

Abstract
The succinate receptor 1 (SUCNR1) is a receptor for the metabolite succinate, which functions as a metabolic stress signal in the liver, kidney, adipose tissue and the retina. However, potent non-metabolite tool compounds are needed to reveal the physiological role and pharmacological potential of SUCNR1. Recently, we published the discovery of a computationally receptor-structure derived non-metabolite SUCNR1 agonist series with high target selectivity. We here report our structure-activity exploration and optimisation that has resulted in the development of agonists with nanomolar potency and excellent solubility and stability properties in a number of in vitro assays. Ligand-guided receptor models with high discriminative power between binding of active and inactive compounds were developed for design of novel chemotypes.
Introduction
The 7-transmembrane G-protein coupled receptor SUCNR1 was first identified in 2001 and found to have close homology with the purinergic receptor P2Y1. The receptor was therefore initially believed to be activated by purinergic compounds1. However, in 2004, SUCNR1 was deorphanised and found to be activated by the citric acid cycle intermediate succinate at micromolar concentrations2. The other citric acid cycle intermediates oxaloacetate and α-ketoglutarate were also found to activate the receptor, albeit with reduced potency3,4. SUCNR1 is highly expressed in liver, kidney, and adipose tissue, but has also been reported in retina, heart, and immune cells2,5–7. During oxidative stress succinate can accumulate and reach local concentrations sufficiently high to activate SUCNR18, leading to a variety of unwanted physiological effects such as hypertension2, hypertrophy of the heart7, inflammation9, inhibition of lipolysis in the white adipose tissue5, activation of hepatic stellate cells10, and vascular growth in the retina6.
Although most studies so far indicate that antagonists for SUCNR1 might be optimal from a therapeutic perspective this has yet to be experimentally confirmed and is mainly based on studies using the natural agonist succinate, a compound that beside its relatively weak potency also is an intermediate in the citric acid cycle and exerts effects unrelated to SUCNR1. In 2011, the first and so far only antagonists for SUCNR1 were reported, consisting of a series of naphthyridines that demonstrated good pharmacokinetic properties in rats11. Small synthetic succinate analogues have recently been reported as full agonists and although the compounds show improved agonistic potency, the best compound, cis-epoxysuccinic acid, has an EC50 of only 2.7 μM3.
Based on the characterisation of the binding site for succinate in SUCNR1 and identification of an empty side-pocket in the receptor, we have recently published the discovery of novel drug-like SUCNR1 agonists which in accordance with both loss- and gain-of-function mutational data exploited this pocket4. Here we report the further development and structure-activity investigations of this agonist series. The generated library of active and inactive compounds together with the original receptor model based on the X-ray structure of the P2Y1 receptor allowed for generation of new SUCNR1 models which could discriminate between active and inactive compounds with high discriminative power. Although the human and mouse receptor orthologues are closely related they are not identical and succinate shows a small increase in potency on the mouse receptor2. All compounds have therefore been studied on both the human and mouse orthologues in order to develop tool compounds for further investigations of SUCNR1.
Results and Discussion
Synthesis
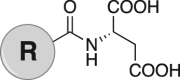
All test compounds were synthesised by the same overall route, starting from L-aspartic acid (1) that was converted to the corresponding dimethyl ester hydrochloride 2 by an acid catalysed esterification (Fig. 1). Coupling of 2 to various carboxylic acids was efficiently achieved via the corresponding acyl fluoride generated in situ by fluoro-N,N,N’,N’-bis(tetramethylene)foramidinium hexafluorophosphate (BTFFH)12. Halogenated aryl and heteroaryl compounds were coupled with various aryl boronic acids by a Suzuki reaction using the 4th generation Pd-XPhos precatalyst. The phenolic compounds were afterwards alkylated by alkylhalides or –tosylates. Finally, base promoted hydrolysis using LiOH gave the desired dicarboxylic acid test compounds.
Figure 1.

General synthetic route. Reagents and conditions: (a) SOCl2, MeOH, 0 °C → reflux. (b) BTFFH, DIPEA, ArCOOH, DCM, 80 °C. (c) ArB(OH)2, Pd-XPhos-G4, 0.5 M K3PO4aq, THF, room temp. (d) For R” = OH: K2CO3, alkylhalide/tosylate, MeCN, 50–55 °C. (e) 0.6 M LiOHaq, THF, room temp.
Exploration of structure activity relationship (SAR)
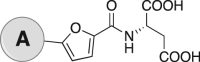
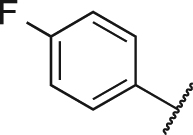
In order to optimise the activity of the lead compound Cmpd (S)−130 (3), we started by exploring larger modifications of the aromatic part of the molecule (selected examples are included in Table 1). Initially, the aromatic part was truncated by exchanging the fluorophenyl with a bromine (4), which resulted in more than one order of magnitude decrease in potency. Exchanging the bromofurane with the halogenated phenyls 5–7 revealed weak activity on both receptor orthologues of the ortho-chlorine and para-bromine analogs. Only the larger meta-iodine substituted compound 6 showed micromolar activity on both receptor orthologues. Inserting a methylene-linker between the amide and the bromophenyl (8) resulted in an inactive compound and exchanging the bromine for the fluorophenyl (9) only gave a weakly active compound, far less potent than the lead 3.
Table 1.
Investigation of miscellaneous amide analogues.
| ||||
|---|---|---|---|---|
| R | hSUCNR1 | mSUCNR1 | ClogPb | |
| pEC50 (efficacy, %)a | ||||
| 3 |
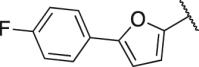
|
5.75 ± 0.08 (72.8 ± 2.6) | 6.46 ± 0.06 (79.3 ± 1.6) | 1.66 |
| 4 |
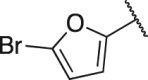
|
4.53 ± 0.13 (78.1 ± 7.5) | 4.93 ± 0.05 (95.8 ± 2.9) | 0.44 |
| 5 |
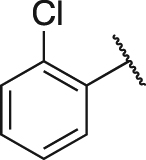
|
22% @10−4M | 13% @10−4M | 0.28 |
| 6 |
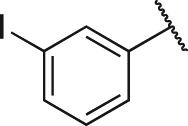
|
50% @10−4M | 64% @10−4M | 1.52 |
| 7 |
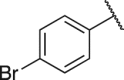
|
8% @10−4M | 15% @10−4M | 1.26 |
| 8 |
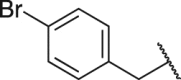
|
5% @10−4M | 11% @10−4M | 1.28 |
| 9 |
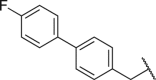
|
25% @10−4M | 15% @10−4M | 2.44 |
apEC50 values were determined from dose-response curves of induction of IP3 turnover in SUCNR1 transfected HEK cells (N = 3), efficacy is determined relative to succinate (100%). bCalculated by BioByte’s algorithm as implemented in ChemDraw Professional 16.0.1.4 (ClogP option).
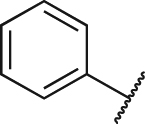
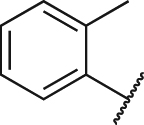
Since the smaller compounds did not show sufficient activity we moved the focus to explorations on the terminal phenyl ring (Table 2). Initially, the original 4-fluoro substituent was removed (10). This only affected the potency marginally on both receptor orthologues, but exposes a potential metabolically labile site. Next, a methyl scan of the ring was performed to investigate the binding site for additional space (11–13). Whereas both the ortho- and meta-methyl were tolerated only the para-methyl 13 led to a more potent compound on both human and mouse SUCNR1. Attempts to pick up hydrogen bond interactions revealed that 2-methoxy (16) was equivalent to the 2-methyl in potency and that 3-hydroxymethyl (14) was less tolerated than the 3-methyl (12) with a 3-fold decrease in potency. Again, the para-position was favoured with the 4-hydroxy (15) being almost equipotent with the 4-methyl compound. Further exploration of the para-position indicated that this part of the binding pocket is able to accommodate polar substituents such as methoxy (17) and nitrile (19) as well as nonpolar substituents such as trifluoromethyl (18), all showing similar potencies in the submicromolar range on the human receptor orthologue and with 17 slightly favoured on the mouse orthologue.
Table 2.
Investigation of ring A.
| ||||
|---|---|---|---|---|
| Ring A | hSUCNR1 | mSUCNR1 | ClogPb | |
| pEC50 (efficacy, %)a | ||||
| 3c |
|
5.75 ± 0.08 (72.8 ± 2.6) | 6.46 ± 0.06 (79.3 ± 1.6) | 1.66 |
| 10 |
|
5.29 ± 0.19 (81.6 ± 8.4) | 6.16 ± 0.11 (81.3 ± 3.6) | 1.47 |
| 11 |
|
5.27 ± 0.18 (94.7 ± 8.7) | 6.13 ± 0.10 (76.7 ± 3.1) | 1.67 |
| 12 |
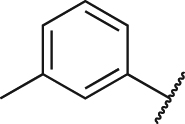
|
5.02 ± 0.10 (91.9 ± 5.3) | 6.07 ± 0.09 (81.6 ± 3.0) | 1.97 |
| 13 |
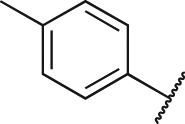
|
6.33 ± 0.14 (69.1 ± 3.9) | 6.83 ± 0.11 (73.2 ± 2.7) | 1.97 |
| 14 |
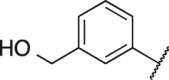
|
4.81 ± 0.20 (74.2 ± 11.8) | 5.71 ± 0.09 (79.6 ± 3.7) | 0.43 |
| 15 |
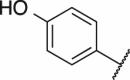
|
6.13 ± 0.14 (71.1 ± 4.2) | 6.82 ± 0.08 (74.8 ± 2.0) | 0.87 |
| 16 |
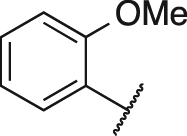
|
5.29 ± 0.15 (103.8 ± 7.9) | 6.14 ± 0.10 (78.1 ± 3.1) | 0.89 |
| 17 |
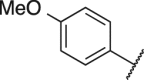
|
6.41 ± 0.14 (85.3 ± 4.5) | 7.39 ± 0.07 (84.2 ± 1.8) | 1.45 |
| 18 |
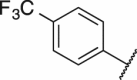
|
6.55 ± 0.25 (81.0 ± 7.2) | 6.81 ± 0.17 (80.6 ± 4.6) | 2.43 |
| 19 |
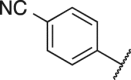
|
6.39 ± 0.11 (72.1 ± 3.2) | 6.79 ± 0.06 (76.0 ± 1.5) | 1.00 |
apEC50 values were determined from dose-response curves of induction of IP3 turnover in SUCNR1 transfected HEK cells (N = 3), efficacy is determined relative to succinate (100%). bCalculated by BioByte’s algorithm as implemented in ChemDraw Professional 16.0.1.4 (ClogP option). cDuplication of data from Table 1.
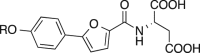
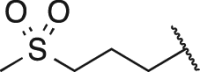
To further explore the 4-methoxy compound, the most potent analogue on the mouse receptor, a sub series of para-alkoxy analogues were investigated (Table 3). The larger and more electronegative trifluoromethoxy (20) showed a 2-fold increase on hSUCNR1 but was accompanied by >4-fold decrease on mSUCNR1, rendering the compound approximately equipotent on both orthologues. Expanding to either ethoxy (21) or i-propoxy (22) resulted in a small increase in potency, indicating that the binding pocket can accommodate more elongated and bulky substituents. To explore whether or not the binding site could also accommodate larger hydrophilic substituents the oxatane 23 was investigated and found tolerable, but led to >2-fold reduction of potency. Finally, the mesylpropoxy analogue 24 was explored. This appendage, which lowers the lipophilicity an order of magnitude, has previously been applied to lower lipophilicity of ligands for the free fatty acid receptor FFA1 and has proven to be a metabolically stable substituent13,14. Lipophilicity of this compound is in the low end of the desired range but it is interesting to note that the compound exhibited a ligand lipophilicity efficiency (LLE) >5 based on ClogP and almost sustained potency on both mSUCNR1 and hSUCNR1.
Table 3.
Alkoxy analogues.
| ||||
|---|---|---|---|---|
| R | hSUCNR1 | mSUCNR1 | ClogPb | |
| pEC50 (efficacy, %)a | ||||
| 17c | Me | 6.41 ± 0.14 (85.3 ± 4.5) | 7.39 ± 0.07 (84.2 ± 1.8) | 1.45 |
| 20 | F3C | 6.99 ± 0.15 (61.7 ± 3.6) | 6.72 ± 0.11 (69.7 ± 2.8) | 3.17 |
| 21 | Et | 6.59 ± 0.14 (56.3 ± 3.1) | 7.44 ± 0.09 (70.2 ± 2.0) | 1.98 |
| 22 | iPr | 6.84 ± 0.14 (69.6 ± 4.3) | 7.29 ± 0.07 (75.6 ± 1.7) | 2.29 |
| 23 |

|
6.27 ± 0.11 (76.5 ± 3.1) | 6.97 ± 0.07 (82.4 ± 1.9) | 1.81 |
| 24 |
|
6.10 ± 0.09 (63.9 ± 2.4) | 7.33 ± 0.09 (74.6 ± 2.1) | 0.53 |
apEC50 values were determined from dose-response curves of induction of IP3 turnover in SUCNR1 transfected HEK cells (N = 3), efficacy is determined relative to succinate (100%). bCalculated by BioByte’s algorithm as implemented in ChemDraw Professional 16.0.1.4 (ClogP option). cDuplication of data from Table 2.
