

Abstract
Biomarkers for α-synuclein are needed for diagnosis and prognosis in Parkinson’s disease (PD). Endogenous auto-antibodies to α-synuclein could serve as biomarkers for underlying synucleinopathy, but previous assessments of auto-antibodies have shown variability and inconsistent clinical correlations. We hypothesized that auto-antibodies to α-synuclein could be diagnostic for PD and explain its clinical heterogeneity. To test this hypothesis, we developed an enzyme-linked immunosorbent assay for measuring α-synuclein auto-antibodies in human samples. We evaluated 69 serum samples (16 healthy controls (HC) and 53 PD patients) and 145 CSF samples (52 HC and 93 PD patients) from our Institution. Both serum and CSF were available for 24 participants. Males had higher auto-antibody levels than females in both fluids. CSF auto-antibody levels were significantly higher in PD patients as compared to HC, whereas serum levels were not significantly different. CSF auto-antibody levels did not associate with amyloid-β1–42, total tau, or phosphorylated tau. CSF auto-antibody levels correlated with performance on the Montreal Cognitive Assessment, even when controlled for CSF amyloidβ1–42. CSF hemoglobin levels, as a proxy for contamination of CSF by blood during lumbar puncture, did not influence these observations. Using recombinant α-synuclein with N- and C-terminal truncations, we found that CSF auto-antibodies target amino acids 100 through 120 of α-synuclein. We conclude that endogenous CSF auto-antibodies are significantly higher in PD patients as compared to HC, suggesting that they could indicate the presence of underlying synucleinopathy. These auto-antibodies associate with poor cognition, independently of CSF amyloidβ1–42., and target a select C-terminal region of α-synuclein.
Keywords: Parkinson’s disease, α-synuclein, biomarker, neurodegeneration, auto-antibody
Graphical Abstract
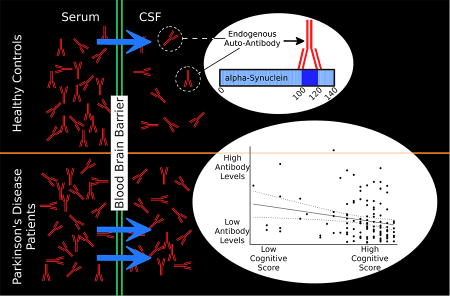
Endogenous auto-antibodies to α-synuclein could be useful biomarkers in Parkinson’s disease (PD), but previous attempts to measure auto-antibodies have been inconclusive. We developed a reliable method to measure α-synuclein auto-antibodies in biofluid samples. We found significantly higher levels of auto-antibodies in the cerebrospinal fluid (CSF) of PD patients than in healthy controls. In the serum, levels of auto-antibodies were similar in both groups. Participants with higher levels of CSF auto-antibodies tended to have worse cognitive performance, as measured by the Montreal Cognitive Assessment. These auto-antibodies recognized the C-terminal domain in α-synuclein. This study suggests that auto-antibodies to α-synuclein could be useful biomarkers for diagnosis in PD and may correlate with clinical disease features.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder affecting motor and cognitive function. PD pathology is thought to begin in the brain long before clinical symptoms appear (Gaig & Tolosa 2009), and most PD patients have already accrued significant neuronal loss by the time they are diagnosed (Fearnley & Lees 1991). For these reasons, new diagnostic biomarkers are critically necessary for earlier and accurate clinical diagnosis.
The protein α-synuclein (α-syn) accumulates as insoluble, misfolded aggregates in neurons of PD patients, which leads to neuronal loss in multiple brain regions, including the substantia nigra (SN) (Lee & Trojanowski 2006). Based on post-mortem studies, α-syn pathology appears to spread spatially and temporally through the brain (Braak et al. 2003). Thus, biomarkers based on α-syn have the potential to address both early detection and prognostication in PD.
Endogenously generated antibody responses to self-antigens, termed auto-antibodies (AAbs), play a role in diagnosis and prognosis of many medical conditions (Hawa et al. 2004). The relevance of neurological disease-associated AAbs can vary from screening for the presence of a disease state (i.e. CSF VDRL test for neurosyphilis) (Kennedy & Creighton 1998) to directly contributing to disease pathogenesis (i.e. CSF neuronal surface antigens in limbic encephalitis) (Irani et al. 2014). In PD brains, α-syn aggregates in the SN appear to co-localize with deposited IgG (Orr et al. 2005), suggesting that pathological α-syn is capable of inducing an in situ AAb response. Further, exogenously applied monoclonal antibodies to α-syn can deter the formation of α-syn aggregates in cellular and animal models of PD (Games et al. 2014, Masliah et al. 2005, Tran et al. 2014), which suggests that endogenous α-syn AAbs have a participatory role in pathogenesis.
There is no single, widely accepted method to measure AAbs to α-syn in patient-derived biological fluid samples. Several studies have used enzyme linked immunosorbent assay (ELISA)-based approaches to measure AAbs to α-syn in serum from PD patients, with mixed results. In two small cohorts, PD patient sera had higher levels of α-syn AAbs than healthy sera, but AAb levels dropped with longer disease duration (Gruden et al. 2011) or with more advanced motor disease (Horvath et al. 2017, Yanamandra et al. 2011). Another study found elevated levels of α-syn AAbs in sera from familial PD but not from sporadic PD (Papachroni et al. 2007). More advanced techniques to measure α-syn AAbs also find higher levels of α-syn AAbs in PD that peak at middle stages of motor impairment (Bryan et al. 2012). In contrast, several other studies found no difference in serum α-syn AAbs between healthy subjects and PD patients (Woulfe et al. 2002, Maetzler et al. 2014) and no association with disease duration or motor disease stage (Besong-Agbo et al. 2013). Fewer studies have examined PD CSF samples for α-syn AAbs. One study did not find elevated α-syn AAbs in PD CSF (Woulfe et al. 2002), whereas another found elevated AAb levels that decreased with advanced motor disease (Horvath et al. 2017).
We hypothesized that endogenous α-syn AAbs could serve as a diagnostic biomarker for PD, and that AAb level could correlate with either disease features or with overall disease severity. Here, we developed a new ELISA method to detect α-syn AAbs in human biological fluids. We assayed serum and CSF samples collected at our Centers wherein patients are followed to assemble a cohort of well-characterized healthy controls and PD patients. We tested hypothetical correlations between AAb level and demographic, clinical, and psychometric characteristics. We also compared CSF AAb levels to amyloid-β1–42 (Aβ1–42), total tau, and phosphorylated tau. Because CSF samples can be contaminated by peripheral blood during the collection procedure, we also measured hemoglobin levels in CSF as a surrogate for blood product contamination and included hemoglobin as a covariate in our analyses. Finally, we also used a panel of recombinant α-syn containing selected truncations as standards to determine the epitope(s) recognized by α-syn AAbs in both PD and control samples.
