes with a high correlation between each other (12 probes for cGAS/6 probes for STING) to the expression levels found using RNAseq (Figure 6b). There was a negative correlation between the methylation level and gene expression level (r=-0.609 (Pearson)/-0.636 (Spearman) for cGAS; r=-0.678 (Pearson)/-0.660 (Spearman) for STING), further suggesting that cGAS and STING expression may be significantly suppressed by methylation in tumors (Figure 6b).
Interestingly, at low level of methylation level there was still a full range of gene expression level, this is not true at high methylation level (Figure 6b). This indicates that when cGAS gene is unmethylated, there are still multiple other types of regulation at play. That the methylation signal comes from cancer cells and not infiltrating immune cells was investigated by comparing the methylation level to the tumor purity as estimated by Aran et al.24. It is clear that there is a full range of methylation β-values for the most pure tumors and that infiltrating cells mutes the methylation signal, all indicating the methylation signal comes from tumor cells (Supplementary Figure 5).
Next, we investigated the methylation level of cGAS or STING gene in individual tumors. we tested the frequency of promoter methylation of both the cGAS and STING genes in 18 human tumors documented in the MethHC (DNA methylation and gene expression in human cancer) database25. This study indicated that the cGAS and STING promoter region was highly methylated in 14 of 18 tumors examined, compared to normal tissues (Supplementary Figure 6). In contrast, significantly less evidence of RIG-I (DDX58), MDA5 (IFIH1), or MAVS promoter methylation was observed which control the dsRNA activated RLH innate immune pathway (Supplementary Figure 6). This result complements our previous studies showing that numerous tumor cell types retain their ability to produce type I IFN in response to cytosolic dsRNA species, but not cytosolic dsDNA (Figure 2b)2, 4. We also noticed that the promoter of TBK1 was not highly methylated either, indicating that tumor cells may want to retain the activity of this kinase that is required by both the RLH and STING-dependent pathways to activate IRF3.
We further investigated the frequency of promoter methylation of both the cGAS and STING genes in 32 human tumors types in TCGA. The average methylation for cGAS and STING gene were compared between normal and tumor tissue (Figure 7a, b). The results indicate larger variation in cGAS methylation in tumor samples compared to in normal tissue (*, Bartlett’s test p<0.05) and that there are more tumor samples with high level of methylation compared to normal tissue (+, 75th percentile Fisher exact test). This is especially clear for BLCA, BRCA, COAD, HNSC, LUAD, LUSC and PRAD. STING showed a similar behavior but was show high degree of methylation in some of the normal samples KIRC, KIRP and LIHC. In contrast, significantly less evidence of RIG-I (DDX58), MDA5 (IFIH1), or MAVS promoter methylation was observed which control the dsRNA activated RLH innate immune pathway (data not shown). Taken together, in a wide variety of tumors, both cGAS and STING expression are epigenetically suppressed, which may be beneficial for tumors to grow and avoid antitumor immunity.
Figure 7.
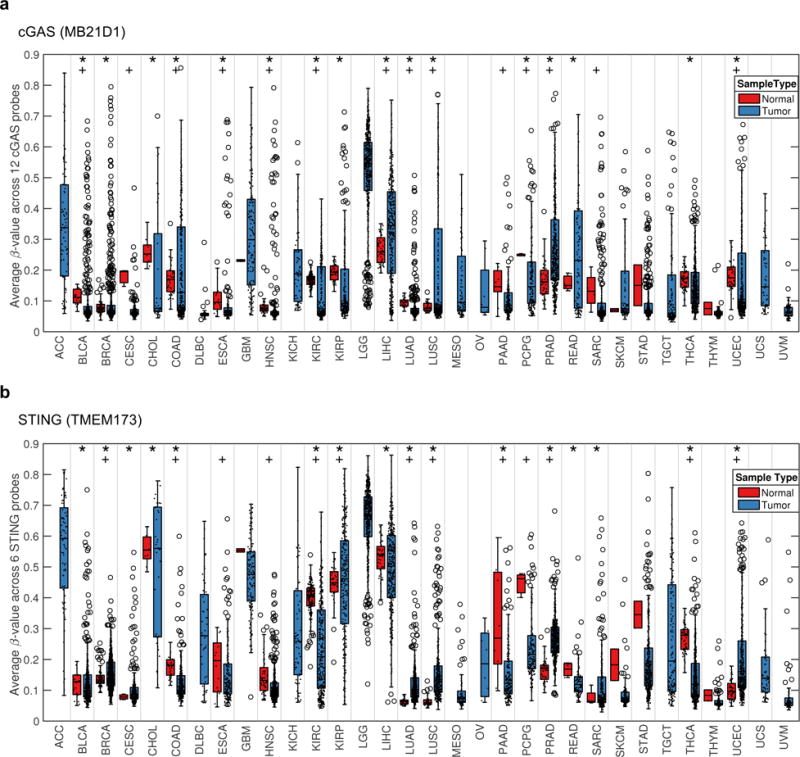
cGAS and STING show a large variability in methylation for a variety of human tumors. (a and b) The average methylation level across 32 tumor types are shown in box plots for cGAS (a) or STING (b). Each individual sample is also represented by a dot (.) and outlier samples are indicated by circles (o). The statistical analysis was performed by Bartlett’s test (*; p<0.05) and Fisher’s exact test for enrichment of tumors in the 75th percentile (+; p<0.05). Abbreviation for each tumor type is shown in Supplementary Information.