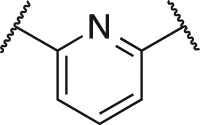
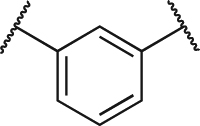
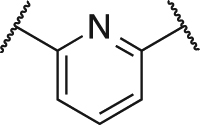
Subsequently, the attention was directed towards the central aromatic ring (Table 4). Replacing the furane of 3 with the 1,3-substituted phenyl 25 improved the potency somewhat on the human orthologue whereas the potency on the mouse orthologue decreased 10-fold. The 1,4-substituted phenyl 26 clearly led to an unfavoured geometry with only trace activity on SUCNR1. The corresponding 2,6-substituted pyridine 27 sustained the potency on hSUCNR1 and almost regained the potency on mSUCNR1.
Table 4.
Investigation of ring B.
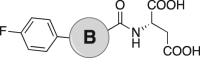
| ||||
|---|---|---|---|---|
| Ring B | hSUCNR1 | mSUCNR1 | ClogPb | |
| pEC50 (efficacy, %)a | ||||
| 3c |

|
5.75 ± 0.08 (72.8 ± 2.6) | 6.46 ± 0.06 (79.3 ± 1.6) | 1.66 |
| 25 |
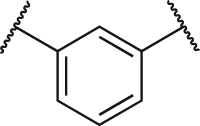
|
5.91 ± 0.11 (49.7 ± 3.9) | 5.40 ± 0.13 (58.0 ± 4.5) | 2.27 |
| 26 |
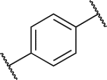
|
4% @10−4M | 7% @10−4M | 2.27 |
| 27 |
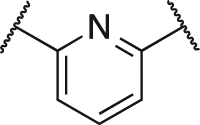
|
6.03 ± 0.25 (78.2 ± 8.0) | 6.10 ± 0.17 (89.3 ± 6.1) | 2.05 |
apEC50 values were determined from dose-response curves of induction of IP3 turnover in SUCNR1 transfected HEK cells (N = 3), efficacy is determined relative to succinate (100%). bCalculated by BioByte’s algorithm as implemented in ChemDraw Professional 16.0.1.4 (ClogP option). cDuplication of data from Table 1.
Next, a small selection of compounds was synthesised to combine the observed SAR of the alkoxy-substituents and alterations of the central ring (Table 5). The 4-methoxy and 4-trifluoromethoxy substituents, being the most potent alkoxy-substituents on the mouse and human receptor orthologues, respectively, were attached to compounds bearing the 1,3-substituted phenyl and the 2,6-substituted pyridine as the central ring. No improvement was observed for the methoxy analogues (28–29). In contrast, the trifluoromethoxy analogues (30–31) were found to be the most potent compounds on hSUCNR1, but unfortunately without high potency on mSUCNR1, especially for 30. The elongated trifluoroethoxy analogue 32 showed improved potency on the mouse orthologue. Still, 31 remained the most potent agonist on hSUCNR1. Finally, the 4-propoxy analogue 33 was found to be approximately twice as potent as the corresponding methoxy analogue 29 and almost equipotent on both orthologues.
Table 5.
Combined analogues.
| |||||
|---|---|---|---|---|---|
| Ring A | Ring B | hSUCNR1 | mSUCNR1 | ClogPb | |
| pEC50 (efficacy, %)a | |||||
| 28 |
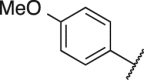
|
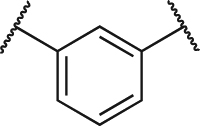
|
6.79 ± 0.12 (58.0 ± 2.7) | 5.92 ± 0.09 (64.0 ± 2.5) | 2.01 |
| 29 |
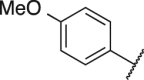
|
|
6.85 ± 0.19 (52.6 ± 3.9) | 6.74 ± 0.13 (71.2 ± 3.2) | 1.07 |
| 30 |
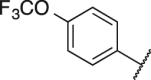
|
|
7.23 ± 0.14 (49.2 ± 2.7) | 5.84 ± 0.16 (45.3 ± 3.4) | 3.11 |
| 31 |
|
|
7.64 ± 0.13 (61.7 ± 2.5) | 6.75 ± 0.09 (69.9 ± 2.2) | 2.18 |
| 32 |
|
|
7.23 ± 0.23 (88.7 ± 6.1) | 7.00 ± 0.09 (68.1 ± 2.5) | 1.86 |
| 33 |
|
|
7.23 ± 0.37 (66.4 ± 7.2) | 7.29 ± 0.18 (61.5 ± 3.8) | 2.13 |
apEC50 values were determined from dose-response curves of induction of IP3 turnover in SUCNR1 transfected HEK cells (N = 3), efficacy is determined relative to succinate (100%). bCalculated by BioByte’s algorithm as implemented in ChemDraw Professional 16.0.1.4 (ClogP option).
To get a better overview of the observed SAR on the human and mouse receptor orthologues a scatterplot of all active compounds was made (Fig. 2a). The compounds were color-coded according to the central or terminal ring, which clearly indicated that compounds with a central meta-substituted phenyl were better tolerated on hSUCNR1. The 2,6-substituted pyridines were in general more potent and especially 31 was favoured on hSUCNR1. Compounds with a furane as central ring were in general equipotent on the two receptor orthologues, but with addition of a phenalkoxy moiety, the compounds became more potent on mSUCNR1 with 21 being the most potent. Furthermore, dose-response curves of succinate, 3, 21, and 31 clearly showed that the non-metabolite compounds were partial agonists, with clear species differences on potency but not efficacy (Fig. 2b,c). The partial agonism was observed for all agonistic compounds (Tables 1–5, Supplementary Fig. S1).
Figure 2.

Summary of in vitro agonist potencies. (a) Scatterplot of in vitro agonist potencies by IP3 accumulation in SUCNR1 transfected HEK-293 cells of the compounds in Tables 1–5 on the hSUCNR1 and mSUCNR1 receptor. (b,c) Dose-response curves for succinate and selected compounds (3, 21 and 31) on hSUCNR1 and mSUCNR1.
Molecular modelling and unbiased ligand guided refinement of SUCNR1 receptor-ligand complexes
To describe the detailed molecular mechanism of action and to rationalise the structure-activity data, we docked the compounds in Tables 1–5 to a model of SUCNR1 in complex with the lead compound (3) supported by a dozen of site directed mutagenesis data as presented earlier4. Despite that we observed binding conformations similar to the lead and with favourable scores, the model - as perhaps could be expected - was unable to rank the compounds in reasonable agreement with the activity data. The interpretation of SAR and structure based design reaches its maximum potential when the receptor displays the structural changes needed for ligand binding, as it has previously been shown in a blind prediction assessment of adenosine A2a receptor complexes (GPCR Dock 2008)15–18. We therefore applied an iterative Automated Ligand-guided Backbone Ensemble Receptor Optimisation protocol (ALiBERO)19, which samples full receptor and ligand flexibility guided by the ligand information gained in this study to validate and build confidence in the model.
In brief, starting from the homology model supported by mutagenesis data4, ALiBERO introduced receptor flexibility via Normal Mode Analysis and Monte Carlo sampling, to generate a small subset of receptor models (pockets). All compounds tested in this study were grouped into an active (EC50 ≤ 10 µM) and a decoy set (EC50 > 10 µM) consisting of 25 compounds each. For a list of all compounds used in the optimisation protocol, see Table S1. Receptor structures were then chosen based on their ability to discriminate actives from inactives in a retrospective virtual screening using the docking protocol and scoring function in ICM (Molsoft L.L.C., San Diego, CA, USA)20, as measured by the normalised square root area under the curve (NSQ_AUC). The best-performing structures from the first generation were consequently selected for the next generation and the steps were repeated in an iterative fashion until maximum docking performance of receptor structures to enrich active compounds was reached. The ALiBERO-optimised receptor ensemble was subsequently validated in a virtual ligand screening using an external test set with a higher active:decoy ratio (~1:50).
Notably, the ligand-guided ALiBERO-based mSUCNR1 models demonstrated a dramatic improvement in retrospective virtual screening performance of the developed compounds compared with the initial homology models and proved successful in separating the majority of active from inactive ligands in docking screens (Fig. 3a, Supplementary Fig. S2). It is interesting to note that the active ligands bind to an extended binding cavity in a very consistent pose compared to the lead compound (3) supported by both loss-of-function but also gain-of-function mutagenesis data as we have reported previously4. For example, the receptor mutants R251:6.58 L and R276:7.39 F showed a potency decrease of more than 100-fold. The binding cavity of SUCNR1 is characterised by a polar network consisting of residues in TM-II (Y79:2.64), TM-III (R95:3.29), TM-VI (R248:6.55, R251:6.58), TM-VII (K269:7.32, Y272:7.35, R276:7.39), ECL2 (D174) as well as a relatively hydrophobic subpocket spanning between TM-I, -II, ECL1 and −2 up towards the extracellular surface of the receptor (residue numbering according to gpcrdb.org). The optimised receptor ensemble (Fig. 3b) accommodates the active compounds in a common binding mode in which the left-hand side carboxylic acids make hydrogen bond interactions with R95:3.29, R251:6.58 and R276:7.39 and where the right-hand side of the ligands adopts an angled conformation that is defined by the linker between ring A and B in good agreement with the observed structure-activity relationship. Due to the shape and direction of the subpocket accommodating ring A, compounds with a bent conformation between the amide and the terminal ring, e.g. compounds having 2,5-substitued furan or meta-substituted phenyl as central ring, are sterically better tolerated than e.g. the para-substituted phenyl 26 and elongated para-substituted phenyl 9. Furthermore, compounds with a pyridine (e.g. 27, 29, 31), or furan as central ring, can stabilise the favoured bent conformation by intramolecular hydrogen bonding between the pyridine nitrogen or furan oxygen and the amide N-H, thereby inducing an optimal low energy conformation.
Figure 3.

Iterative SUCNR1 receptor optimisation based on ligand information gained in this study. (a) Small-scale virtual ligand screening (VLS) results at different stages of the optimisation process (generations GEN_1 - GEN_4). Receiver operating characteristic (ROC) curves and normalised square root area under the curve (NSQ_AUC) values are shown for each generation of receptor pockets. The diagonal corresponds to a random VLS performance. (b) Best-performing receptor pocket ensemble (GEN_4), consisting of three receptor structures shown with the best-scored docking poses of all 25 active compounds investigated in this study (dark green lines). Best-scored docking poses of compound 24 (b), 31 (c), 3 (d) and 21 (e) are shown in green sticks. Polar receptor-ligand interactions are indicated by yellow spheres. Homology models are based on the x-ray crystal structure of the P2Y1 receptor (PDB 4XNW) which shares several key side-chains in the binding site with SUCNR1, i.e. Y79:2.64, D174 and R276:7.39. Figure was made using PyMol.
The subpocket that is occupied by ring A, contains W10:1.42 which allows for parallel displaced aromatic stacking interactions that are present in all binding poses for the active ligands. As this pocket spans all the way to the extracellular tips of TM-I and -II, it is able to accomodate longer substituents, such as the mesylate 24 (Fig. 3b), which likely interacts with solvent water molecules on the extracellular surface of the receptor cavity. Compounds that contain O-alkyl, hydroxyl, or nitrile groups in the para-position on ring A, such as 15, 17, 19–24 and 28–31, can form hydrogen bond interactions with K83 (Asn87 in hSUCNR1) and backbone NH of Glu170 in ECL1 and −2b (Fig. 3c). Due to the geometry of this interaction, ortho- and meta-subtitutions (14 + 16) are less favorable.
While the polar residues that coordinate the dicarbocylic acid moiety are highly conserved between mouse and human SUCNR1, the hydrophobic subpocket accommodating ring A is more diverse with several differences in ECL1 (Lys83/Asn87), ECL2 (Lys162/Thr166, Glu163/Asp166, Glu164/Asn166, Asn167/Thr171) and TM-VII (Lys269/Asn274) between mSUCNR1 and hSUCNR1, respectively. The Lys83/Asn87 difference between mSUCNR1 and hSUCNR1 might explain the higher potencies of the trifluoromethoxy-substituted compounds 30 and 31 on hSUCNR1, as these bulky moities likely form more favorable interactions with the shorter Asn side-chain (Fig. 3c).
In conclusion, the developed ligand-guided SUCNR1 models are consistent with the SAR, and they are sufficiently accurate to separate actives from decoys, suggesting that the models will be valuable in prospective studies to support structure-based drug design of additional chemotypes directly related to drug discovery applications.