Methods
Preparation of molecular biology reagents
Recombinant human α-syn was expressed and purified in E. coli BL21(DE3)-RIL Stratagene cells (Agilent, cat. 230245) using a PRK172/HuWT α-syn plasmid vector as previously described.(Giasson et al. 2001, Volpicelli-Daley et al. 2014) Analogous plasmid vectors expressing N-terminal (a.a. 58–140) and C-terminal (a.a. 1–120 and a.a. 1–99) truncations were used to generate truncated α-syn as previously described (Giasson et al. 2000). Aliquots of α-syn were stored at 5 mg/mL at −80°C and thawed on ice immediately before use. Mouse monoclonal antibody Syn 211 (RRID:AB_310817) and affinity-purified rabbit polyclonal antibodies SNL-1 and SNL-4 to α-syn were used as controls to confirm purity of recombinant truncated α-syn given their respective putative epitopes (a.a. 121–125, a.a. 104–119, and a.a. 2–12, respectively), as previously described (Giasson et al. 2000).
Participants
Healthy controls and PD patients were recruited from the Penn Udall Center, the Parkinson’s Disease and Movement Disorder Center, and the Penn Memory Center of the University of Pennsylvania. All clinical data was captured by movement disorder neurologists, behavioral neurologists, or a senior behavioral neurology research nurse. Healthy subjects were included if they were free of clinical symptoms of neurodegenerative disease, able to provide informed consent, and able to participate in biological fluid collection procedures. PD participants were included if they met U.K. Brain Bank clinical criteria for PD (Hughes et al. 1992) were able to provide informed consent, and were able to participate in biological fluid collection procedures. Disease duration was defined by patient report of initial symptoms and supplemented by chart review. Each participant was administered the Montreal Cognitive Assessment (MoCA) to measure global cognitive function (Nasreddine et al. 2005). Disease severity was assessed by the Unified Parkinson’s Disease Rating Scale (UPDRS) motor score in the participant’s “ON” state and the Hoehn & Yahr (H&Y) scale. PD patients were additionally phenotyped as tremor-dominant (TD) type or postural instability – gait dysfunction (PIGD) type by calculating a TD / PIGD ratio as previously described (Stebbins et al. 2013). Total levodopa equivalent daily dose (LEDD) was calculated as previously described (Tomlinson et al. 2010). All study protocols were approved by the University of Pennsylvania Institutional Review Board (Protocol #820710). Our study was not pre-registered. Informed consent was obtained from each participant prior to enrollment, in concordance with the Declaration of Helsinki.
Experimental Design
All clinical data and samples were handled in a de-identified manner as previously described (Toledo et al. 2014). Briefly, each participant was assigned a random and unique six-digit number. All biological samples were tagged by this six-digit number and only this number was used to track samples by authors performing benchtop experiments (RSA and JPL). These investigators remained blinded to participant level data (e.g. demographic information, diagnostic group, clinical measures) until after all assays were completed. Samples were placed in random order on each assay plate.
Biological fluid collection and Alzheimer disease CSF analyses
Serum and CSF samples were collected and processed according to established procedures (Toledo et al. 2014, Trojanowski et al. 2010, Kang et al. 2013). Briefly, serum samples were collected in serum separation tubes and centrifuged at 3000 × g for 15 minutes at 4°C prior to storage at −80°C. CSF samples were stored neat in 0.5 mL aliquots at −80°C. Samples were thawed on ice immediately before use. Twenty-four participants had matched serum and CSF samples. CSF Aβ1–42, total tau, and phosphorylated tau were measured using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) and research use-only reagents from Innogenetics (INNO-BIA AlzBio3, cat. 80584, Ghent, Belgium) as previously described (Shaw et al. 2011, Shaw et al. 2009).
ELISA for α-syn AAb measurement
ELISA was performed in clear, flat-bottom 384-multiwell polystyrene plates (MaxiSorp, cat. 12565347, Thermo Scientific). Recombinant α-syn was diluted in 0.2 M sodium carbonate / bicarbonate buffer, pH 9.6 and adsorbed at 100 ng α-syn per well (233.1 nM) overnight at 4°C. Non-specific binding was blocked using 100 µl of blocking solution (see Results for optimization of the blocking protocol) per well, overnight at 4°C. Samples were diluted in phosphate buffered saline (PBS) to 1:5 for CSF and 1:500 for serum (see Results) and applied using 30 µl per well overnight at 4°C. Secondary antibodies used were horseradish peroxidase (HRP)-conjugated anti-human IgG antibodies raised in rabbit (RRID:AB_2339653, Jackson Immunoresearch, cat. 309-035-082, West Grove, PA), donkey (RRID:AB_2340495, Jackson Immunoresearch, cat. 709-035-149), or goat (RRID:AB_257868, Sigma-Aldrich, cat. A0170, St. Louis, MO). Each secondary antibody was diluted in 30 µl PBS and incubated overnight at 4°C. Prior to each incubation, plates were centrifuged at 1000 × g for 1 minute at 25°C. In between each incubation, plates were washed three times in PBS with 0.05% Tween using an automated plate washer (ELx405 Select CW, BioTek Instruments). To detect bound HRP activity, 30 µl of 3,3’,5,5’-tetramethylbenzidine substrate (Pierce 1-Step Ultra, cat. 34029, Thermo Scientific) was added per well at room temperature and quenched with 30 µl 10% phosphoric acid after exactly 10 minutes. Absorbance at 450 nm was measured using an automated plate reader (Spectramax M5, Molecular Devices, Sunnyvale, CA). For assay optimization, wells that lacked α-syn were used as negative controls.
Hemoglobin quantification assay
Hemoglobin contamination of CSF samples was determined using sandwich ELISA according to manufacturer’s instructions (Bethyl Labs, cat. E80-135, Montgomery, TX). 384-multiwell plates were coated with 60 pg of affinity purified goat anti-human hemoglobin antibody (Bethyl Labs, cat. A80-134A) and non-specific binding was blocked with 100 µl 1% cold fish gelatin (Sigma-Aldrich, cat. G7765) in PBS (v/v) per well overnight at 4°C. CSF samples were diluted 1:5 in PBS and incubated overnight at 4°C. Hemoglobin was detected using a polyclonal HRP-conjugated human hemoglobin detection antibody (1 pg/well; RRID:AB_67041, Bethyl Labs, cat. A80-134P). All CSF samples were measured in triplicate over three separate assays. A standard curve (1 µg/mL to 48.8 pg/mL) using a human hemoglobin calibrator accompanied each assay. Assay data was fit to a four-parameter logistic dose-response curve to quantify absolute hemoglobin levels. Curves were constrained by absorbance for 1 µg/mL calibrator (for top) and absorbance for no calibrator (for bottom). A pooled serum sample was analyzed at a 1:5000 dilution to allow for normalization between assays.
APOE genotyping
Genomic DNA was isolated from peripheral blood samples using manufacturer’s protocols (FlexiGene, QIAGEN, Valencia CA or QuickGene, Autogen, Holliston, MA). Genotyping for the APOE ε4 allele was performed using real-time allelic discrimination PCR using TaqMan reagents and ABI assays C_904973_10 (for single nucleotide polymorphism rs7412) and C_3084793_20 (for rs429358) on an ABI 7500 real-time PCR instrument (Applied Biosystems, Foster City, CA) using standard conditions.