Discussion
The STING-dependent innate immune signaling pathway is critically important for responding to microbial infection and triggering the production of host defense genes26. Intrinsic STING signaling can be activated in response to DNA damaging events likely through chromatin escaping from the nucleus of the cell to generate CDN’s3. Thus, STING may play an important role in the DNA-damage response (DDR) pathway by alerting the immune system to the region of damaged cell. Perhaps as a consequence, we have found that STING signaling is defective in a variety of cancers such as colon cancer and melanoma2, 4 (Supplementary Figure 1e). In part, loss of signaling was seen to involve epigenetic silencing of the cGAS or STING promoters to repress transcription and expression of the corresponding proteins2, 4. Thus, suppression of STING signaling may enable cells having undergone mutagenic events to evade immune related elimination through phagocytosis. In this study, we have found evidence that the STING or cGAS coding region has undergone missense mutation in a number of cases, and that these variants can exert a dominant-negative phenotype to impede STING signaling. The frequency of these events is small, however (less than 1% in pan-cancer). Although it is unclear why the cGAS mutants exhibit relatively weak dominant-negative activity, the STING mutants do significantly affect STING function which is essential to transmit signals from cGAS. While R376 is important for cGAMP-binding, E383 is adjacent to DNA-binding amino acid residue (K384)27, 28. According to published crystal structures, cGAS forms a 2:2 complex with DNA23. Therefore, when the complex includes a R376Q variant, the efficiency of cGAMP production may decrease. Alternatively, the E383K mutant may be slightly defective in its ability to bind with DNA (Figure 3d) perhaps due to the negative charge on E383 changing to a positive charge (K). While E383K was expressed greater compared to wild-type cGAS, the amount of E383K protein precipitated with DNA was lower than that of the wild-type molecule (Figure 3d).
In contrast, we observed a high incidence of cGAS and STING gene methylation in a variety of tumors, compared to normal tissue. Thus, suppression of STING or cGAS signaling through epigenetic silencing may commonly occur in a wide variety of tumor types. Indeed, impeding STING signaling may be a predominant requirement for the transformation process to proceed and may be required to enable cells to escape the immunosurveillance system. STING-deficient mice are known to be defective in their ability to mount effective antitumor T cell responses7, 8. Moreover, loss of STING facilitates the development of CAC in response to AOM/DSS treatment11. Possibly, STING senses DNA damaging events and triggers cytokine production to alert the immune system. This promotes wound repair processes. In addition, mutagenic cells may be removed via phagocytosis, to prevent the further accrual of genetic changes which could enable the damaged cell to become transformed.
In addition, extrinsic STING signaling plays an important role in facilitating antitumor T cells responses. That is, damaged or infected cells are phagocytosed and the nucleic acid from the engulfed cell (viral or self) triggers STING signaling within the phagocyte resulting in the production of type I IFN and pro-inflammatory cytokines that facilitates the cross-priming of T cells. STING agonists are presently being evaluated as therapeutics to enhance antitumor T cells responses and likely function by stimulating professional APC’s.