Physicochemical and in vitro stability properties
Based on the SAR exploration, the original lead (3) and the most potent agonists on hSUCNR1 (31) and mSUCNR1 (21) were selected for further property investigations. All compounds showed excellent chemical stability (>98% after 70 hours) and kinetic solubility (>200 μM) in 10 mM PBS7.4. However, the high aqueous solubility made logD7.4 determinations very challenging and only the most lipophilic compound 31 could be quantified (logD7.4 = −2.13). The stability of the compounds was further examined in a selection of simulated gastrointestinal fluids (FaSSGF, FaSSIF and FeSSIF) and all compounds were found to be stable for 2 hours, with exception of 31 in FeSSIF, which could not be determined due to overlapping UV-absorption of the compound and media. Finally, the stability in mouse liver microsomes was investigated and all compounds were found to be stable for 1 hour, possibly partly due to the hydrophilic nature of the compounds.
Conclusion
We here report the structure-activity investigations of a series of non-metabolite SUCNR1 agonists that was originally identified from a computational, receptor-structure derived agonistic lead. ALiBERO optimised homology models of the mouse SUCNR1 were developed based on the x-ray structure of the closely related P2Y1 receptor and found to be sufficiently accurate to discriminate between actives and inactives and could explain the majority of the SAR observations. The exploration led to development of potent drug-like, non-metabolite tool compounds with nanomolar potency on both the human and murine receptor orthologues and excellent physicochemical and in vitro stability properties. We believe these compounds will be useful for further investigations of SUCNR1 as a potential therapeutic target and pharmacokinetic studies in mice are currently ongoing and will reveal if absorption of the compounds might be challenged by their hydrophilic nature and if the excellent stability properties are conserved in vivo.
Methods
General
Commercial starting materials and solvents were used without further purification, unless otherwise stated. THF was freshly distilled from sodium/benzophenone. DCM was distilled and stored over 3 Å sieves. MeCN and N,N-diisopropylethylamine were dried over 3 Å sieves. K2CO3 was dried and stored in an oven. TLC was performed on TLC silica gel 60 F254 plates and visualised at 254 or 365 nm or by staining with phosphomolybdic acid, ninhydrin, or KMnO4 stains. Purification by flash chromatography was carried out using silica gel 60 (0.040–0.063 mm, Merck). 1H and 13C NMR spectra were recorded at 400 and 101 MHz, respectively, on a Bruker Avance III 400 at 300 K. Spectra were calibrated relative to the internal standard TMS or residual solvent peak: CDCl3 (δC = 77.16 ppm, δH = 7.26 ppm), DMSO-d6 (δC = 39.52 ppm, δH = 2.50 ppm) and acetone-d6 (δC = 29.84 ppm, δH = 2.05 ppm).
HPLC analysis was performed using a Gemini C18 column (5 μm, 4.6 × 150 mm); flow: 1 mL/min; 10% MeCN in water (0–1 min), 10–100% MeCN in water (1–10 min), 100% MeCN (11–15 min), with both solvents containing 0.1% HCOOH as modifier; UV detection at 254 nm (or 280 nm, 304 nm and 315 nm depending on test compound absorption maximum for solubility and stability studies). UPLC analysis was performed using a 100 Å C18 column (1.7 μm, 2.1 × 100 mm); flow: 0.3 mL/min; 70% MeOH in water (0–6 min), the water containing 0.1% HCOOH as modifier; UV detection at 254 nm, 280 nm, 304 nm or 315 nm depending on test compound absorption maximum. High-resolution mass spectra (HRMS) were obtained on a Bruker micrOTOF-Q II (ESI). Optical rotation was measured on an Anton Paar MCP 100 Polarimeter (Anton Paar Cell 100 mm, CL. 0.01, Ø 5 mm). Purity was determined by HPLC (254 nm) and confirmed by inspection of NMR spectra. The purity of all test compounds were >95%.
Amide coupling
An oven dried microwave vial under argon atmosphere was charged with the acid (1.3 equiv), dry DCM (2 mL/mmol), N,N-diisopropylethylamine (5.5 equiv) and BTFFH (1.5 equiv). The reaction mixture was stirred at rt for 30 min before the HCl salt of the amine (1 equiv) was added. After addition, the vial was capped and heated to 80 °C overnight. The reaction was cooled to rt and diluted with water and extracted with EtOAc (x3). The organic phases were combined, washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, EtOAc:petroleum ether).
Suzuki coupling
A schlenck flask under argon was charged with boronic acid (1.1 equiv), aryl/pyridyl halide (1 equiv) and Pd-XPhos-G4 (2 mol%). The flask was evacuated and backfilled with argon (x3). THF (5 mL/mmol) and aqueous 0.5 M K3PO4 (2 equiv) was added, and the reaction was stirred at rt. After completion, the reaction mixture was diluted with water and extracted with EtOAc (x3). The organic phases were combined, washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, EtOAc:petroleum ether).
Alkylation
The phenol (1 equiv) was dissolved in dry MeCN (~6 mL/mmol) in a dry vial under argon atmosphere. The alkyl halide/tosylate (2–7 equiv) and dry K2CO3 (2 equiv) were added and the reaction was stirred at 50–55 °C until consumption of the phenol as monitored by TLC. After completion, the reaction was diluted with water and extracted with EtOAc (x3). The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, EtOAc:petroleum ether).
Ester hydrolysis
The ester (1 equiv) was dissolved in THF (~6 mL/mmol), and aqueous 0.6 M LiOH (3 equiv) was added. The reaction was stirred at rt until consumption of the ester as monitored by TLC. After completion, the reaction was diluted with water, acidified with aqueous 1 M HCl and extracted with EtOAc (x3). The organic phases were combined, washed with brine, and dried over Na2SO4. The residue was concentrated in vacuo to give the pure title compounds.
(5-Bromofuran-2-carbonyl)-L-aspartic acid (4)
Dimethyl (5-bromofuran-2-carbonyl)-L-aspartate (4e) was synthesised from 5-bromo furoic acid (126 mg, 0.66 mmol) and 2 (100 mg, 0.51 mmol) according to the general amide coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:2) gave 136 mg (81%) of a yellow oil: Rf = 0.30 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 7.9 Hz, 1H), 7.09 (d, J = 2.5 Hz, 1H), 6.45 (d, J = 3.5 Hz, 1H), 5.02 (dt, J = 8.6, 4.5 Hz, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.13 (dd, J = 17.3, 4.5 Hz, 1H), 2.94 (dd, J = 17.3, 4.5 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 171.4, 170.8, 156.8, 148.9, 125.1, 117.3, 114.2, 53.0, 52.1, 48.3, 36.1; ESI-HRMS: calcd for C11H12BrNNaO6 (M + Na)+ 355.9740, found 355.9757; [α]20D − 16.5° (c 0.2, MeOH).
4 was synthesised from 4e (35 mg, 0.10 mmol) according to the general ester hydrolysis procedure to give 28 mg (88%) of a white solid: tR = 7.68 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 7.91 (d, J = 7.9 Hz, 1H), 7.13 (d, J = 3.5 Hz, 1H), 6.67 (d, J = 3.5 Hz, 1H), 5.02–4.94 (m, 1H), 3.02 (d, J = 5.8 Hz, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.4, 172.3, 157.4, 150.7, 125.2, 117.4, 115.1, 49.4, 36.4; ESI-HRMS calcd for C8H9BrNNaO6 (M + Na)+ 327.9427, found 327.9431; [α]20D + 7.2° (c 0.2, MeOH).
(2-Chlorobenzoyl)-L-aspartic acid (5)
Dimethyl (2-chlorobenzoyl)-L-aspartate (5e) was synthesised from 2-chlorobenzoic acid (103 mg, 0.66 mmol) and 2 (99 mg, 0.50 mmol) according to the general amide coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 121 mg (80%) of a pale yellow solid: Rf = 0.65 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.70–7.66 (m, 1H), 7.44–7.30 (m, 3H), 7.25–7.23 (m, 1H), 5.11–5.05 (m, 1H), 3.80 (s, 3H), 3.70 (s, 3H), 3.19–2.98 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.0, 166.2, 134.4, 131.8, 131.2, 130.5, 130.4, 127.2, 53.1, 52.2, 49.3, 36.2; ESI-HRMS calcd for C13H14ClNNaO5 (M + Na)+ 322.0453, found 322.0461; [α]20D − 22.5° (c 0.2, MeOH).
5 was synthesised from 5e (25 mg, 0.08 mmol) according to the general ester hydrolysis procedure to give 22 mg (97%) of a white solid: tR = 7.34 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 10.80 (br s, 2H), 7.88–7.80 (m, 1H), 7.60–7.55 (m, 1H), 7.50–7.44 (m, 2H), 7.44–7.36 (m, 1H), 5.08–4.98 (m, 1H), 3.09–2.96 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.3, 172.2, 166.9, 136.8, 132.1, 131.6, 130.8, 130.4, 127.9, 50.0, 36.4; ESI-HRMS calcd for C11H9ClNO5 (M − H)− 270.0175, found 270.0177; [α]20D + 2.0° (c 0.2, MeOH).
(3-Iodobenzoyl)-L-aspartic acid (6)
Dimethyl (3-iodobenzoyl)-L-aspartate (6e) was synthesised from 3-iodobenzoic acid (161 mg, 0.65 mmol) and 2 (99 mg, 0.50 mmol) according to the general amide coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 164 mg (84%) of a pale yellow sticky oil: Rf = 0.33 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 8.17–8.12 (m, 1H), 7.88–7.83 (m, 1H), 7.78–7.72 (m, 1H), 7.22–7.14 (m, 2H), 5.08–4.98 (m, 1H), 3.80 (s, 3H), 3.71 (s, 3H), 3.20–2.91 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 171.8, 171.2, 165.5, 141.0, 136.4, 135.8, 130.4, 126.4, 94.4, 53.1, 52.3, 49.2, 36.1; ESI-HRMS calcd for C13H14INNaO5 (M + Na)+ 413.9809, found 413.9801; [α]20D − 22.0° (c 0.2, MeOH).
6 was synthesised from 6e (27 mg, 0.07 mmol) according to the general ester hydrolysis procedure to give 24 mg (97%) of a white solid: tR = 8.93 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.25 (t, J = 1.7 Hz, 1H), 8.13–8.07 (m, 1H), 7.94–7.89 (m, 2H), 7.30 (t, J = 7.8 Hz, 1H), 5.06–4.99 (m, 1H), 3.09–2.93 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.3, 172.2, 165.9, 141.2, 137.3, 137.1, 131.4, 127.6, 94.4, 50.3, 36.4; ESI-HRMS calcd for C11H9INO5 (M − H)− 361.9531, found 361.9531; [α]20D + 4.0° (c 0.2, MeOH).
(4-Bromobenzoyl)-L-aspartic acid (7)
Dimethyl (4-bromobenzoyl)-L-aspartate (7e) was synthesised from 4-bromobenzoic acid (133 mg, 0.66 mmol) and 2 (99 mg, 0.50 mmol) according to the general amide coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 137 mg (80%) of a beige solid: Rf = 0.35 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.71–7.66 (m, 2H), 7.61–7.56 (m, 2H), 7.23–7.17 (m, 1H), 5.07–5.00 (m, 1H), 3.80 (s, 3H), 3.71 (s, 3H), 3.19–2.93 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 171.9, 171.2, 166.1, 132.7, 132.0, 128.9, 126.8, 53.1, 52.3, 49.1, 36.1; ESI-HRMS calcd for C13H14BrNNaO5 (M + Na)+ 365.9948, found 365.9937; [α]20D −22.5° (c 0.2, MeOH).
7 was synthesised from 7e (25 mg, 0.07 mmol) according to the general ester hydrolysis procedure to give 21 mg (93%) of a white solid: tR = 8.73 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 10.69 (br s, 2H), 8.09–8.01 (m, 1H), 7.89–7.82 (m, 2H), 7.72–7.65 (m, 2H), 5.07–4.99 (m, 1H), 3.09–2.93 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.4, 172.3, 166.5, 134.3, 132.5, 130.2, 126.5, 50.3, 36.4; ESI-HRMS calcd for C11H9BrNO5 (M − H)− 313.9670, found 313.9674; [α]20D + 4.0° (c 0.2, MeOH).
(2-(4-Bromophenyl)acetyl)-L-aspartic acid (8)
A dry flask under an argon atmosphere was charged with 4-bromophenylacetic acid (300 mg, 1.4 mmol), 2 (276 mg, 1.4 mmol), DMF (5 mL), N,N-diisopropylethylamine (0.85 mL), 1-hydroxybenzotriazole (256 mg, 1.67 mmol), and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (321 mg, 1.67 mmol). The reaction was stirred overnight at rt. After completion, the solvent was evaporated and the residue was extracted with EtOAc (x3). The organic phases were combined, washed with aqueous 1 M HCl, saturated aqueous NaHCO3, brine, and dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc:petroleum ether, 1:1) to give 317 mg (63%) of dimethyl (2-(4-bromophenyl)acetyl)-L-aspartate (8e) as a white solid: Rf = 0.14 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 2H), 7.19–7.13 (m, 2H), 6.47 (d, J = 7.5 Hz, 1H), 4.86–4.79 (m, 1H), 3.74 (s, 3H), 3.64 (s, 3H), 3.54 (s, 2H), 3.04–2.96 (m, 1H), 2.86–2.78 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.3, 170.9, 170.0, 133.4, 132.0, 131.0, 121.4 52.8, 52.0, 48.7, 42.8, 35.9; ESI-HRMS calcd for C14H17BrNO5 (M + H)+ 358.0285, found 358.0295; [α]20D − 6.0° (c 0.2, MeCN).