Statistical analysis
The absorbance from three wells per sample and per assay was averaged and a standard deviation (SD) and coefficient of variance (CV, defined by SD divided by average absorbance) were calculated. Samples with greater than 15% CV were re-assayed. Only assay data with ≤ 15% CV were used in subsequent statistical analysis. At least three independent assays were used to calculate a grand average (mean of each individual assay absorbance) and grand SD (square root of the mean squared SDs). A final CV was calculated using the grand average and grand SD.
For univariate correlations with AAb levels, we used two-sample t-test, Kruskal-Wallis rank test, or linear regression for categorical, normally distributed ordinal, or continuous variables. We used two-way ANOVA or multiple regression to look for interactions between ordinal and continuous variables. We used forward selection linear regression modeling to confirm univariate associations with AAb levels.
All statistical analyses were performed using Stata/IC (13.1, College Station, TX). Figures were prepared with GraphPad Prism (6.0, La Jolla, CA) and GNU Image Manipulation Program (GIMP 2.8.22-1, https://www.gimp.org/). For figures, each point depicts mean ± standard deviation unless otherwise specified. For all figures, each point represents at least three assays per sample and three technical replicates per assay unless otherwise specified. All statistical tests were two-sided. An alpha-level of p < 0.05 was used to determine statistical significance. Because this study was exploratory using a newly developed ELISA, sample size and power calculations were not performed. No randomization was performed in this study.
Results
Development of ELISA for α-syn AAbs in patient samples
We modified an indirect ELISA approach to detect α-syn AAbs (see Methods), wherein recombinant α-syn was first applied directly to a solid surface, followed by application of a blocking solution to occupy any remaining unbound areas. Then, either serum or CSF samples were applied to the surface to allow any AAbs within these samples to bind to the affixed α-syn. Finally, AAb binding was detected using a HRP-conjugated secondary antibody to human immunoglobulin, and HRP activity was measured by colorimetric assay.
To optimize our ELISA for α-syn AAbs, we tested multiple blocking agents, multiple anti-human IgG HRP-conjugated antibodies, and variable blocking time. There was a dilution-dependent, non-specific HRP activity when serum samples were tested on wells that lacked α-syn coating (Figure 1). This nonspecific signal was higher when plates were blocked with 5% bovine serum albumin (Sigma-Aldrich, cat. A7906) and lower when blocked with 1% cold fish gelatin (Figure 1A, top row vs bottom row). Nonspecific signals were optimal when using a goat anti-human IgG HRP-conjugate (Figure 1A, top right). Blocking time beyond overnight paradoxically worsened the nonspecific background (Figure 1B).
Figure 1. Optimization of ELISA for α-syn AAb detection.

Dilute serum was applied to 384-well ELISA plates either coated with recombinant α-syn or not coated. (A) Plates were blocked with either 1% cold fish gelatin (top row) or 5% bovine serum albumin (middle row) overnight at 4 °C. Binding was detected using anti-human IgG HRP-conjugates raised in rabbit (left column), donkey (middle column), or goat (right column).
(B) Plates were blocked with 1% cold fish gelatin at 4°C for either 6 days (left), 4 days (middle), or 1 day (right). Additional blocking solutions (1% bovine serum albumin, 5% milk in PBS, several commercial blocking solutions) did not demonstrate any improvement over 1% cold fish gelatin. Each data point represents three technical replicates (n=3 wells). Wells coated with α-syn denoted by closed circles. Uncoated wells denoted by closed triangles. BSA = bovine serum albumin; CFG = cold fish gelatin; HRP = horseradish peroxidase conjugate.
To confirm that our ELISA detected immunoglobulins, we tested the heat stability of our samples in our assay. There was no significant effect of heating to 50°C for 10 minutes for either serum or CSF samples (Figure 2). In contrast, heating to 90°C for 10 minutes completely abrogated the detected signal consistent with the denaturation of immunoglobulins within the sample.
Figure 2. Heat inactivates immunoglobulins detected in α-syn AAb ELISA.

Diluted serum or CSF of two HC and two PD patients were prepared in PBS and heated to 50°C, 90°C, or kept on ice for 10 minutes prior to addition to ELISA. Activity on ice was similar to activity following incubation at 50°C for 10 minutes. In contrast, incubation at 90°C for 10 minutes denatured the immunoglobulins in the sample and ELISA activity was lost. Each column represents mean ± standard deviation of three technical replicates (n=3 wells). HC = healthy control; PD = Parkinson’s disease.
To further confirm that our ELISA detected immunoglobulins that recognize α-sn, we performed competitive ELISA by pre-incubating samples with soluble α-syn monomer prior to assay. Pre-incubation with equimolar amounts of soluble α-syn to that adsorbed for ELISA (100 ng) led to a reduction of 45% and 40% activity using two PD serum samples (Figure 3). Ten-fold excess soluble α-syn led to a reduction of 57% and 42% ELISA activity. These data suggested that the AAbs detected in our ELISA were of low-affinity and capable of exchanging with immobilized α-syn.
Figure 3. Pre-incubation with soluble α-syn reveals low-affinity AAb interactions.

Diluted serum of two PD patients were prepared in PBS along with increasing concentrations of soluble α-syn monomer. This preparation was incubated at room temperature for two hours on an orbital shaker before analysis in ELISA. Activity was reduced to 92% (PD1) and 97% (PD2) with 1 ng soluble α-syn monomer, 82% and 83% with 10 ng, 56% and 60% with 100 ng, and 43% and 58% with 1000 ng α-syn in the pre-incubation step. Each column represents mean ± standard deviation of three technical replicates (n=3 wells). PD = Parkinson’s disease.
Levels of α-syn AAbs are higher in males but not associated with age or APOE ε4 status
We analyzed serum and CSF samples from two separate cohorts of healthy controls (HC) and PD patients for α-syn AAbs, and evaluated for correlations between AAb level and clinical phenotypes. The overall demographics for each cohort are listed in Table 1. There were no significant differences in age or education between HC and PD patients in either sample cohort. Similar numbers of male and female serum samples were tested. However, there were significantly more CSF samples from male subjects than from female subjects (Table 1). For this reason, we included gender in all multivariable models. Presence of an APOE ε4 allele was similar in both sample cohorts. Cognitive performance on the MoCA was better in HC than PD in both sample cohorts (Table 1). Mean CSF total tau was significantly lower in PD cases as compared to HC (Table 1). There was no significant difference in CSF Aβ1–42 or phosphorylated tau between HC and PD (Table 1).
Table 1.