Conversely, while transient STING signaling has been shown to be critical for anti-microbial and antitumor responses, chronic STING signaling has been shown to be responsible for a wide variety of autoinflammatory disease5. Thus, critical control of STING signaling is required to avoid these events. It is possible that chronic STING signaling may play key role in promoting the progression of certain inflammation aggravated cancers. For example, DMBA recurrently painted onto the skin of mice gives rise to driven skin cancer and polyp formation that is dependent on the production of pro-inflammatory cytokines and growth factors. Recently, STING-signaling has been shown to play a key role in this process since STING-deficient mice were greatly resistant to polyp development3. Plausibly, DMBA induces DNA-damaged triggered pro-inflammatory cytokine production that requires STING-signaling, which fuels cancer development. Thus, transient STING signaling may be a potent mechanism to trigger host defense and antitumor responses, but may drive cancer progression if chronically stimulated.
Finally, loss of STING signaling may help explain mechanisms of viral oncolysis, especially in the case of DNA based oncolytic microbes. Evaluation of whether STING or cGAS is expressed in a tumor may help predict responses relating to oncolytic therapy but also to other forms of cancer treatment including radiation. Using compounds that stimulate STING signaling may significantly enhance antitumor immune responses, especially when used in conjunction with other treatments and immunoncological therapies.
Materials and Methods
Reagents, Cells, and Viruses
All reagents were purchased from SIGMA unless specified. Primary Sting−/− MEF cells were prepared as previously described26. Primary cGas−/− MEF cells were isolated from cGas−/− mice that were kindly gifted from Dr. Herbert “Skip” Virgin, Washington University School of Medicine in St. Louis. Poly I:C was purchased from American Biosciences. dsDNA (ISD90-mer) was prepared as previously described29. To make biotin-labeled dsDNA (ISD90-mer), 5′-biotin labeled ISD90-mer sense DNA and unlabeled ISD90-mer antisense DNA that were synthesized at SIGMA were annealed in a water bath at 70 °C for 30 min and then cooled in the water bath to room temperature. cGAMP and cyclic di-GMP were purchased from BioLog. HSV-1 (KOS strain) was purchased from ATCC. HSV-1 γ34.5 was kindly provided by Bernard Roizman, University of Chicago. The viral titer was determined by plaque assay using Vero cells as previously described29. Anti-STING rabbit polyclonal antibody was prepared as previously described26. Other antibodies used in this paper are as follows: anti-β-actin (SIGMA, A5441); anti-FLAG (SIGMA, F1804); anti-calreticulin (Abcam, ab14234); anti-IRF3 (Cell Signaling, 4302); anti-phospho-IRF3 (Cell Signaling, 4947); anti-TBK1 (Abcam, ab40676); anti-phospho-TBK1 (Cell Signaling, 5483); anti-cGAS (Cell Signaling, 15102).
Database analysis
The data for mutations in STING and cGAS genes in human tumors were obtained from cBioPortal (http://www.cbioportal.org), COSMIC (http://cancer.sanger.ac.uk/cosmic), and ICGC (http://icgc.org) databases. The methylation data were obtained from MethHC (http://methhc.mbc.nctu.edu.tw/php/index.php) and TCGA (https://cancergenome.nih.gov) databases. BoxPlotR (http://shiny.chemgrid.org/boxplotr/) was used to make box plot30. The SNP data were obtained from dbSNP (https://www.ncbi.nlm.nih.gov/SNP/index.html), 1000 Genomes Project (Phase III, http://browser.1000genomes.org/index.html), and ExAC (http://exac.broadinstitute.org). TCGA RNAseqV2 level 3 data was downloaded March 2016 from the TCGA data portal. Data was log2 transformed prior to analysis. The TCGA Level 1 methylation data were downloaded between April and May of 2016 using the former Data Matrix accessed through the now defunct web address (http://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.html). The Level 1 data were retrieved as binary IDAT files, which consist of raw, probe-level signal intensities generated with Illumina’s Infinium HumanMethylation450 BeadChip technology. Preprocessing the data included normalization via internal controls followed by background subtraction using the methylumi R package from Bioconductor (Davis S, Du P, Bilke S, Triche T, Bootwalla M. methylumi: Handle Illumina methylation data. R package version 2.12.0 2014). The calculated β-values were then extracted from the MethyLumiSet object following preprocessing. To compare the variation within probe between normal and tumor samples we used the used Bartlett’s test as previous used for methylation data by Teschendorff et al.31. To compare highly methylated samples, we used the 75th percentile and a Fisher’s Exact test to compare normal and tumor samples. These statistical tests and Figures 6 and 7 was generated using MATLAB 9.2 and Statistics and Machine Learning Toolbox 11.1, The MathWorks, Inc., Natick, Massachusetts, USA.