8 was synthesised from 8e (50 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 45 mg (98%) of a white solid: tR = 8.88 min (HPLC); 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 7.9 Hz, 1H), 7.54–7.41 (m, 2H), 7.21 (d, J = 8.4 Hz, 2H), 4.55–4.45 (m, 1H), 3.45 (s, 2H), 2.73–2.64 (m, 1H), 2.62–2.53 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 177.5, 176.8, 174.8, 140.9, 136.5, 136.2, 124.7, 54.0, 46.3, 41.3; ESI-HRMS calcd for C12H12BrNNaO5 (M + Na)+ 351.9791, found 351.9775; [α]20D + 3.0° (c 0.2, MeOH).
(2-(4′-Fluoro-[1,1′-biphenyl]-4-yl)acetyl)-L-aspartic acid (9)
Dimethyl (2-(4′-fluoro-[1,1′-biphenyl]−4-yl)acetyl)-L-aspartate (9e) was synthesised from 8e (100 mg, 0.28 mmol) and 4-fluorophenylboronic acid (43 mg, 0.31 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:4) gave 80 mg (77%) of a white solid: Rf = 0.22 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.57–7.49 (m, 4H), 7.34 (d, J = 8.4 Hz, 2H), 7.15–7.08 (m, 2H), 6.51 (d, J = 7.6 Hz, 1H), 4.89–4.82 (m, 1H), 3.73 (s, 3H), 3.65–2.60 (m, 5H), 3.04–2.96 (m, 1H), 2.88–2.80 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.3, 171.0, 170.5, 160.7 (d, J = 292.3 Hz), 139.3, 136.8 (d, J = 3.3 Hz), 133.5, 129.8, 128.6 (d, J = 8.0 Hz), 127.5, 115.7 (d, J = 21.5 Hz), 52.8, 52.0, 48.7, 43.1, 36.0; ESI-HRMS calcd for C20H20FNNaO5 (M + Na)+ 396.1218, found 396.1224; [α]20D − 5.0° (c 0.2, MeCN).
9 was synthesised from 9e (73 mg, 0.20 mmol) according to the general ester hydrolysis procedure to give 65 mg (96%) of a white solid: tR = 10.25 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 7.71–7.65 (m, 2H), 7.58–7.54 (m, 2H), 7.42 (d, J = 8.1 Hz, 2H), 7.25–7.18 (m, 2H), 4.85–4.79 (m, 1H), 3.65 (s, 2H), 2.90–2.86 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 176.6, 176.5, 175.6, 167.6 (d, J = 244.4 Hz), 143.4, 142.3 (d, J = 3.2 Hz), 140.4, 135.0, 133.8 (d, J = 8.1 Hz), 131.9, 120.7 (d, J = 21.6 Hz), 53.9, 47.1, 40.8; ESI-HRMS calcd for C18H15FNO5 (M + H)+ 344.0940, found 344.0949; [α]20D + 14.6° (c 0.2, MeCN).
(5-Phenylfuran-2-carbonyl)-L-aspartic acid (10)
Dimethyl (5-phenylfuran-2-carbonyl)-L-aspartate (10e) was synthesised from 4e (100 mg, 0.30 mmol) and phenylboronic acid (40 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 67 mg (67%) of a clear oil: Rf = 0.25 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.76–7.72 (m, 2H), 7.46–7.33 (m, 4H), 7.22 (d, J = 3.6 Hz, 1H), 6.75 (d, J = 3.6 Hz, 1H), 5.10–5.04 (m, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.20–3.12 (m 1H), 3.03–2.95 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.1, 158.0, 156.0, 146.3, 129.6, 128.9, 128.8, 124.6, 117.2, 107.3, 53.0, 52.1, 48.3, 36.3; ESI-HRMS calcd for C17H18NO6 (M + H)+ 332.1129, found 332.1118; [α]20D − 7.5° (c 0.2, MeCN).
10 was synthesised from 10e (50 mg, 0.15 mmol) according to the general ester hydrolysis procedure to give 44 mg (96%) of a white solid: tR = 9.38 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.08 (d, J = 8.1 Hz, 1H), 7.87–7.80 (m, 2H), 7.49–7.42 (m, 2H), 7.40–7.33 (m, 1H), 7.21 (d, J = 3.6 Hz, 1H), 7.01 (d, J = 3.6 Hz, 1H), 5.09–5.01 (m, 1H), 3.12–2.97 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.4, 172.3, 158.5, 156.3, 148.0, 130.7, 129.8, 129.5, 125.2, 117.1, 108.2, 49.4, 36.5; ESI-HRMS calcd for C15H13NNaO6 (M + Na)+ 326.0635, found 326.0645; [α]20D + 3.9° (c 0.2, MeCN).
(5-(o-Tolyl)furan-2-carbonyl)-L-aspartic acid (11)
Dimethyl (5-(o-tolyl)furan-2-carbonyl)-L-aspartate (11e) was synthesised from 4e (100 mg, 0.30 mmol) and o-tolylboronic acid (45 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 80 mg (77%) of a clear oil: Rf = 0.29 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.71–7.76 (m, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.32–7.27 (m, 3H), 7.25 (d, J = 3.6 Hz, 1H), 6.64 (d, J = 3.6 Hz), 5.09–5.03 (m, 1H), 3.80 (s, 3H), 3.71 (s, 3H), 3.19–3.11 (m, 1H), 3.02–3.94 (m, 1H), 2.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.5, 171.1, 158.0, 155.8, 146.0, 135.4, 131.4, 129.0, 128.8, 127.8, 126.2, 116.6, 110.6, 52.9, 52.1, 48.2, 36.3, 21.7; ESI-HRMS calcd for C18H19NNaO6 (M + Na)+ 368.1105, found 368.1120; [α]20D + 23.7° (c 0.2, DCM).
11 was synthesised from 11e (50 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 45 mg (98%) of a pale yellow solid: tR = 9.85 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 7.99 (d, J = 8.3 Hz, 1H), 7.77–7.73 (m, 1H), 7.34–7.26 (m, 3H), 7.23 (d, J = 3.6 Hz, 1H), 6.83 (d, J = 3.6 Hz, 1H), 5.08–5.01 (m, 1H), 3.05 (d, J = 5.7 Hz, 2H), 2.52 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.3, 158.5, 156.1, 147.7, 136.2, 132.2, 130.0, 129.5, 128.4, 127.1, 116.7, 111.6, 49.2, 36.4, 21.9; ESI-HRMS calcd for C16H14NO6 (M + H)+ 316.0827, found 316.0816; [α]20D + 30.5° (c 0.2, MeCN).
(5-(m-Tolyl)furan-2-carbonyl)-L-aspartic acid (12)
Dimethyl (5-(m-tolyl)furan-2-carbonyl)-L-aspartate (12e) was synthesised from 4e (100 mg, 0.30 mmol) and m-tolylboronic acid (45 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 70 mg (68%) of a clear oil: Rf = 0.23 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.56–7.52 (m, 2H), 7.37 (d, J = 8.1Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.22 (d, J = 3.6 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 6.73 (d, J = 3.6 Hz, 1H), 5.11–5.04 (m, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.20–3.12 (m, 1H), 3.04–2.96 (m, 1H), 2.42 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.1, 158.0, 156.2, 146.2, 138.6, 129.7, 129.5, 128.8, 125.2, 121.9, 117.2, 107.2, 53.0, 52.1, 48.3, 36.3, 21.5; ESI-HRMS calcd for C18H19NaNO6 (M + Na)+ 368.1105, found 368.1103; [α]20D + 19.3° (c 0.2, MeCN).
12 was synthesised from 12e (50 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 46 mg (quant.) of a white solid: tR = 9.88 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.04 (d, J = 8.1 Hz, 1H), 7.68–7.61 (m, 2H), 7.34 (t, J = 7.7 Hz, 1H), 7.22–7.17 (m, 2H), 6.99 (d, J = 3.6 Hz, 1H), 5.08–5.00 (m, 1H), 3.10–2.96 (m, 2H), 2.37 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.34, 172.27, 156.5, 154.2, 148.0, 139.4, 130.7, 130.3, 129.7, 125.8, 122.5, 117.0, 108.1, 49.4, 36.43, 21.4; ESI-HRMS calcd for C16H14NO6 (M + H)+ 316.0827, found 316.0826; [α]20D + 7.0° (c 0.2, MeCN).
(5-(p-Tolyl)furan-2-carbonyl)-L-aspartic acid (13)
Dimethyl (5-(p-tolyl)furan-2-carbonyl)-L-aspartate (13e) was synthesised from 4e (100 mg, 0.30 mmol) and p-tolylboronic acid (45 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 80 mg (77%) of a clear oil: Rf = 0.26 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.66–7.59 (m, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.23 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 3.6 Hz, 1H), 6.69 (d, J = 3.6 Hz, 1H), 5.10–5.03 (m, 1H), 3.81 (s, 3H), 3.71 (s, 3H), 3.19–3.11 (m, 1H), 3.03–2.95 (m, 1H), 2.39 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.1, 158.0, 156.2, 145.9, 138.9, 129.6, 126.9, 124.6, 117.2, 106.6, 52.9, 52.1, 48.3, 36.3, 21.4; ESI-HRMS calcd for C18H19NaNO6 (M + Na)+ 368.1105, found 368.1104; [α]20D + 15.5° (c 0.2, DCM).
13 was synthesised from 13e (50 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 45 mg (98%) of a white solid: tR = 9.89 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 7.74–7.69 (m, 2H), 7.27 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 3.6 Hz, 1H), 6.94 (d, J = 3.6 Hz, 1H), 5.03 (t, J = 5.9 Hz, 1H), 3.11–2.96 (m, 2H), 2.35 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.48, 172.45, 158.5, 156.6, 147.7, 139.6, 130.4, 128.0, 125.3, 117.2, 107.5, 49.3, 36.6, 21.3; ESI-HRMS calcd for C14H16NO6 (M + H)+ 316.0827, found 316.0821; [α]20D + 10.5° (c 0.2, MeCN).
(5-(3-(Hydroxymethyl)phenyl)furan-2-carbonyl)-L-aspartic acid (14)
Dimethyl (5-(3-(hydroxymethyl)phenyl)furan-2-carbonyl)-L-aspartate (14e) was synthesised from 4e (107 mg, 0.32 mmol) and 3-hydroxymethylphenylboronic acid (54 mg, 0.35 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 2:1) gave 55 mg (47%) of a clear oil: Rf = 0.29 (EtOAc:petroleum ether, 2:1); 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.46–7.33 (m, 3H), 7.21 (d, J = 3.6 Hz, 1H), 6.76 (d, J = 3.6 Hz, 1H), 5.09–5.03 (m, 1H), 4.76 (s, 2H), 3.81 (s, 3H), 3.73 (s, 3H), 3.19–3.11 (m, 1H), 3.03–2.95 (m, 1H), 1.99 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.1, 157.9, 155.8, 146.3, 141.7, 129.8, 129.1, 127.3, 123.8, 123.0, 117.2, 107.5, 65.0, 52.9, 52.1, 48.3, 36.2; ESI-HRMS calcd for C18H20NO7 (M + H)+ 362.1234, found 362.1246; [α]20D − 8.0° (c 0.2, MeCN).
14 was synthesised from 14e (50 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 44 mg (95%) of a white solid: tR = 8.04 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.11 (d, J = 8.3 Hz, 1H), 7.82 (s, 1H), 7.70 (d, J = 7.5 Hz, 1H), 7.45–7.33 (m, 2H), 7.21 (d, J = 3.6 Hz, 1H), 7.00 (d, J = 3.6 Hz, 1H), 5.09–5.01 (m, 1H), 4.68 (s, 2H), 3.10–2.97 (m, 2H), 1.31–1.25 (m, 1H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.4, 158.6, 156.6, 147.9, 144.2, 130.6, 129.7, 127.8, 123.8, 123.4, 117.2, 108.2, 64.5, 49.4, 36.5; ESI-HRMS calcd for C16H14NO7 (M + H)+ 332.0776, found 332.0760; [α]20D + 14.5° (c 0.2, MeCN).
(5-(4-Hydroxyphenyl)furan-2-carbonyl)-L-aspartic acid (15)
Dimethyl (5-(4-hydroxyphenyl)furan-2-carbonyl)-L-aspartate (15e) was synthesised from 4e (100 mg, 0.30 mmol) and 4-hydroxyphenylboronic acid (45 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:2) gave 30 mg (29%) of a clear oil: Rf = 0.23 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.64–7.56 (m, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H), 6.94–6.87 (m, 2H), 6.59 (d, J = 3.6 Hz, 1H), 5.79 (s, 1H), 5.10–5.04 (m, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.20–3.12 (m, 1H), 3.03–2.95 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.7, 171.2, 158.2, 156.6, 156.3, 145.5, 126.4, 122.5, 117.5, 115.9, 105.8, 53.0, 52.1, 48.3, 36.3; ESI-HRMS calcd for C17H18NO7 (M + H)+ 348.1078, found 348.1083; [α]20D − 3.5° (c 0.2, MeCN).