Demographic and Biomarker Information
| Serum | CSF | |||||
|---|---|---|---|---|---|---|
| HC | PD | HC | PD | |||
| Samples, n. | 16 | 53 | 52 | 93 | ||
| Age, y | 73.7 ± 11.5 | 70.9 ± 7.0 | n.s. | 69.4 ± 9.8 | 67.1 ± 9.4 | n.s. |
| Education, y | 16.4 ± 2.6 | 16.2 ± 2.4 | n.s. | 15.8 ± 4.6 | 16.3 ± 2.3 | n.s. |
| Sex (m / f) | 7 / 9 | 34 / 19 | n.s. | 20 / 32 | 66 / 27 | * |
| APOE ε4 | ||||||
| Absent | 15 | 37 | n.s. | 32 | 61 | n.s. |
| Present | 1 | 15 | 15 | 29 | ||
| MoCA | 27.4 ± 2.3 (13) | 23.9 ± 5.6 (49) | ** | 26.7 ± 2.8 (33) | 24.5 ± 4.8 (88) | ** |
| Aβ1–42 (pg/mL) | n.a. | n.a. | 269 ± 80 (43) | 246 ± 65 (82) | n.s. | |
| t-tau (pg/mL) | n.a. | n.a. | 52.8 ± 25 (43) | 44.6 ± 19 (80) | ** | |
| p-tau (pg/mL | n.a. | n.a. | 21.8 ± 13 (43) | 21.3 ± 8.9 (82) | n.s. | |
All data are mean ± standard deviation unless otherwise specified.
Fisher’s exact < 0.001.
p < 0.05.
Abbreviations: HC, healthy controls; PD, Parkinson’s disease; n.s., not significant; MoCA, Montreal Cognitive Assessment; Aβ, amyloid-β; n.a. not applicable.
For all samples, mean α-syn AAb levels were lower in CSF (0.596 ± 0.47) than in serum (1.332 ± 0.75). During our assay optimization, CSF dilution of 1:5 and serum dilution of 1:500 led to optimal signal-to-noise. Thus, serum α-syn AAb levels were over 200-fold higher than CSF α-syn levels. We hypothesized that if α-syn AAb levels directly reflected underlying α-synucleinopathy, AAb levels would increase over time. However, there was no linear correlation between age and α-syn AAb levels in either serum (p = 0.55) or CSF (p = 0.15). In both serum and CSF, males had significantly higher levels of α-syn AAbs than females (Figure 4). There was no linear correlation between years of education and α-syn AAb levels in either serum (p = 0.93) or CSF (p = 0.95). AAb levels in serum were similar in APOE ε4 allele carriers (1.437 ± 0.70) as compared to non-carriers (1.316 ± 0.77) (p = 0.58). Similarly, CSF AAb levels were similar in ε4 carriers (0.654 ± 0.53) vs non-carriers (0.575 ± 0.45) (p = 0.37).
Figure 4. Auto-antibodies to α-syn are higher in males than females.

Males had significantly higher CSF levels of α-syn AAbs (0.664 ± 0.51) than females (0.497 ± 0.40) (t143 = 2.12, p = 0.035). Similarly, levels of AAbs were higher in serum in males (1.481 ± 0.74) than in females (1.114 ± 0.72) (t67 = 2.04, p = 0.046). CSF samples were diluted 1:5 and serum samples were diluted 1:500. Each data point depicts mean ± standard deviation of three samples per assay, each assay having three technical replicates (n=9 wells). Horizontal lines indicate group mean and standard deviations.
Disease characteristics are listed in Table 2. Disease duration, PD motor subtype, H&Y stage, and total UPDRS motor score were similar in the serum cohort and the CSF cohort (Table 2).
Table 2.
Parkinson’s disease Clinical Information
| Serum | CSF | ||
|---|---|---|---|
| Samples, n. | 53 | 93 | |
| Disease duration, y | 7.9 ± 5.0 | 8.5 ± 5.7 | n.s. |
| LEDD | 809 ± 557 | 696 ± 462 | n.s. |
| Motor subtype | |||
| Tremor predominant | 11 | 20 | n.s. |
| PIGD | 25 | 59 | |
| Indeterminant | 4 | 3 | |
| H&Y stage | |||
| 1 | 6 | 6 | n.s. |
| 2 | 18 | 25 | |
| 3 | 27 | 52 | |
| 4 | 2 | 4 | |
| 5 | 0 | 1 | |
| UPDRS motor score | 22.4 ± 11.5 | 23.2 ± 12.0 | n.s. |
All data are mean ± standard deviation unless otherwise specified.
Fisher’s exact < 0.001.
p < 0.05.
Abbreviations: n.s., not significant; LEDD, levodopa equivalent daily dose; H&Y, Hoehn and Yahr; UPDRS, Unified Parkinson’s Disease Rating Scale.
PD patients have higher CSF levels of α-syn AAbs than HC subjects
We hypothesized that α-syn AAb levels would be higher in PD samples as compared to HC samples and thus act as a diagnostic biomarker. CSF α-syn AAb levels were significantly higher in PD patients as compared to HC (Figure 5A). Additionally, significantly more PD participants had CSF α-syn AAb levels equal to or above the overall mean (41.5%) as compared to HC participants (23.1%) (Fisher’s exact = 0.029). Because males had significantly higher levels of AAbs in both biofluids, we used two-way ANOVA to determine the contribution of gender and PD diagnosis to AAb levels. AAb levels remained significantly higher in PD samples as compared to HC samples (F1,142 = 5.91, p = 0.016). We found no significant main effect of gender (F1,142 = 1.63, p = 0.20; overall effect F2,142 = 5.29, p = 0.006 by two-way ANOVA). Univariate analyses also found no difference between male and female samples, within either HC (Figure 5B) or PD patients (Figure 5C).
Figure 5. CSF auto-antibodies to α-syn are higher in Parkinson’s disease patients as compared to healthy controls.

(A) Levels of α-syn AAbs were significantly higher in CSF from PD patients (0.681 ± 0.50) as compared to CSF from HC (0.444 ± 0.36) (t143 = 2.98, p = 0.003). (B) Male HC had higher levels (0.537 ± 0.41) as compared to female HC (0.385 ± 0.33), which was not statistically significant (p = 0.14). (B) Male PD patients also had higher levels (0.702 ± 0.53) as compared to female PD patients (0.629 ± 0.44), which was not statistically significant (p = 0.53). Each data point depicts mean ± standard deviation of three samples per assay, each assay having three technical replicates (n=9 wells). Horizontal lines indicate mean and standard deviations.
In contrast to CSF, α-syn AAb levels in serum were similar between HC and PD patients (Figure 6A), suggesting that α-syn AAb levels in this compartment could not function as a diagnostic biomarker. However, activity in two HC samples and fourteen PD samples exceeded an absorbance value of 2.0 (Figure 6A, samples above dotted line) and many appeared to fall outside the dynamic range of the assay when diluted 1:500. We hypothesized that this problem might contribute to a lower the mean PD AAb level relative to HC since the majority of these samples were from PD patients. These out-of-range samples were re-analyzed at a dilution of 1:2000 and AAb levels were imputed given the additional dilution factor. When analyzed this way, we continued to see no significant difference between HC and PD α-syn AAb levels in serum (Figure 6B). There was no significant main effect of gender (p = 0.14) or diagnosis (p = 0.19) to serum levels of AAb when analyzed using two-way ANOVA (overall p = 0.097). However, univariate analysis found significantly higher levels of serum AAb in male HC as compared to female HC (Figure 6C). Original, non-imputed values were also significantly different between male and female HC by two-sample t-test (t14 = 2.77, p = 0.015). There was no difference in univariate analysis between male and female PD patients (Figure 6D), including when using non-imputed values (p = 0.33).