Plasmids, Mutagenesis, and Cloning
Reporter plasmids (IFNβ-luc and TK-luc) were prepared as described before26. pcDNA3-hSTING, pcDNA3-mSTING, pBabe-hSTING-puro, pBabe-hSTING-hygro, and pcDNA3-RIG-I-FLAG were made as described before26, 29. pCMV-cGAS-myc-DDK was purchased from OriGene. pBabe-hcGAS-puro was made by the ligation of human cGAS gene that was amplified using PrimeSTAR GXL DNA Polymerase (Clontech) at Bam HI and Sal I sites of pBabe-puro. The primers used for cloning are shown in Supplementary Information. To make mutants of STING and cGAS, QuickChange II XL site directed mutagenesis kit (Stratagene) was used with primer sets that are also shown in Supplementary Information.
Luciferase assay
HEK293T cells were transfected with pcDNA3-STING and/or pCMV-cGAS-myc-DDK, and reporter plasmid (IFNβ-luc and TK-luc) using lipofectamine 2000 (Invitrogen). TK-luc (a plasmid encoding renilla luciferase gene under the control of the thymidine kinase (TK) promoter) was used for normalization. After 24 hr, the cells were lysed by Cell Culture Lysis Reagent (Promega) and then luciferase activity was measured with Luciferase assay substrate (Promega) for firefly luciferase activity and Renilla luciferase assay system (Promega) for renilla luciferase activity. HEK293T-hSTING cells that stably express human STING were made by using pBabe-hSTING-puro.
ELISA
The reconstituted primary Sting−/− or cGas−/− MEF cells using retrovirus were made as previously described29. The reconstituted MEF cells were transfected with poly I:C or dsDNA using lipofectamine 2000. The supernatants were collected and the amount of IFNβ was measured using IFNβ ELISA kit (PBL Interferon Source).
Immunofluorescence staining
The cells were seeded on poly-D-Lysine coated round coverslips (BD bioscience). After treatment with dsDNA, the cells were fixed with 4% paraformaldehyde in PBS for 15 min and then permeabilized with 0.2% Triton X-100 in PBS for 5 min. After blocking cells with 1% bovine serum albumin (BSA) in PBS for 30 min, the coverslips were incubated with the indicated antibodies in 1% BSA/PBS solution. After washing with PBS three times, the coverslips were incubated with Alexa Fluor 488-goat anti-rabbit IgG and Alexa Fluor-647 goat anti-chicken IgG (Invitrogen). After washing with PBS three times, the coverslips were mounted onto the glass slides with ProLong Gold anti-fade reagent (Invitrogen) and then observed under SP5 confocal microscope (Leica).
Western blot
After boiled in SDS-sample buffer, samples were separated in an acrylamide gel and then transferred to Immobilon-P membrane (Millipore). The membrane was treated with 5% Blotting-Grade Blocker (BIO-RAD) in TBS-T (TBS with 0.1% Tween20) for blocking and then incubated with the indicated primary antibodies. The membrane was washed with TBS-T buffer three times and then incubated with HRP-conjugated anti-rabbit or mouse IgG (Promega). After the incubation and washing, the bands were developed with Super Signal West Pico or Femto (Thermo) and then exposed to Premium X-ray film (Phenix).
DNA-pulldown assay
HEK293T cells that were transfected with the indicated plasmids for 24 hr were lysed in the lysis buffer (50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10% glycerol, 0.5 mM EDTA, 0.5 mM EGTA, 0.5% NP-40). 500 μl of the cell lysates was incubated with 200 pmol of biotin-labeled dsDNA and 10 μl of neutravidin-conjugated beads (Thermo) overnight at 4 °C. After washing with the lysis buffer three times, the precipitants were boiled in SDS-sampled buffer to elute the proteins.
Realtime PCR
RNA was extracted using RNeasy Mini Kit (QIAGEN) and cDNA was synthesized using QuantiTect Reverse Transcription Kit (QIAGEN) according to the manufacture’s protocol. Realtime PCR was performed using StepOnePlus Real-Time PCR system (Applied Biosystems). Probes for Cxcl10, Ccl5, Stat1, and Irf7 were purchased from Applied Biosystems. Gapdh was used for normalization.