15 was synthesised from 15e (27 mg, 0.08 mmol) according to the general ester hydrolysis procedure to give 24 mg (97%) of a pale brown solid: tR = 8.15 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 7.99 (d, J = 8.2 Hz, 1H), 7.71–7.64 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 6.94–6.88 (m, 2H), 6.80 (d, J = 3.6 Hz, 1H), 5.07–4.99 (m, 1H), 3.09–2.97 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.41, 172.36, 159.0, 158.6, 156.9, 147.1, 127.0, 122.5, 117.2, 116.7, 106.1, 49.3, 36.5; ESI-HRMS calcd for C15H12NO7 (M + H)+ 318.0619, found 318.0615; [α]20D + 5.4° (c 0.2, MeCN).
(5-(2-Methoxyphenyl)furan-2-carbonyl)-L-aspartic acid (16)
Dimethyl (5-(2-methoxyphenyl)furan-2-carbonyl)-L-aspartate (16e) was synthesised from 4e (100 mg, 0.30 mmol) and o-methoxyphenylboronic acid (40 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 61 mg (56%) of a clear oil: Rf = 0.26 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.89 (dd, J = 7.8, 1.7 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.33 (ddd, J = 8.4, 7.4, 1.7 Hz, 1H), 7.23 (d, J = 3.6 Hz, 1H), 7.07 (td, J = 7.7, 1.0 Hz, 1H), 7.03 (d, J = 3.6 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 5.12–5.01 (m, 1H), 3.96 (s, 3H), 3.81 (s, 3H), 3.73 (s, 3H), 3.20–3.12 (m, 1H), 3.03–2.95 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.2, 158.1, 156.3, 152.5, 145.2, 129.6, 126.6, 120.9, 118.6, 117.2, 112.1, 111.2, 55.5, 52.9, 52.1, 48.3, 36.3; ESI-HRMS calcd for C18H20NO7 (M + H)+ 362.1234, found 362.1249; [α]20D − 5.9° (c 0.2, MeCN).
16 was synthesised from 16e (49 mg, 0.13 mmol) according to the general ester hydrolysis procedure to give 40 mg (89%) of a white solid: tR = 9.59 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (dd, J = 7.8, 1.7 Hz, 1H), 7.35 (ddd, J = 8.4, 7.4, 1.7 Hz, 1H), 7.19 (d, J = 3.6 Hz, 1H), 7.14 (d, J = 8.4, 0.8 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 7.04 (td, J = 7.7, 1.1 Hz, 1H), 5.08–5.02 (m, 1H), 3.99 (s, 3H), 3.10–2.98 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.4, 158.6, 157.3, 152.9, 146.9, 130.5, 127.0, 121.5, 119.4, 117.1, 112.8, 112.5, 55.9, 49.4, 36.5; ESI-HRMS calcd for C16H15NNaO7 (M + Na)+ 356.0741, found 356.0743; [α]20D + 3.0° (c 0.2, MeCN).
(5-(4-Methoxyphenyl)furan-2-carbonyl)-L-aspartic acid (17)
Dimethyl (5-(4-methoxyphenyl)furan-2-carbonyl)-L-aspartate (17e) was synthesised from 4e (100 mg, 0.30 mmol) and p-methoxyphenylboronic acid (40 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 89 mg (82%) of a clear oil: Rf = 0.20 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.69–7.64 (m, 2H), 7.35 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 3.60 Hz, 1H), 6.99–6.92 (m, 2H), 6.61 (d, J = 3.6 Hz, 1H), 5.09–5.03 (m, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.71 (s, 3H), 3.19–3.11 (m, 1H), 3.02–2.94 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.2, 160.2, 158.0, 156.1, 145.7, 126.2, 122.5, 117.3, 114.4, 105.8, 55.4, 52.9, 52.1, 48.2, 36.3; ESI-HRMS calcd for C18H20NO7 (M + H)+ 362.1234, found 362.1249, [α]20D − 6.0° (c 0.2, MeCN).
17 was synthesised from 17e (75 mg, 0.21 mmol) according to the general ester hydrolysis procedure to give 66 mg (96%) of a white solid: tR = 9.48 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.05 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 3.6 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 3.6 Hz, 1H), 5.09–5.00 (m, 1H), 3.83 (s, 3H), 3.11–2.98 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.4, 161.2, 158.7, 156.7, 147.3, 126.9, 123.5, 117.4, 115.3, 106.6, 55.8, 49.4, 36.5; ESI-HRMS calcd for C16H15NNaO7 (M + Na)+ 356.0741, found 356.0755; [α]20D + 2.5° (c 0.2, MeCN).
(5-(4-(Trifluoromethyl)phenyl)furan-2-carbonyl)-L-aspartic acid (18)
Dimethyl (5-(4-(trifluoromethyl)phenyl)furan-2-carbonyl)-L-aspartate (18e) was synthesised from 4e (100 mg, 0.30 mmol) and 4-trifluoromethylphenylboronic acid (63 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 67 mg (56%) of a clear oil: Rf = 0.59 (EtOAc:petroleum ether, 2:1); 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 6.86 (d, J = 3.6 Hz, 1H), 5.09–5.03 (m, 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.21–3.13 (m, 1H), 3.03–2.95 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.0, 157.7, 154.2, 147.2, 132.7, 130.5 (q, J = 32.7 Hz), 126.0 (q, J = 3.8 Hz), 125.3, 124.7, 117.1, 109.1, 53.0, 52.2, 48.3, 36.2; ESI-HRMS calcd for C18H16F3NaNO6 (M + Na)+ 422.0822, found 422.0832; [α]20D − 5.0° (c 0.2, MeCN).
18 was synthesised from 18e (50 mg, 0.13 mmol) according to the general ester hydrolysis procedure to give 46 mg (99%) of a white solid: tR = 10.73 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.19 (d, J = 8.2 Hz, 1H), 8.02 (t, J = 9.7 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 7.26 (d, J = 3.6 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H), 5.10–5.01 (m, 1H), 3.08–3.00 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.4, 172.3, 158.4, 154.6, 148.9, 134.2, 130.4 (q, J = 32.3 Hz), 126.8 (q, J = 3.8 Hz), 125.7, 117.2, 110.5, 49.5, 36.5; ESI-HRMS calcd for C16H11F3NO6 (M + H)+ 370.0544, found 370.0528; [α]20D + 3.5° (c 0.2, MeCN).
(5-(4-Cyanophenyl)furan-2-carbonyl)-L-aspartic acid (19)
Dimethyl (5-(4-cyanophenyl)furan-2-carbonyl)-L-aspartate (19e) was synthesised from 4e (100 mg, 0.30 mmol) and 4-cyanophenylboronic acid (48 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 70 mg (66%) of a clear oil: Rf = 0.21 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.85–7.79 (m, 2H), 7.75–7.69 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 3.7 Hz, 1H), 6.90 (d, J = 3.7 Hz, 1H), 5.08–5.02 (m, 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.21–3.13 (m, 1H), 3.03–2.95 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 170.9, 157.5, 153.6, 147.6, 133.3, 132.8, 124.9, 118.5, 117.1, 112.0, 110.0, 53.0, 52.2, 48.3, 36.1; ESI-HRMS calcd for C18H16N2NaO6 (M + Na)+ 379.0901, found 379.0892; [α]D20 – 9.5° (c 0.2, MeCN).
19 was synthesised from 19e (57 mg, 0.16 mmol) according to the general ester hydrolysis procedure to give 50 mg (95%) of a pale yellow solid: tR = 9.16 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.18 (d, J = 8.2 Hz, 1H), 8.06–8.12 (m, 2H), 7.89–7.85 (m, 2H), 7.27 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 5.06–5.00 (m, 1H), 3.07–2.98 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.3, 172.2, 158.2, 154.2, 145.9, 134.5, 133.7, 125.7, 119.1, 117.2, 112.4, 111.2, 49.5, 36.4; ESI-HRMS calcd for C16H11N2O6 (M + H)+ 327.0623, found 327.0628; [α]D20 + 15.8° (c 0.2, MeCN).
(5-(4-(Trifluoromethoxy)phenyl)furan-2-carbonyl)-L-aspartic acid (20)
Dimethyl (5-(4-(trifluoromethoxy)phenyl)furan-2-carbonyl)-L-aspartate (20e) was synthesised from 4e (100 mg, 0.30 mmol) and 4-trifluoromethoxyphenylboronic acid (68 mg, 0.33 mmol) according to the general Suzuki coupling procedure. Purification by flash chromatography (EtOAc:petroleum ether, 1:3) gave 54 mg (43%) of a clear oil: Rf = 0.32 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.78–7.73 (m, 2H), 7.40 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 3.4 Hz, 1H), 6.75 (d, J = 3.6 Hz, 1H), 5.09–5.03 (m, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.20–3.12 (m, 1H), 3.02–2.94 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 171.6, 171.1, 157.8, 154.6, 149.4, 146.7, 135.5, 128.3, 126.1, 121.4, 117.2, 107.9, 53.0, 52.1, 48.3, 36.2; ESI-HRMS calcd for C18H16F3NNaO7 (M + Na)+ 438.0771, found 438.0782; [α]20D − 8.0° (c 0.2, MeCN).
20 was synthesised from 20e (46 mg, 0.11 mmol) according to the general ester hydrolysis procedure to give 41 mg (95%) of a white solid: tR = 10.88 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.00–7.92 (m, 2H), 7.46–7.38 (m, 2H), 7.22 (d, J = 3.6 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 5.03 (t, J = 5.9 Hz, 1H), 3.07–2.99 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.39, 172.36, 158.4, 154.8, 149.8, 148.5, 129.9, 127.1, 122.4, 117.2, 109.1, 49.4, 36.5; ESI-HRMS calcd for C16H11F3NO7 (M + H)+ 386.0493, found 386.050; [α]20D + 6.5° (c 0.2, MeCN).
(5-(4-Ethoxyphenyl)furan-2-carbonyl)-L-aspartic acid (21)
Dimethyl (5-(4-ethoxyphenyl)furan-2-carbonyl)-L-aspartate (21e) was synthesised from 15e (61 mg, 0.18 mmol) and ethyl iodide (50 µL, 0.62 mmol) according to the general alkylation procedure. After one day of stirring, additional ethyl iodide (50 µL, 0.62 mmol) was added. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 55 mg (95%) of a pale yellow solid: Rf = 0.22 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.09–8.02 (m, 1H), 7.78–7.73 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 7.03–6.98 (m, 2H), 6.85 (d, J = 3.6 Hz, 1H), 5.07–4.99 (m, 1H), 4.10 (q, J = 7.0 Hz, 2H), 3.71 (s, 3H), 3.67 (s, 3H), 3.09–2.91 (m, 3H), 1.38 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.7, 160.6, 158.4, 156.7, 147.3, 126.9, 123.4, 117.3, 115.8, 106.5, 64.3, 52.8, 52.1, 49.6, 36.7, 15.0; ESI-HRMS calcd for C19H21NNaO7 (M + Na)+ 398.1210, found 398.1223; [α]20D − 4.5° (c 0.2, MeOH).
21 was synthesised from 21e (44 mg, 0.12 mmol) according to the general ester hydrolysis procedure to give 37 mg (89%) of a pale yellow foam: tR = 10.01 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 10.93 (br s, 2H), 8.05–7.97 (m, 1H), 7.80–7.71 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 7.04–6.97 (m, 2H), 6.85 (d, J = 3.6 Hz, 1H), 5.08–5.00 (m, 1H), 4.10 (q, J = 7.0 Hz, 2H), 3.12–2.96 (m, 2H), 1.38 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.42, 172.38, 160.6, 158.6, 156.6, 147.4, 126.9, 123.4, 117.2, 115.8, 106.5, 64.3, 49.4, 36.5, 15.1; ESI-HRMS calcd for C17H16NO7 (M − H)− 346.0932, found 346.0945; [α]20D + 6.5° (c 0.2, MeOH).
(5-(4-Isopropoxyphenyl)furan-2-carbonyl)-L-aspartic acid (22)
Dimethyl (5-(4-isopropoxyphenyl)furan-2-carbonyl)-L-aspartate (22e) was synthesised from 15e (60 mg, 0.17 mmol) and 2-bromopropane (50 µL, 0.53 mmol) according to the general alkylation procedure. In addition, KI (6 mg, 0.03 mmol) was added to promote the reaction. After one day of stirring, additional 2-bromopropane (50 µL, 0.53 mmol) was added. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 11 mg (16%) of a pale yellow solid: Rf = 0.25 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.08–8.00 (m, 1H), 7.77–7.71 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 7.03–6.96 (m, 2H), 6.85 (d, J = 3.6 Hz, 1H), 5.08–4.98 (m, 1H), 4.74–4.64 (m, 1H), 3.71 (s, 3H), 3.67 (s, 3H), 3.09–2.91 (m, 2H), 1.32 (d, J = 6.0 Hz, 6H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.7, 159.5, 158.5, 156.7, 147.2, 126.9, 123.2, 117.3, 116.9, 106.5, 70.6, 52.8, 52.1, 49.6, 36.7, 22.3; ESI-HRMS calcd for C20H23NNaO7 (M + Na)+ 412.1367, found 412.1387.