Figure 6. Serum auto-antibodies to α-syn are similar between Parkinson’s disease patients and healthy controls.

(A) In serum samples diluted 1:500, α-syn AAb levels were not significantly different between HC (1.114 ± 0.58) and PD patients (1.398 ± 0.79) (p = 0.19). (B) Sixteen serum samples re-analyzed at 1:2000 dilution and AAb levels were imputed (closed triangles). Although PD α-syn AAb levels were higher (2.190 ± 2.30) than HC levels (1.250 ± 0.91) when substituting imputed values, this difference was still not statistically significant (p = 0.12). (C) Male HC had significantly higher imputed levels (1.805 ± 1.11) as compared to female HC (0.818 ± 0.37) (t14 = 2.51, p = 0.025). (D) Male PD patients had higher levels (2.440 ± 2.55) than female PD patients (1.744 ± 1.73) that was not significantly different (p = 0.295). Each data point depicts mean ± standard deviation of three samples per assay, each assay having three technical replicates (n=9 wells). Horizontal lines indicate mean and standard deviations. Closed triangles indicate imputed values.
We hypothesized that α-syn AAb levels could reflect motor disease features. There was no linear correlation between α-syn AAb levels in serum or CSF and motor disease duration (p = 0.67 and p = 0.58). There was no difference in serum AAb levels among different H&Y stages (p = 0.79). Similarly, there was no difference in CSF AAb levels among different H&Y stages (p = 0.77). There was no linear correlation between AAb levels in serum or CSF and total UPDRS motor scores (p = 0.49 and (p = 0.81). There was also no correlation between AAb levels in serum or CSF and total daily LEDD (p = 0.29 and p = 0.42).
Both serum and CSF were available from twenty-four individuals (four HC and twenty PD). In these cases, CSF α-syn AAb levels were significantly correlated to serum α-syn AAb levels, when compared to either non-imputed serum values (r = 0.77, p < 0.0001) or imputed serum values (r = 0.76, p < 0.0001).
Levels of CSF α-syn AAbs do not correlate with Aβ1–42 or tau CSF biomarkers
CSF Aβ1–42, total tau, and phosphorylated tau are supportive diagnostic biomarkers for Alzheimer’s disease (McKhann et al. 2011). We hypothesized that CSF α-syn AAb levels reflected a separate underlying pathophysiology and thus would be not correlated with these other CSF biomarkers. We did not find a significant correlation between CSF α-syn AAb level and CSF Aβ1–42, when analyzed for all participants (p = 0.44) or by diagnosis (for HC, p = 0.10; for PD, p = 0.16). In addition, mean CSF α-syn AAb levels were not significantly different in cases with positive CSF Aβ1–42 as determined by cut-off value of 192 pg/mL (p = 0.38).(Shaw et al. 2009) There was no significant correlation between CSF α-syn AAb level and CSF total tau (for all participants, p = 0.29; for HC, p = 0.24; for PD, p = 0.95). Because both CSF total tau and CSF α-syn AAbs were independently significantly lower in HC than PD (Table 1 and Figure 5), we tested the contribution of each variable to diagnosis while controlling for the other using partial correlation. Higher CSF α-syn AAbs levels continued to correlate with a diagnosis of PD when total tau was controlled (p = 0.005). However, when controlling for α-syn AAb level, total tau no longer correlated with diagnosis (p = 0.073). For phosphorylated tau, we found no correlation with CSF α-syn AAb levels among all participants (p = 0.99), or within HC (p = 0.31) or PD (p = 0.39).
Higher CSF levels of α-syn AAbs correlate with lower MoCA scores
We next hypothesized that CSF α-syn AAb levels could reflect cognitive disease severity. Using linear regression, we found a significant, albeit modest, correlation between higher levels of CSF α-syn AAbs and lower total score on the MoCA (Figure 7). In a step-wise, forward linear regression model, CSF AAb level still significantly predicted total MoCA score when controlled for age and education (Table 3, Regression 1). However, once diagnosis was included as a predictor, CSF AAb levels no longer significantly predicted MoCA score (Table 3, Regression 1), owing to the significant difference in MoCA scores between the two groups (Table 1). In contrast to CSF α-syn AAbs, we did not find a significant correlation between serum α-syn AAbs and MoCA (Figure 7).
Figure 7. Higher levels of α-syn auto-antibodies in CSF correlate with lower global cognitive performance.

Higher levels of CSF α-syn AAbs significantly correlated with lower total score on the MoCA (r = −0.22, p = 0.014), which remained significant when controlled for age and education (see text). In contrast, there was no significant correlation between serum α-syn AAb level and total MoCA score (p = 0.21). Each data point depicts mean of three samples per assay, each assay having three technical replicates (n=9 wells). Each serum data point depicts un-adjusted values. Regression line and 95% confidence band shown.
Table 3.
CSF α-syn AAb level contributes to total MoCA score
| Regression 1 | Model 1 | Model 2 | Model 3 | Model 4 | ||||
| Predictor | β | (SE) | β | (SE) | β | (SE) | β | (SE) |
| t | (p) | t | (p) | t | (p) | t | (p) | |
| Age, y | −0.146 | (0.05) | −0.136 | (0.05) | −0.170 | (0.05) | −0.160 | (0.04) |
| −3.19 | (0.002) | −3.02 | (0.003) | −3.81 | (0.000) | −3.58 | (0.001) | |
| Education, y | 0.297 | (0.15) | 0.284 | (0.14) | 0.318 | (0.14) | 0.307 | (0.14) |
| 2.04 | (0.043) | 1.98 | (0.050) | 2.27 | (0.025) | 2.20 | (0.030) | |
| CSF α-syn AAb level | -- | −1.784 | (0.80) | -- | −1.213 | (0.80) | ||
| −2.24 | (0.027) | −1.51 | (0.133) | |||||
| Diagnosis (HC vs PD) | -- | -- | −2.77 | (0.84) | −2.441 | (0.87) | ||
| −3.29 | (0.001) | −2.81 | (0.006) | |||||
| Regression 2 | Model 1 | Model 2 | Model 3 | Model 4 | ||||
| Age, y | −0.132 | (0.05) | −0.124 | (0.05) | −0.156 | (0.04) | −0.148 | (0.05) |
| −2.92 | (0.004) | −2.78 | (0.006) | −3.52 | (0.001) | −3.32 | (0.001) | |
| Education, y | 0.273 | (0.14) | 0.263 | (0.14) | 0.295 | (0.14) | 0.286 | (0.14) |
| 1.91 | (0.059) | 1.86 | (0.065) | 2.14 | (0.035) | 2.08 | (0.040) | |
| CSF α-syn AAb level | -- | −1.618 | (0.79) | -- | −1.094 | (0.79) | ||
| −2.05 | (0.042) | −1.38 | (0.170) | |||||
| Diagnosis (HC vs PD) | -- | -- | −2.59 | (0.83) | −2.301 | (0.86) | ||
| −3.11 | (0.002) | −2.68 | (0.008) | |||||
| Aβ1–42 (− vs +) | −2.497 | (1.03) | 2.301 | (1.02) | −2.19 | (1.00) | −2.093 | (1.00) |
| −2.43 | (0.017) | −2.26 | (0.026) | −2.20 | (0.030) | −2.10 | (0.038) | |
| Regression 3 | Model 1 | Model 2 | Model 3 | Model 4 | ||||
| Age, y | −0.146 | (0.05) | −0.135 | (0.05) | −0.168 | (0.05) | −0.158 | (0.05) |
| −3.16 | (0.002) | −2.96 | (0.004) | −3.73 | (0.000) | −3.49 | (0.001) | |
| Education, y | 0.297 | (0.15) | 0.283 | (0.14) | 0.316 | (0.14) | 0.305 | (0.14) |
| 2.03 | (0.044) | 1.97 | (0.051) | −2.25 | (0.026) | 2.18 | (0.032) | |
| CSF α-syn AAb level | -- | −1.801 | (0.81) | -- | −1.241 | (0.81) | ||
| −2.24 | (0.027) | −1.54 | (0.126) | |||||
| Diagnosis (HC vs PD) | -- | -- | −2.84 | (0.86) | −2.510 | (0.88) | ||
| −3.32 | (0.001) | −2.86 | (0.005) | |||||
| CSF hgb (− vs +) | 0.000 | (0.79) | 0.167 | (0.78) | 0.383 | (0.76) | 0.452 | (0.76) |
| 0.00 | (0.997) | 0.21 | (0.830) | 0.50 | (0.617) | 0.59 | (0.553) | |
Step-wise, forward linear regression models were performed for total MoCA score as an outcome variable and predictor variables as shown. Constant term not shown. Significant effects are shown in bold-face type.