Mass spectrometry
HEK293T cells were transfected with the indicated plasmids for 24 hr. In total 1×107 cells were pelleted and snap-frozen in liquid nitrogen. The frozen cell pellets were stored at −80 °C before further processing. The frozen cells were thawed on ice and lysed in cold 80% methanol with 2% acetic acid, and then homogenized using a homogenizer. The lysates were centrifuged at 10000xg for 10 min at 4 °C. The supernatants were kept in different tubes and the cell pellets were re-lysed in cold 20% methanol with 2% acetic acid twice. The supernatants from three times extraction were pooled in the same tube. 50 ng of cyclic di-GMP was added to the supernatants to validate the following purification process. cGAMP in the pooled supernatants was enriched by solid-phase extraction (SPE) using HyperSep Aminopropyl SPE column (Thermo). The columns were activated by 100% methanol and washed with 2% acetic acid twice. After adding the supernatants, the columns were washed with 2% acetic acid twice and then with 80% methanol once, and finally eluted with 2% ammonium hydroxide in 80% methanol. The eluents were dried by a centrifugal evaporator and then reconstituted in liquid chromatography (LC)/mass (MS)-grade water, and stored at −20 °C before LC/MS analysis. The liquid chromatography was performed using Thermo Scientific Surveyor MS Pump Plus with Micro AS auto sampler. The separation was isocratic on a Water’s XBridge Amide column (3.5 μm, 2.1×100 mm) at 200 μl/min using 18:82 water:acetonitrile with 6.3 mM ammonium hydroxide and 6.3 mM ammonium bicarbonate. The samples were introduced into Thermo Scientific LTQ-FT, a hybrid mass spectrometer consisting of a linear ion trap and a Fourier transform ion cyclotron resonance mass spectrometer. The standard electrospray source was used in negative ion mode. cGAMP was quantitated using the m/z 522 product ion from the collision-induced dissociation of the deprotonated parent ion at m/z 673. An external calibration curve derived from eight standards was used in the quantitation and acquired before and after the samples were analyzed. The cyclic-di-GMP component was quantitated from the m/z 344 product ion originating from the deprotonated m/z 689 parent. The LC/MS was performed at the R. Marshall Wilson Mass Spectrometry Facility, University of Cincinnati.
Statistical analysis
All data are representative of multiple independent experiments and they are presented as the average ± SD. We used two-tailed Student’s t-test to evaluate the differences between two groups. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by grants from NCI (R01CA194404) (G. N. Barber) and NCI-NIH (1R01 CA148995, 1R01 CA184845, P30 CA076292, P50 CA168536), Cindy and Jon Gruden Fund, Chris Sullivan Fund, and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (J. J. Mulé). We thank the staff of the Moffitt Cancer Center Cancer Informatics and Collaborative Data Services.
Footnotes
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- 2.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell reports. 2016;14:282–297. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nature communications. 2014;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer research. 2016;76:6747–6759. doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 5.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:15408–15413. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segawa K, Nagata S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Ahn J, Konno H, Barber GN. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene. 2015;34:5302–5308. doi: 10.1038/onc.2014.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Scientific reports. 2017;7:39858. doi: 10.1038/srep39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic acids research. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Science signaling. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolodkin-Gal D, Edden Y, Hartshtark Z, Ilan L, Khalaileh A, Pikarsky AJ, et al. Herpes simplex virus delivery to orthotopic rectal carcinoma results in an efficient and selective antitumor effect. Gene Ther. 2009;16:905–915. doi: 10.1038/gt.2009.44. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, Jin H, Valyi-Nagy T, Cao Y, Yan Z, He B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. Journal of virology. 2012;86:2188–2196. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nature communications. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, et al. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic acids research. 2015;43:D856–861. doi: 10.1093/nar/gku1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kranzusch PJ, Lee ASY, Wilson SC, Solovykh MS, Vance RE, Berger JM, et al. Structure-guided reprogramming of human cGAS dinucleotide linkage specificity. Cell. 2014;158:1011–1021. doi: 10.1016/j.cell.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell reports. 2014;6:421–430. doi: 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nat Methods. 2014;11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, et al. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4:24. doi: 10.1186/gm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.