22 was synthesised from 22e (11 mg, 0.03 mmol) according to the general ester hydrolysis procedure to give 9 mg (86%) of a pale yellow solid: tR = 10.50 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.19 (br s, 2H), 8.02–7.97 (m, 1H), 7.78–7.71 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 7.03–6.96 (m, 2H), 6.85 (d, J = 3.6 Hz, 1H), 5.09–4.98 (m, 1H), 4.74–4.63 (m, 1H), 3.10–2.95 (m, 2H), 1.32 (d, J = 6.0 Hz, 6H); 13C NMR (101 MHz, Acetone-d6) δ 172.41, 172.38, 159.5, 158.6, 156.7, 147.4, 126.9, 123.3, 117.2, 116.9, 106.5, 70.6, 49.4, 36.5, 22.3; ESI-HRMS calcd for C18H18NO7 (M − H)− 360.1089, found 360.1103; [α]20D + 57.0° (c 0.2, MeOH).
(5-(4-((3-Methyloxetan-3-yl)methoxy)phenyl)furan-2-carbonyl)-L-aspartic acid (23)
Dimethyl (5-(4-((3-methyloxetan-3-yl)methoxy)phenyl)furan-2-carbonyl)-L-aspartate (23e) was synthesised from 15e (55 mg, 0.35 mmol) and (3-methyloxetan-3-yl)methyl 4-methylbenzenesulfonate (89 mg, 0.53 mmol) according to the general alkylation procedure. In addition, KI (10 mg, 0.06 mmol) was added to promote the reaction. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 38 mg (55%) of a yellow solid: Rf = 0.17 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.10–8.03 (m, 1H), 7.82–7.75 (m, 2H), 7.17 (d, J = 3.6 Hz, 1H), 7.12–7.06 (m, 2H), 6.88 (d, J = 3.6 Hz, 1H), 5.09–4.98 (m, 1H), 4.55 (d, J = 5.8 Hz, 2H), 4.35 (d, J = 5.8 Hz, 2H), 4.16 (s, 2H), 3.71 (s, 3H), 3.67 (s, 3H), 3.09–2.89 (m, 2H), 1.43 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 171.9, 171.7, 160.8, 158.4, 156.6, 147.4, 126.9, 123.8, 117.3, 116.0, 106.7, 79.7, 73.9, 52.8, 52.1, 49.6, 40.4, 36.7, 21.5; ESI-HRMS calcd for C22H26NO8 (M + H)+ 432.1653, found 432.1668; [α]20D + 0.5° (c 0.2, MeOH).
23 was synthesised from 23e (30 mg, 0.07 mmol) according to the general ester hydrolysis procedure to give 26 mg (92%) of a pale yellow foam: tR = 9.55 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 10.93 (br s, 2H), 8.06–7.98 (m, 1H), 7.83–7.75 (m, 2H), 7.17 (d, J = 3.6 Hz, 1H), 7.13–7.05 (m, 2H), 6.88 (d, J = 3.6 Hz, 1H), 5.07–5.00 (m, 1H), 4.56 (d, J = 5.8 Hz, 2H), 4.35 (d, J = 5.8 Hz, 2H), 4.15 (s, 2H), 3.12–2.97 (m, 2H), 1.43 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.41, 172.38, 160.8, 158.6, 156.5, 147.5, 126.9, 123.8, 117.2, 116.0, 106.7, 79.7, 73.8, 49.4, 40.4, 36.5, 21.5; ESI-HRMS calcd for C20H20NO8 (M − H)− 402.1194, found 402.1197; [α]20D + 10.0° (c 0.2, MeOH).
(5-(4-(3-(Methylsulfonyl)propoxy)phenyl)furan-2-carbonyl)-L-aspartic acid (24)
Dimethyl (5-(4-(3-(methylsulfonyl)propoxy)phenyl)furan-2-carbonyl)-L-aspartate (24e) was synthesised from 15e (40 mg, 0.12 mmol) and 3-(methylsulfonyl)propyl 4-methylbenzenesulfonate (69 mg, 0.24 mmol) according to the general alkylation procedure. In addition, KI (16 mg, 0.09 mmol) was added to promote the reaction. Purification by flash column chromatography (EtOAc:petroleum ether, 2:1) gave 35 mg (65%) of a white solid: Rf = 0.11 (EtOAc:petroleum ether, 2:1); 1H NMR (400 MHz, Acetone-d6) δ 8.09–8.01 (m, 1H), 7.80–7.74 (m, 2H), 7.16 (d, J = 3.6 Hz, 1H), 7.08–7.02 (m, 2H), 6.87 (d, J = 3.6 Hz, 1H), 5.07–4.99 (m, 1H), 4.23 (t, J = 6.1 Hz, 2H), 3.71 (s, 3H), 3.67 (s, 3H), 3.35–3.27 (m, 2H), 3.07–2.92 (m, 2H), 2.99 (s, 3H), 2.35–2.25 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 171.9, 171.7, 160.2, 158.4, 156.5, 147.4, 126.9, 123.8, 117.3, 115.9, 106.7, 66.9, 52.8, 52.1, 51.9, 49.6, 40.8, 36.7, 23.5; ESI-HRMS calcd for C21H26NO9S (M + H)+ 468.1323, found 468.1339; [α]20D − 9.0° (c 0.2, MeOH).
24 was synthesised from 24e (25 mg, 0.05 mmol) according to the general ester hydrolysis procedure to give 24 mg (99%) of a white solid: tR = 8.76 min (HPLC); 1H NMR (400 MHz, DMSO-d6) δ 12.66 (br s, 2H), 8.72–8.64 (m, 1H), 7.90–7.81 (m, 2H), 7.18 (d, J = 3.6 Hz, 1H), 7.05 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 3.6 Hz, 1H), 4.82–4.73 (m, 1 H), 4.15 (t, J = 6.2 Hz, 2H), 3.38–3.25 (m, 2H), 3.03 (s, 3H), 2.94–2.68 (m, 2H), 2.24–2.11 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 172.3, 171.8, 158.6, 157.4, 154.8, 145.9 126.0, 122.3, 116.3, 114.9, 106.0, 65.8, 50.5, 48.5, 40.2, 35.8, 21.9; ESI-HRMS calcd for C19H20NO9S (M − H)− 438.0864, found 438.0871; [α]20D + 15.0° (c 0.2, MeOH).
(4′-Fluoro-[1,1′-biphenyl]-3-carbonyl)-L-aspartic acid (25)
Dimethyl (4′-fluoro-[1,1′-biphenyl]-3-carbonyl)-L-aspartate (25e) was synthesised from (4-fluorophenyl)boronic acid (36 mg, 0.26 mmol) and 6e (79 mg, 0.20 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 25 mg (35%) of a pale yellow sticky oil: Rf = 0.36 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.22–8.15 (m, 1H), 8.15–8.12 (m, 1H), 7.91–7.86 (m, 1H), 7.84–7.80 (m, 1H), 7.77–7.70 (m, 2H), 7.57 (t, J = 7.8 Hz, 1H), 7.30–7.22 (m, 2H), 5.10–5.01 (m, 1H), 3.71 (s, 3H), 3.66 (s, 3H), 3.08–2.90 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.6, 167.2, 164.9, 141.1, 137.5 (d, J = 3.4 Hz), 135.8, 130.8, 130.0, 129.9 (d, J = 8.2 Hz), 127.2, 126.6, 116.6 (d, J = 21.6 Hz), 52.7, 52.1, 50.5, 36.7; ESI-HRMS calcd for C19H18FNNaO5 (M + Na)+ 382.1061, found 382.1053.
25 was synthesised from 25e (25 mg, 0.07 mmol) according to the general ester hydrolysis procedure to give 20 mg (87%) of a white foam: tR = 10.03 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 8.18–8.10 (m, 2H), 7.89 (dd, J = 7.7, 1.1 Hz, 1H), 7.85–7.79 (m, 1H), 7.78–7.71 (m, 2H), 7.61–7.54 (m, 1H), 7.31–7.20 (m, 1H), 5.11–5.02 (m, 1H), 3.10–2.94 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.3, 167.3, 163.6 (d, J = 245.2 Hz), 141.1, 137.5 (d, J = 3.1 Hz), 135.9, 130.8, 130.0, 129.9 (d, J = 8.2 Hz), 127.2, 126.6, 116.6 (d, J = 21.7 Hz), 50.3, 36.5; ESI-HRMS calcd for C17H13FNO5 (M − H)− 330.0783, found 330.0796; [α]20D + 9.0° (c 0.2, MeOH).
(4′-Fluoro-[1,1′-biphenyl]-4-carbonyl)-L-aspartic acid (26)
Dimethyl (4′-fluoro-[1,1′-biphenyl]-4-carbonyl)-L-aspartate (26e) was synthesised from (4-fluorophenyl)boronic acid (37 mg, 0.26 mmol) and 7e (81 mg, 0.34 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 71 mg (84%) of a white solid: Rf = 0.31 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.10–8.04 (m, 1H), 8.00–7.95 (m, 2H), 7.80–7.72 (m, 4H), 7.30–7.22 (m, 2H), 5.09–5.02 (m, 1H), 3.72 (s, 3H), 3.67 (s, 3H), 3.08–2.93 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.1, 171.7, 166.9, 163.8 (d, J = 245.6 Hz), 143.9, 137.2 (d, J = 3.1 Hz), 133.9, 129.9 (d, J = 8.2 Hz), 128.9, 127.7, 116.6 (d, J = 21.7 Hz), 52.7, 52.1, 50.4, 36.7; ESI-HRMS calcd for C19H18FNNaO5 (M+Na)+ 382.1061, found 382.1056; [α]20D − 27.0° (c 0.2, MeOH).
26 was synthesised from 26e (56 mg, 0.15 mmol) according to the general ester hydrolysis procedure to give 48 mg (94%) of a white solid: tR = 10.02 min (HPLC); 1H NMR (400 MHz, DMSO-d6) δ 12.58 (br s, 2H), 8.83–8.75 (m, 1H), 8.02–7.92 (m, 2H), 7.85–7.75 (m, 4H), 7.39–7.28 (m, 2H), 4.83–4.74 (m, 1H), 2.93–2.69 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 172.5, 171.7, 165.6, 161.0, 141.8, 135.6 (d, J = 3.1 Hz), 132.6, 128.9 (d, J = 8.3 Hz), 128.0, 126.4, 115.8 (d, J = 21.4 Hz), 49.4, 35.8; ESI-HRMS calcd for C17H13FNO5 (M − H)− 330.0783, found 330.0768; [α]20D + 1.0° (c 0.2, MeOH).
(6-(4-Fluorophenyl)picolinoyl)-L-aspartic acid (27)
Dimethyl (6-(4-fluorophenyl)picolinoyl)-L-aspartate (27e) was synthesised from (4-fluorophenyl)boronic acid (40 mg, 0.28 mmol) and dimethyl (6-bromopicolinoyl)-L-aspartate (88 mg, 0.26 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 81 mg (88%) of a pale yellow sticky oil: Rf = 0.62 (EtOAc); 1H NMR (400 MHz, Acetone-d6) δ 9.18–9.11 (m, 1H), 8.29–8.23 (m, 2H), 8.18–8.13 (m, 1H), 8.12–8.07 (m, 2H), 7.35–7.26 (m, 2H), 5.13–5.04 (m, 1H), 3.74 (s, 3H), 3.69 (s, 3H), 3.11–3.04 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.9, 164.7 (d, J = 247.5 Hz), 164.5, 155.7, 150.5, 139.8, 135.5 (d, J = 3.1 Hz), 130.0 (d, J = 8.5 Hz), 123.8, 121.4, 116.5 (d, J = 21.9 Hz), 52.9, 52.2, 49.7, 36.8; ESI-HRMS calcd for C18H17FN2NaO5 (M + Na)+ 383.1014, found 383.1011; [α]20D + 4.5° (c 0.2, MeOH).
27 was synthesised from 27e (70 mg, 0.19 mmol) according to the general ester hydrolysis procedure to give 57 mg (88%) of a pale yellow solid: tR = 9.77 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.25 (br s, 2H), 9.27–9.16 (m, 1H), 8.35–8.21 (m, 2H), 8.21–8.07 (m, 3H), 7.34–7.22 (m, 2H), 5.13–5.03 (m, 1H), 3.19–3.01 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.6, 172.3, 164.7 (d, J = 247.3 Hz), 164.5, 155.6, 150.6, 139.7, 135.5 (d, J = 3.2 Hz), 129.9 (d, J = 8.5 Hz), 123.7, 121.4, 116.5 (d, J = 21.8 Hz), 49.5, 36.5; ESI-HRMS calcd for C16H12FN2O5 (M − H)− 331.0736, found 331.0745; [α]20D + 19.0° (c 0.2, MeOH).