Abbreviations: MoCA, Montreal Cognitive Assessment; AAb, auto-antibody; HC, healthy controls; PD, Parkinson’s disease; hgb, hemoglobin; --, not included.
CSF Aβ1–42 is known to contribute to cognitive impairment in PD.(Siderowf et al. 2010, Alves et al. 2014, Compta et al. 2013) Therefore, we repeated our step-wise linear regression analysis to determine if the contribution of CSF α-syn AAb to MoCA score was influenced by CSF Aβ1–42 positivity. Positive CSF Aβ1–42 significantly predicted lower MoCA score as did age (p = 0.017, Table 3, Regression 2). Education was no longer significantly associated with lower MoCA score when CSF Aβ1–42 was included in the model. CSF α-syn AAb level continued to significantly predict MoCA score when controlled for CSF Aβ1–42, age, and education (p = 0.042, Table 3, Regression 2). Addition of diagnosis (HC vs PD) as a co-variate did not alter the significant association between CSF Aβ1–42 with MoCA score, when controlled for age and education (Table 3, Regression 2). However, once diagnosis was included as a predictor, CSF AAb levels no longer significantly correlated with MoCA score, while CSF Aβ1–42 remained significantly correlated (Table 3, Regression 2).
Serum contamination of CSF samples do not influence CSF α-syn AAb findings
Because absolute levels of α-syn AAbs were higher in serum than CSF by over two-hundred fold, we reasoned that serum AAbs could contaminate our CSF samples since peripheral blood can contaminate CSF during lumbar puncture. To assess the presence and significance of peripheral blood contamination in the CSF samples, we measured free hemoglobin in each CSF sample (Figure 8).
Figure 8. Hemoglobin measures in CSF using ELISA.

CSF diluted 1:5 and a pooled serum sample diluted 1:5000 for normalization were analyzed using a commercially available ELISA for hemoglobin. (A) Representative standard curve with a hemoglobin calibrator fit with four-parameter logistic dose-response curve (Sy.x. = 0.072). (B) Given a narrow dynamic range of this assay, an arbitrary hemoglobin level of 0.558 absorbance units (horizontal dotted line) was selected to dichotomize samples as hemoglobin-absent vs hemoglobin-present. Each data point represents three technical replicates (n=3 wells). Hgb = hemoglobin.
In this analysis, similar numbers of male and female CSF samples were positive for hemoglobin (44.2% and 42.4%) (p = 0.83). Consistent with our previous univariate analysis (Figure 4), CSF α-syn AAb level was still higher in males as compared to females when presence of hemoglobin was included as a variable using two-way ANOVA (F1,142 = 4.46, p = 0.036). Hemoglobin was present in similar numbers of HC (36.5%) and PD (47.3%) samples (p = 0.21). Presence of hemoglobin did not alter our finding that PD CSF had higher levels of α-syn AAbs than HC CSF (F1,142 = 8.75, p = 0.004), as seen with univariate analyses (Figure 5). When controlling for the presence of hemoglobin, there remained no association between CSF α-syn AAb level and PD motor disease duration (p = 0.56), UPDRS motor score (p = 0.81), or LEDD (p = 0.40). H&Y stage did not significantly predict CSF α-syn levels with (p = 0.70) or without (p = 0.72) inclusion of hemoglobin status.
However, higher CSF α-syn AAb levels continued to predict lower MoCA score when presence of hemoglobin was included as a co-variate in multiple linear regression (Table 3, Regression 3). As seen in Regression 2, CSF α-syn AAb levels no longer significantly contributed to MoCA score when both disease status and hemoglobin contamination status were included in the model.
CSF α-syn AAb binding is reduced by select C-terminal truncation of α-syn
Previous studies suggest that the C-terminal region of α-syn is recognized by α-syn AAbs.(Woulfe et al. 2014) To identify the epitope(s) bound by CSF α-syn AAb and confirm and extend these findings, we modified our ELISA to incorporate various truncated α-syn proteins and compared AAb recruitment to that of full length α-syn. Mouse monoclonal antibody Syn-211 detected full-length α-syn (a.a. 1–140) and N-terminal truncated (a.a. 58–140) α-syn but did not detect either C-terminal preparation (Figure 9A). Rabbit polyclonal antibody SNL-1 detected all preparations except for the C-terminal truncated (a.a. 1–99) α-syn. Rabbit polyclonal antibody SNL-4 detected all preparations except for the N-terminal truncated preparation. Each antibody detected approximately equivalent levels of full-length or truncated α-syn, suggesting that equivalent amounts of each preparation were adsorbed.
Figure 9. Recruitment of CSF α-syn auto-antibodies is reduced by select C-terminal truncation of α-syn.