(4′-Methoxy-[1,1′-biphenyl]-3-carbonyl)-L-aspartic acid (28)
Dimethyl (4′-methoxy-[1,1′-biphenyl]-3-carbonyl)-L-aspartate (28a) was synthesised from (4-methoxyphenyl)boronic acid (43 mg, 0.28 mmol) and 7e (88 mg, 0.26 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 72 mg (75%) of a pale yellow sticky oil: Rf = 0.29 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.20–8.14 (m, 1H), 8.13–8.09 (m, 1H), 7.85–7.76 (m, 2H), 7.67–7.60 (m, 2H), 7.57–7.50 (m, 1H), 7.08–7.02 (m, 2H), 5.11–5.01 (m, 1H), 3.85 (s, 3H), 3.71 (s, 3H), 3.66 (s, 3H), 3.09–2.90 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.1, 171.6, 167.3, 160.7, 141.9, 135.7, 133.4, 130.4, 129.9, 129.0, 126.5, 126.2, 115.3, 55.7, 52.7, 52.1, 50.5, 36.7; ESI-HRMS calcd for C20H21NNaO6 (M + Na)+ 394.1261, found 394.1268; [α]20D − 23.5° (c 0.2, MeOH).
28 was synthesised from 28e (64 mg, 0.17 mmol) according to the general ester hydrolysis procedure to give 57 mg (97%) of a pale yellow solid: tR = 9.92 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.15 (br s, 2H), 8.16–8.08 (m, 2H), 7.87–7.82 (m, 1H), 7.82–7.76 (m, 1H), 7.68–7.61 (m, 2H), 7.56–7.50 (m, 1H), 7.09–7.01 (m, 2H), 5.12–5.02 (m, 1H), 3.85 (s, 3H), 3.13–2.95 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.3, 167.5, 160.7, 141.9, 135.8, 133.4, 130.3, 129.9, 129.0, 126.4, 126.2, 115.3, 55.7, 50.3, 36.5; ESI-HRMS calcd for C18H16NO6 (M − H)− 342.0983, found 342.0989; [α]20D − 1.5° (c 0.2, MeOH).
(6-(4-Methoxyphenyl)picolinoyl)-L-aspartic acid (29)
Dimethyl (6-(4-methoxyphenyl)picolinoyl)-L-aspartate (29e) was synthesised from (4-methoxyphenyl)boronic acid (42 mg, 0.28 mmol) and dimethyl (6-bromopicolinoyl)-L-aspartate (86 mg, 0.25 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:1) gave 63 mg (67%) of a milky sticky oil: Rf = 0.25 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 9.20–9.11 (m, 1H), 8.20–8.13 (m, 2H), 8.11–7.98 (m, 3H), 7.12–7.04 (m, 2H), 5.12–5.04 (m, 1H), 3.89 (s, 3H), 3.74 (s, 3H), 3.69 (s, 3H), 3.13–3.01 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 171.94, 171.93, 164.6, 162.2, 156.5, 150.3, 139.4, 131.4, 129.2, 123.1, 120.6, 115.1, 55.8, 52.8, 52.2, 49.7, 36.8; ESI-HRMS calcd for C19H20N2NaO6 (M + Na)+ 395.1214, found 395.1217; [α]20D + 4.0° (c 0.2, MeOH).
29 was synthesised from 29e (54 mg, 0.14 mmol) according to the general ester hydrolysis procedure to give 48 mg (96%) of a pale yellow foam: tR = 9.69 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.26 (br s, 2H), 9.29–9.16 (m, 1H), 8.22–8.13 (m, 2H), 8.11–7.99 (m, 3H), 7.12–7.00 (m, 2H), 5.13–5.01 (m, 1H), 3.88 (s, 3H), 3.24–3.01 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.7, 172.4, 164.7, 162.1, 156.4, 150.4, 139.4, 131.5, 129.1, 123.0, 120.6, 115.1, 55.8, 49.4, 36.6; ESI-HRMS calcd for C17H15N2O6 (M − H)− 343.0936, found 343.0932; [α]20D + 18.0° (c 0.2, MeOH).
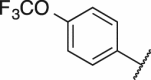
(4′-(Trifluoromethoxy)-[1,1′-biphenyl]-3-carbonyl)-L-aspartic acid (30)
Dimethyl (4′-(trifluoromethoxy)-[1,1′-biphenyl]-3-carbonyl)-L-aspartate (30e) was synthesised from (4-(trifluoromethoxy)phenyl)boronic acid (58 mg, 0.28 mmol) and 7e (87 mg, 0.25 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 2:3) gave 77 mg (72%) of a pale yellow solid: Rf = 0.40 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.23–8.18 (m, 1H), 8.18–8.15 (m, 1H), 7.94–7.90 (m, 1H), 7.89–7.85 (m, 1H), 7.85–7.80 (m, 2H), 7.63–7.57 (m, 1H), 7.49–7.43 (m, 2H), 5.11–5.02 (m, 1H), 3.72 (s, 3H), 3.66 (s, 3H), 3.09–2.91 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.6, 167.1, 140.7, 140.4, 135.9, 131.0, 130.1, 129.7, 127.6, 126.8, 122.4, 52.7, 52.1, 50.5, 36.7; ESI-HRMS calcd for C20H18F3NNaO6 (M + Na)+ 448.0978, found 448.0981; [α]20D − 22.5° (c 0.2, MeOH).
30 was synthesised from 30e (68 mg, 0.16 mmol) according to the general ester hydrolysis procedure to give 60 mg (94%) of a white solid: tR = 11.38 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.21 (br s, 2H), 8.21–8.18 (m, 1H), 8.18–8.11 (m, 1H), 7.97–7.90 (m, 1H), 7.90–7.80 (m, 3H), 7.64–7.57 (m, 1H), 7.49–7.42 (m, 2H), 5.12–5.02 (m, 1H), 3.11–2.94 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.5, 172.3, 167.2, 149.7, 140.7, 140.4, 136.1, 130.9, 130.1, 129.7, 127.6, 126.8, 122.4, 50.4, 36.5; ESI-HRMS calcd for C18H13F3NO6 (M − H)− 396.0700, found 396.0716; [α]20D − 1.0° (c 0.2, MeOH).
(6-(4-(Trifluoromethoxy)phenyl)picolinoyl)-L-aspartic acid (31)
Dimethyl (6-(4-(trifluoromethoxy)phenyl)picolinoyl)-L-aspartate (31e) was synthesised from (4-(trifluoromethoxy)phenyl)boronic acid (60 mg, 0.29 mmol) and dimethyl (6-bromopicolinoyl)-L-aspartate (86 mg, 0.25 mmol) according to the general Suzuki coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:2) gave 76 mg (72%) of a milky sticky oil: Rf = 0.38 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 9.19–9.11 (m, 1H), 8.36–8.31 (m, 2H), 8.23–8.18 (m, 1H), 8.15–8.11 (m, 2H), 7.53–7.47 (m, 2H), 5.13–5.06 (m, 1H), 3.74 (s, 3H), 3.69 (s, 3H), 3.10–3.04 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.0, 171.9, 164.4, 155.3, 151.0, 150.6, 139.9, 138.2, 129.7, 124.2, 122.2, 121.9, 52.9, 52.2, 49.8, 36.7; ESI-HRMS calcd for C19H17F3N2NaO6 (M + Na)+ 449.0931, found 449.0949; [α]20D + 2.0° (c 0.2, MeOH).
31 was synthesised from 31e (65 mg, 0.15 mmol) according to the general ester hydrolysis procedure to give 58 mg (95%) of a white solid: tR = 10.98 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.14 (br s, 2H), 9.25–9.18 (m, 1H), 8.37–8.30 (m, 2H), 8.24–8.17 (m, 1H), 8.17–8.10 (m, 2H), 7.51–7.45 (m, 2H), 5.12–5.03 (m, 1H), 3.21–3.00 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.6, 172.3, 164.4, 155.2, 151.0, 150.7, 139.9, 138.2, 129.7, 124.0, 122.1, 121.8, 49.5, 49.4, 36.5; ESI-HRMS calcd for C17H12F3N2O6 (M − H)− 397.0653, found 397.0665; [α]20D + 14.0° (c 0.2, MeOH).
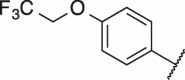
(6-(4-(2,2,2-Trifluoroethoxy)phenyl)picolinoyl)-L-aspartic acid (32)
Diethyl (6-chloropicolinoyl)-L-aspartate (32a) was synthesised from 6-chloropicolinic acid (615 mg, 3.90 mmol) and diethyl L-aspartate hydrochloride (679 mg, 3.01 mmol) according to the general amide coupling procedure. Purification by flash column chromatography (EtOAc:petroleum ether, 1:3) gave 840 mg (85%) of an orange oil: Rf = 0.31 (EtOAc:petroleum ether, 1:1); 1H NMR (400 MHz, Acetone-d6) δ 8.73 (d, J = 7.5 Hz, 1H), 8.13–8.05 (m, 2H), 7.70 (dd, J = 7.4, 1.4 Hz, 1H), 5.07–4.99 (m, 1H), 4.28–4.07 (m, 4H), 3.12–2.96 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 171.2, 171.1, 163.1, 151.2, 150.7, 141.9, 128.3, 122.1, 62.1, 61.3, 49.9, 36.9, 14.5, 14.4.
A Schlenck flask under an argon atmosphere was charged with Pd-XPhos-G4 (8 mg, 0.01 mmol), XPhos (9 mg, 0.02 mmol), diboronic acid (181 mg, 2.02 mmol), and KOAc (198 mg, 2.02 mmol). The flask was evacuated and backfilled with argon, before addition of degassed EtOH (7 mL) and 1-chloro-4-(2,2,2-trifluoroethoxy)benzene (141 mg, 0.67 mmol, prepared according to21). The flask was evacuated and backfilled with argon and stirred at 80 °C until the reaction turned yellow (30 min). Degassed aqueous K2CO3 (1.8 M, 1.1 mL) and 32a (222 mg, 0.67 mmol) dissolved in degassed EtOH (1 mL) were added, and the flask was once again evacuated and backfilled with argon and stirred at 80 °C for 16 hours. After completion, the reaction was cooled to rt, diluted with water and extracted with EtOAc (x3). The organic phases were combined, washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc:petroleum ether:AcOH, 1:2:0→99:0:1) to give 73 mg (25%) of (S)-4-ethoxy-4-oxo-2-(6-(4-(2,2,2-trifluoroethoxy)phenyl)picolinamido)butanoic acid (32b) as a brown sticky solid: Rf = 0.22 (EtOAc + 1% AcOH); 1H NMR (400 MHz, Acetone-d6) δ 9.16 (d, J = 8.5 Hz, 1H), 8.25–8.20 (m, 2H), 8.15–8.09 (m, 1H), 8.09–8.03 (m, 2H), 7.25–7.17 (m, 2H), 5.06 (dt, J = 8.5, 5.3 Hz, 1H), 4.77 (q, J = 8.6 Hz, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.07 (qd, J = 16.7, 5.3 Hz, 2H), 1.22 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.4, 172.0, 171.5, 164.6, 159.6, 156.0, 150.4, 139.6, 133.2, 129.3, 122.1 (d, J = 238.1 Hz), 116.1, 66.2 (d, J = 35.1 Hz), 61.3, 49.6, 37.1, 14.5; ESI-MS m/z 439.1 (M − H)−; [α]20D − 0.5° (c 0.2, MeOH).
32 was synthesised from 32b (40 mg, 0.09 mmol) according to the general ester hydrolysis procedure to give 29 mg (77%) of a white solid: tR = 10.84 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 11.24 (br s, 2H), 9.21 (d, J = 8.4 Hz, 1H), 8.26–8.18 (m, 2H), 8.11 (dd, J = 8.7, 4.2 Hz, 1H), 8.09–8.02 (m, 2H), 7.26–7.14 (m, 2H), 5.11–5.01 (m, 1H), 4.77 (q, J = 8.6 Hz, 2H), 3.21–3.00 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 172.6, 172.4, 164.6, 159.6, 156.0, 150.5, 139.5, 133.2, 129.4, 124.9 (q, J = 276.5 Hz), 123.3, 121.0, 116.1, 66.2 (q, J = 35.1 Hz), 49.5, 36.6; ESI-HRMS calcd for C18H14F3N2O6 (M − H)− 411.0809, found 411.0812; [α]20D + 29.9° (c 0.2, MeOH).
(6-(4-Propoxyphenyl)picolinoyl)-L-aspartic acid (33)
1-Bromo-4-propoxybenzene (33a) was synthesised from 4-bromophenol (250 mg, 1.44 mmol) and 1-iodopropane (0.45 mL, 4.61 mmol) according to the general alkylation procedure. Purification by flash column chromatography (petroleum ether) gave 248 mg (80%) of a clear oil: Rf = 0.62 (EtOAc:petroleum ether, 1:6); 1H NMR (400 MHz, Acetone-d6) δ 7.44–7.38 (m, 2H), 6.92–6.86 (m, 2H), 3.94 (t, J = 6.5 Hz, 2H), 1.83–1.70 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 159.5, 133.0, 117.5, 112.8, 70.4, 23.2, 10.7.