Full-length or truncated α-syn were adsorbed for ELISA. (A) Mouse monoclonal antibody Syn 211 or rabbit polyclonal antibodies SNL-1 and SNL-4 did not detect α-syn in which truncations removed the binding epitope of each antibody. There was no significant difference in absorbance between the two preparations (full length and a.a 58–140) detected by Syn 211 (p = 0.69). SNL-1 detected slightly less of the N-terminal truncated α-syn than either the full-length or the a.a. 1–120 preparation (p = 0.016 and p = 0.008) (overall effect F2,6 = 12.84, p = 0.007 by one-way ANOVA when excluding a.a. 1–99). SNL-4 detected slightly more of the a.a. 1–99 preparation than full-length (p = 0.013) but there was otherwise no difference between full-length and a.a. 1–120 or between a.a. 1–120 and a.a. 1–99 (overall effect F2,6 = 9.23, p = 0.015 by one-way ANOVA when excluding a.a. 58–140). Each column represents mean ± standard deviation of three technical replicates (n=3 wells). (B) CSF α-syn AAb levels were significantly lower when C-terminal truncated (a.a. 1–99) α-syn was used in ELISA as compared to full-length α-syn (p < 0.001), partial C-terminal truncated α-syn (a.a. 1–120, p < 0.001), or N-terminal truncated α-syn (a.a. 58–140, p < 0.001), (overall effect F3,564 = 27.32, p < 0.0001 by one-way ANOVA). There was no significant difference in average CSF α-syn AAb levels detected by full-length vs a.a 1–120 (p = 0.75), full-length vs a.a. 58–140 (p = 0.96), or a.a 1–120 vs 58–140 (p = 0.44). Each data point depicts mean of three samples per assay, each assay having three technical replicates (n=9 wells). Horizontal lines depict mean and standard deviations
Full-length or truncated α-syn were used to capture CSF α-syn AAbs. For all samples, CSF α-syn AAb levels were significantly lower when residues 100 to 140 were removed from α-syn (Figure 9B). However, removing only residues 121 to 140 did not affect the levels of CSF α-syn AAbs detected, as compared to full-length α-syn. There was no significant effect of N-terminal truncation on CSF α-syn AAb detection.
Discussion
We optimized an ELISA-based assay for α-syn AAbs and measured AAbs in serum and CSF samples from HC and PD patients. For both fluids, α-syn AAb level was higher in males than females. Levels of α-syn AAbs in serum were relatively higher than CSF for all participants. AAb level in CSF, but not serum, predicted a diagnosis of PD. In addition, higher CSF α-syn AAb levels correlated with poorer global cognitive performance. CSF α-syn AAb levels did not correlate with the CSF Alzheimer’s disease biomarkers Aβ1–42, total tau, or phosphorylated tau. The effects we found for CSF AAb and cognitive performance were not altered by CSF hemoglobin values, and were seen in both uni-variate and multi-variate models. By comparing full-length α-syn to truncated α-syn, we determined that CSF α-syn AAb appeared to detect a region between a.a. 100 and a.a. 120 in the C-terminus of α-syn. We did not find an association between α-syn AAb level and measures of motor severity or disease duration. Taken together, our results suggest that CSF α-syn AAbs could serve as a biomarker for underlying α-synucleinopathy in PD, and associate with cognitive impairment, but are independent of PD motor disease severity. Further, the relevance of CSF α-syn AAb to cognitive performance is independent of Aβ1–42. The residues between a.a. 100 and 120 appear to be important epitopes for CSF AAb.
Our study suggests that both HC and PD patients harbor similar amounts of peripherally circulating α-syn AAbs. This degree of overlap suggests that serum α-syn AAb levels may not be useful as a diagnostic biomarker for clinical PD, as defined by loss of striatal dopaminergic tone and the presumed presence of Lewy bodies in the brain (Hughes et al. 1992). However, an emerging hypothesis posits that the initial pathogenesis of PD, and early pathologic α-syn accumulation, might begin in the peripheral nervous system before the onset of motor symptoms (Akhtar & Stern 2012, Olanow & Obeso 2012). In this context, we speculate that peripheral α-syn AAb levels might be useful to identify individuals who have peripheral α-syn deposits, and several peripheral sites, including parasympathetic ganglia, olfactory epithelium, submandibular gland, and colonic mucosa, have demonstrated pathologic α-syn accumulation (Beach et al. 2013, Beach et al. 2010, Beach et al. 2009, Lebouvier et al. 2010, Shannon et al. 2012). These peripheral deposits could be complete asymptomatic (in the case of healthy individuals), or be associated with constipation and hyposmia (in the case of an individual with pre-motor PD) (Doty et al. 1992, Edwards et al. 1992). Serum α-syn AAbs could be the response to these peripheral deposits and AAb levels might change as pathologic deposits accrue. In our study, two HC had relatively higher serum AAb levels than the other HC participants, and these individuals might already harbor a significant peripheral α-synucleinopathy. Alternatively, they represent the extreme end of the natural variation in α-syn AAb levels found in disease-free samples. These exploratory hypotheses will require testing in future studies with samples ideally collected before and after motor symptom onset.
We found significantly higher levels of CSF α-syn AAbs in PD patients as compared to HC subjects. Our observations suggest that CSF α-syn AAb levels could be reliable trait biomarkers for PD, although our results require replication in additional cohorts. Our data also suggests that AAbs may not be state biomarkers for motor disease progression, since they do not associate with measures of motor severity. Previous studies have suggested that serum α-syn AAb levels decrease in patients with disease stage greater than 2.5 on the modified H&Y scale (Horvath et al. 2017, Yanamandra et al. 2011). Among our 53 serum cases, over 50% of cases were H&Y stage 3 and only two had H&Y stages greater than 3. Thus, our serum cohort may have underestimated the association of serum α-syn AAb level with more advanced motor disease stage due to a selection bias. We note that the LEDD for participants in this study was moderate, and UPDRS motor scores demonstrated moderate disease burden, suggesting that the sample was representative of typical PD patients managed at a tertiary care center. It remains to be determined whether α-syn AAb levels in the CSF are seen in α-synucleinopathies more broadly, which include multiple system atrophy and dementia with Lewy bodies.
A small number of HC in our study had elevated α-syn AAbs in their CSF relative to the overall HC group. When dichotomizing using the overall average AAb levels (including both HC and PD), twelve of fifty-two HC cases had CSF α-syn AAb levels equal to or above the mean. These observations could be explained by the fact that some HC probably have incidental α-syn deposits in the brain. In autopsy studies, a small proportion of clinically healthy individuals have incidental Lewy body pathology (ILB) that is α-syn positive (Adler et al. 2010, Markesbery et al. 2009, Fearnley & Lees 1991). Therefore, if higher CSF α-syn AAb levels indicate the presence of CNS α-synucleinopathy, these twelve cases could either represent ILB cases or have pre-manifest PD. Another explanation is that these individuals harbor early α-synucleinopathy in the setting of Alzheimer’s disease (Kotzbauer et al. 2001), which has a long prodrome of Alzheimer’s disease related neuropathologic changes (Dubois et al. 2016). These possibilities require future study since our participants’ diagnoses were clinical and not autopsy-confirmed. However, the consequence for Alzheimer’s disease is significant since co-existing α-syn pathology appears to influence clinical features including cognition (Brenowitz et al. 2017, Chung et al. 2015), and likely influences the degree of underlying Aβ and tau pathology (Walker et al. 2015). Thus, in Alzheimer’s disease, CSF α-syn AAb levels might be useful to identify individuals with co-existing Lewy body pathology.