A Schlenck flask under an argon atmosphere was charged with Pd-XPhos-G4 (4 mg, 1 mol%), XPhos (6 mg, 2 mol%), diboronic acid (135 mg, 1.50 mmol), and KOAc (147 mg, 1.50 mmol). The flask was evacuated and backfilled with argon, before addition of degassed EtOH (2.5 mL) and 33a (114 mg, 0.53 mmol). The flask was evacuated and backfilled with argon and stirred at 80 °C until the reaction turned orange (80 min). Degassed aqueous K2CO3 (1.8 M, 0.85 mL) and dimethyl (6-chloropicolinoyl)-L-aspartate (151 mg, 0.50 mmol) dissolved in degassed EtOH (0.25 mL) were added, and the flask was once again evacuated and backfilled with argon and stirred at 80 °C for 3 hours. After completion, the reaction was cooled to rt, diluted with water and extracted with EtOAc (x3). The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography to give a mixture of methyl and ethyl esters of (6-(4-propoxyphenyl)picolinoyl)-L-aspartic acid after trans-esterification with the reaction solvent. The crude was therefore used directly in the hydrolysis following the general procedure to give 27 mg (15% over two steps) of 33 as a sticky pale yellow foam: tR = 11.09 min (HPLC); 1H NMR (400 MHz, Acetone-d6) δ 10.89 (br s, 2H), 9.22 (d, J = 8.4 Hz, 1H), 8.19–8.10 (m, 2H), 8.10–7.99 (m, 3H), 7.09–7.01 (m, 2H), 5.06 (dt, J = 8.6, 5.1 Hz, 1H), 4.04 (t, J = 6.5 Hz, 2H), 3.20–2.97 (m, 2H), 1.88–1.72 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 172.6, 172.4, 164.7, 161.6, 156.5, 150.4, 139.4, 131.3, 129.1, 123.0, 120.5, 115.6, 70.3, 49.4, 36.6, 23.3, 10.7; ESI-HRMS calcd for C19H19N2O6 (M − H)− 371.1249, found 371.1240; [α]20D + 49.5° (c 0.2, MeOH).
Kinetic aqueous solubility
Duplicates of a 200 μM solution of the dicarboxylic acids were prepared from 0.01 M PBS7.4 and a 10 mM stock solution in DMSO. The samples were incubated in an Eppendorf® Thermomixer (25 °C, 800 rpm) for 24 h. Afterwards, the samples were centrifuged for 5 min at 11,000 rpm and the supernatant was filtered (0.45 μm PTFE membrane) before analysis by HPLC. The solubility was calculated based on a concentration-absorption curve.
Chemical stability
Triplicates of a 50 μM solution of the test compounds were prepared from 0.01 M PBS7.4 and a 10 mM stock solution in DMSO. The samples were incubated in an Eppendorf® Thermomixer (37 °C, 650 rpm). The samples were briefly vortexed and 50 μL aliquots were withdrawn at the time points 0 h, 24 h, 48 h etc. and analysed immediately on UPLC. The chemical stability in PBS7.4 was determined at every time point in percentage relative to the 0 h time point.
Stability in simulated gastric and intestinal fluids
FaSSIF, FeSSGF and FaSSGF were prepared in accordance to the manufacturers procedure, Biorelevant.com. Triplicates of a 50 μM solution of the test compounds were prepared from a 10 mM stock solution in DMSO diluted with FaSSGF/FaSSIF/FeSSIF. The samples were incubated in an Eppendorf® Thermomixer (37 °C, 650 rpm). Samples were withdrawn at 0 min and 120 min, centrifuged (10 min at 10,000 rpm) and the supernatant analysed by HPLC/UPLC. The stability was calculated based on peak area of a 0 point sample.
logD7.4 determination
A glass vial with screw cap (8 mL) was charged with test compound (40 μL, 10 mM in DMSO), PBS7.4 (0.01 M, 1980 μL), and 1-octanol (1980 μL). The vial was capped and sealed with parafilm and shaken at 700 rpm using an IKA® KS 125 basic shaker for 24 h at room temperature. The parafilm was removed and the sample was allowed to equilibrate for 1 h before analysis. 100 μL of the octanol phase was withdrawn and diluted 1:10 with MeOH(+0.1% formic acid)/MilliQ water (4:1, v/v) and analysed by HPLC/UPLC. The interface was removed and the PBS7.4 phase analysed directly by HPLC/UPLC. All analysis was performed in duplicates and logD values were calculated from the peak areas (mAU*min) and adjusted for difference in injection volume and concentration-absorption effects from the solvents, using two calibration points per compound per solvent, and dilution of the octanol phase. All compounds were analysed in triplicates.
Metabolic stability
Microsomal stability was studied in mouse liver microsomes (0.5 mg/mL) at a final test compound concentration of 1 μM and performed in triplicates in accordance to the published protocol22. In short: Prewarmed (37 °C) 0.1 M PBS7.4, 10 mM NADPH in PBS7.4 and test compound (1 mM in DMSO) were added to an Eppendorf® Tube. The samples were incubated for 5 min at 37 °C before addition of newly thawned microsomes. The samples were mixed by gentle vortexing and incubated for 1 h at 37 °C, 300 rpm in an Eppendorf® Thermomixer. Samples were quenched by addition of ice-cold MeOH/MeCN (1:1) and centrifuged for 5 min at 10,000 g. The supernatant was transferred to HPLC vials and stored in the freezer until analysis by HPLC/UPLC. The metabolic stability was calculated based on a 0 min sample. All compounds were analysed in triplicates.
Molecular biology, cell culture, and transfection
Receptor constructs for mSUCNR1 and hSUCNR1 were bought from Origene and cloned into the eukaryotic expression vector pCMV-Tag(2B) (Stratagene).
HEK-293 cells were cultured in Dulbecco’s modified Eagle’s medium 1885 (DMEM) supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. Transient transfection of the HEK-293 cells was done with Lipofectamine-2000 according to manufacturer’s protocol. Cells were supplemented with fresh medium after 5 h.
IP3 turnover assay
96-well plates were coated with poly-D-lysine and HEK-293 cells were plated (35.000 cells/well). Cells were transfected for 5 h the following day and subsequently incubated O/N with 0.5 μCi/mL myo[3H]inositol (Perkin Elmer) in 100 μL growth medium. On day 3 cells were washed with 200 µL HBSS/well (Gibco, Life Technologies) followed by a pre-incubation (30 min, 37 °C) with 100 µL HBSS supplemented with 10 mM LiCl. Cells were stimulated with ligand (120 min, 37 °C) and lysed with 50 µL 10 mM formic acid (30 min on ice). In a white 96 well plate 20 µL cell extracts and 80 µL 1:8 diluted YSi scintillation beads (Perkin Elmer) were mixed. The plate was spun down, and a Packard Top Count NXT counter recorded light emission (scintillation) after an 8 h delay.
Automated Ligand-guided Backbone Ensemble Receptor Optimization protocol (ALiBERO)
ALiBERO is an iterative sampling-selection protocol for receptor optimisation that relies on the use of ligand information for selecting the best-performing receptor conformations19. Homology models of the mouse SUCNR1 receptor were constructed according to Trauelsen et al.4 and loaded into ICM (Molsoft L.L.C., San Diego, CA, USA). The structure was converted into an ICM object, thereby assigning protein atom types, optimising hydrogens and His, Pro, Asn, Gly and Cys side-chain conformations. The explored chemical compounds were used as a training set by dividing all compounds into an active (EC50 ≤ 10 µM) and an inactive (EC50 > 10 µM) group, consisting of 25 compounds each. For a list of all compounds used in the optimisation protocol, see Table S1. ALiBERO was performed using the prepared receptor structure and ligand training set as input. Binding site residues were manually selected based on proximity to the position of MRS2500 in the superimposed structure of the P2Y1 receptor (PDB 4XNW). 100 elastic network normal mode analysis derived conformers were built in order to recreate backbone and side-chain flexibility (T = 300 K). Next, a flexible-ligand static-receptor small-scale virtual screening was performed on each of the receptor conformers, from which several pockets were selected for the following generation. The ligand and decoy molecules were docked into mSUCNR1, represented as pre-calculated potential grids and then sorted according to their ICM VLS scores. The maximum number of complementary pockets for each generation was set to 5 with a maximum of 10 generations. Receptor models were selected based on their combined screening performance, as determined by the normalised square root area under the curve, NSQ_AUC. After each round of virtual screening, an all-atom Monte Carlo side-chain refinement was performed to account for induced-by-ligand changes. NSQ_AUC values were calculated according to Katritch et al.16.
The optimised model ensemble was validated in a virtual ligand screening with an external test set, consisting of the 25 active compounds used in the ALiBERO optimisation and 1247 decoy molecules that were selected in a similarity search with a Daylight-type fingerprint threshold of Tc > 0.6 with the active compound 3 as query molecule and subsequent structural clustering with a Tc threshold of 0.15. The test set was docked into the best-performing receptor ensemble from generation 10 of the ALiBERO refinement, using ICM 4D docking. The ROC-plot for the combined performance of the optimised receptor ensemble was computed by taking the best ICL VLS docking score of the receptor models for each docked compound.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
ERU is supported by a fellowship from the Lundbeck Foundation (R181–2014–3247). The NNF Center for Basic Metabolic Research is supported by an unconditional grant (NNF10CC1016515) from the Novo Nordisk Foundation to University of Copenhagen. The work on metabolite receptors including SUCNR1 is further supported by an Immunometabolism grant NNF15CC0018346 from the Novo Nordisk Foundation to Oxford University, University of Copenhagen and Karolinska Instituttet, Stockholm, Sweden.
Author Contributions
E.R.U., M.B. and L.K.I.J. synthesized the compounds. L.Ø.B. performed stability studies. M.T. performed in vitro assays. M.L. and T.M.F. performed molecular modeling. E.R.U., T.W.S. and T.M.F. designed the overall project. E.R.U. and T.M.F. wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-28263-7.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wittenberger T, Schaller HC, Hellebrand S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J. Mol. Biol. 2001;307:799–813. doi: 10.1006/jmbi.2001.4520. [DOI] [PubMed] [Google Scholar]
- 2.He W, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 3.Geubelle P, et al. Identification and pharmacological characterization of succinate receptor agonists. Br. J. Pharmacol. 2017;174:796–808. doi: 10.1111/bph.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trauelsen M, et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol. Metab. 2017;6:1585–1596. doi: 10.1016/j.molmet.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sapieha P, et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 2008;14:1067–1076. doi: 10.1038/nm.1873. [DOI] [PubMed] [Google Scholar]
- 7.Aguiar CJ, et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun Signal. 2014;12:78. doi: 10.1186/s12964-014-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ariza AC, Deen PM, Robben JH. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012;3:22. doi: 10.3389/fendo.2012.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008;9:1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 10.Correa PR, et al. Succinate is a paracrine signal for liver damage. J Hepatol. 2007;47:262–269. doi: 10.1016/j.jhep.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhuniya D, et al. Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg. Med. Chem. Lett. 2011;21:3596–3602. doi: 10.1016/j.bmcl.2011.04.091. [DOI] [PubMed] [Google Scholar]
- 12.Due-Hansen ME, et al. A protocol for amide bond formation with electron deficient amines and sterically hindered substrates. Org. Biomol. Chem. 2016;14:430–433. doi: 10.1039/C5OB02129D. [DOI] [PubMed] [Google Scholar]
- 13.Negoro N, et al. Discovery of TAK-875: A Potent, Selective, and Orally Bioavailable GPR40 Agonist. ACS Med. Chem. Lett. 2010;1:290–294. doi: 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christiansen E, et al. Free fatty acid receptor 1 (FFA1/GPR40) agonists: mesylpropoxy appendage lowers lipophilicity and improves ADME properties. J. Med. Chem. 2012;55:6624–6628. doi: 10.1021/jm3002026. [DOI] [PubMed] [Google Scholar]
- 15.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat. Rev. Drug Discov. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katritch V, Rueda M, Lam PC, Yeager M, Abagyan R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins. 2010;78:197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luckmann M, Holst B, Schwartz TW, Frimurer TM. In Silico Investigation of the Neurotensin Receptor 1 Binding Site: Overlapping Binding Modes for Small Molecule Antagonists and the Endogenous Peptide Agonist. Mol Inform. 2016;35:19–24. doi: 10.1002/minf.201500080. [DOI] [PubMed] [Google Scholar]
- 18.Norn C, et al. Mutation-Guided Unbiased Modeling of the Fat Sensor GPR119 for High-Yield Agonist Screening. Structure. 2015;23:2377–2386. doi: 10.1016/j.str.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Rueda M, Totrov M, Abagyan R. ALiBERO: evolving a team of complementary pocket conformations rather than a single leader. J. Chem. Inf. Model. 2012;52:2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abagyan, R., Totrov, M. & Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem.15, 488–506, 10.1002/jcc.540150503 (1994).
- 21.Gogsig TM, Sobjerg LS, Lindhardt AT, Jensen KL, Skrydstrup T. Direct vinylation and difluorovinylation of arylboronic acids using vinyl- and 2,2-difluorovinyl tosylates via the Suzuki-Miyaura cross coupling. J. Org. Chem. 2008;73:3404–3410. doi: 10.1021/jo7027097. [DOI] [PubMed] [Google Scholar]
- 22.Hill, J. R. In vitro drug metabolism using liver microsomes. Current Protocols in Pharmacology (John Wiley & Sons, Inc. 2001). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.