Irrespective of underlying diagnosis, we found that high CSF α-syn AAbs significantly correlated with poor cognitive performance, although the magnitude of correlation was modest on univariate analysis. However, the correlation persisted after controlling for age and years of education. As expected, the underlying diagnosis also made a significant contribution to cognitive performance, such that inclusion of diagnosis in our regression models negated the effect of AAb level. It is established that increased burden of cortical Lewy bodies in PD associate with cognitive impairment (Hurtig et al. 2000, Irwin et al. 2012), but the relevance of ILB in HC to cognition is less clear (Adler et al. 2010, Markesbery et al. 2009). It is possible that either the entry of peripheral AAb into the CNS, or the increased interthecal synthesis of AAb, are deleterious to cognition. Alternatively, CSF α-syn AAbs may only signify the presence of a parallel process that portends poor cognition. These potential interactions should be examined in future studies.
Importantly, the association between CSF α-syn AAb and cognitive performance is independent of CSF Aβ1–42, a known contributor to cognitive impairment in PD (Alves et al. 2014, Compta et al. 2013, Siderowf et al. 2010). The association we found was also independent of APOE ε4 status. These observations suggest that the pathological processes leading to aberrant Aβ1–42 could be separate from those processes leading to α-syn AAb generation. To our knowledge, no previous studies examined the relationship between CSF α-syn AAb and these traditional biomarkers.
The etiological mechanism behind α-syn AAbs generation is unknown. Neither age or overall motor disease duration (in the PD sample) were correlated with α-syn AAb level, suggesting that α-syn AAbs are not simply associated with increased overall pathological α-syn burden. Molecular mimicry, which has been implicated in some antibody-mediated diseases, offers one possible explanation for α-syn AAb generation (Woulfe et al. 2014). Previous reports from Woulfe and colleagues suggest that serum AAbs in PD patients could cross react with Epstein-Barr virus (EBV) proteins, and a C-terminal region is important for this activity (Woulfe et al. 2000, Woulfe et al. 2016). EBV causes a latent infection in lymphocytes and typically leads to self-limited syndrome of infectious mononucleosis (Dunmire et al. 2015). However, prevalence of EBV infection in the community is high and far out of proportion to the prevalence of PD, suggesting that prior exposure to EBV alone is not sufficient to cause PD. Nevertheless, we find that this region (between a.a. 100 and 120) is also important for CSF α-syn AAb recruitment. A recent study found that peptides derived from the N-terminal region of α-syn, and from the C-terminal region to a lesser extent, are capable of inducing T-cell responses in peripheral blood mononuclear cells from PD patients (Sulzer et al. 2017). We cannot exclude the possibility that multiple, distinct pools of AAbs are present in our samples, including AAbs that recognize portions of the N-terminus. It is possible that only those AAbs which recognize a.a. 100 to 120 have sufficiently high affinity to be detected in our assay. Nevertheless, binding of AAbs to α-syn could influence the propensity of α-syn to misfold and aggregate, possibly when α-syn is extra-cellular during putative cell-to-cell transmission events (Guo & Lee 2014). For these reasons, more precise identification of the epitope or epitopes within this region recognized by α-syn AAbs might reveal how these AAbs are generated.
The presence of α-syn AAbs in biological fluids could also potentially confound attempts at detecting monomeric α-syn in both serum and CSF. It is plausible that high levels of endogenous AAbs could interfere with assays that leverage monoclonal α-syn antibodies in their design, since many detect α-syn in this same C-terminal region (Kasuga et al. 2012). Interference from non-specific heterophile antibodies is also known to complicate α-syn measurements (Ishii et al. 2015). Future studies would benefit from simultaneous assessment of both α-syn and α-syn AAbs to look for any interaction between the two.
Our study had several limitations. We only had matched serum and CSF samples for a small proportion (twenty-four) of the participants, the majority of whom were PD patients. The data from these matched samples suggest that the α-syn AAb levels correlate between the two compartments, but additional matched samples could allow us to determine the likelihood that peripheral α-syn AAbs enter the CSF rather than be interthecally synthesized. We also did not test longitudinal samples or determine the effect of AAb level on longitudinal measures of motor or cognitive function. Since we may be detecting multiple pools of α-syn AAbs, it is difficult to know if all of the AAbs act uniformly with respect to protection vs pathogenicity.
Using a newly developed assay to measure α-syn AAbs in serum and CSF, we found higher levels of AAbs in males vs females and higher levels in CSF from PD patients as compared to CSF from HC. Levels of CSF AAbs also correlated with cognitive performance and were not influenced by hemoglobin contamination. CSF AAb level did not correlate with traditional Alzheimer’s disease CSF biomarkers, and the association with cognitive performance was independent of CSF Aβ1–42. A discrete region in the C-terminus appears to be important for AAb generation. These studies suggest that measures of α-syn AAb levels could be useful as adjunctive biomarkers in PD, and help extend our understanding of immune mechanisms behind α-synucleinopathy.
Acknowledgments
We thank the patients and their families for participation in this study. We also thank Drs. Dustin Covell, Eddie Lee, and Kurt Brunden for helpful discussions, Marianne Watson for participant recruitment, and Chi Li for technical assistance. This study was funded or supported by the National Institutes of Health: P50-NS053488 (to J.Q.T. and V.M.L.), P30-AG010124 (to J.Q.T.), and K08-NS093127 (to R.S.A.), by the American Brain Foundation, and by the Keefer Family.
Abbreviations
- α-syn
α-synuclein
- Aβ
amyloid-β
- AAb
auto-antibody
- CV
coefficient of variance
- ELISA
enzyme-linked immunosorbent assay
- HC
healthy controls
- H&Y
Hoehn & Yahr
- HRP
horseradish peroxidase
- ILB
incidental Lewy body pathology
- LEDD
levodopa equivalent daily dose
- MoCA
Montreal Cognitive Assessment
- n.s.
not significant
- PBS
phosphate buffered saline
- PIGD
postural-instability gait dysfunction
- SD
standard deviation
- TD
tremor-dominant
- UPDRS
Unified Parkinson Disease Rating Scale
Footnotes
Conflicts of Interest
The authors declare that they have no competing interests.
Author contributions
RSA, KCL, and VML conceptualized the study. RSA and JPL designed and executed experiments. RSA, JPL, KCL, LMS, JQT, and VML analyzed and interpreted the data. RSA performed statistical analysis and wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
References
- Adler CH, Connor DJ, Hentz JG, Sabbagh MN, Caviness JN, Shill HA, Noble B, Beach TG. Incidental Lewy body disease: clinical comparison to a control cohort. Movement disorders : official journal of the Movement Disorder Society. 2010;25:642–646. doi: 10.1002/mds.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar RS, Stern MB. New concepts in the early and preclinical detection of Parkinson's disease: therapeutic implications. Expert review of neurotherapeutics. 2012;12:1429–1438. doi: 10.1586/ern.12.144. [DOI] [PubMed] [Google Scholar]
- Alves G, Lange J, Blennow K, Zetterberg H, Andreasson U, Forland MG, Tysnes OB, Larsen JP, Pedersen KF. CSF Abeta42 predicts early-onset dementia in Parkinson disease. Neurology. 2014;82:1784–1790. doi: 10.1212/WNL.0000000000000425. [DOI] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Dugger BN, et al. Submandibular gland biopsy for the diagnosis of Parkinson disease. Journal of neuropathology and experimental neurology. 2013;72:130–136. doi: 10.1097/NEN.0b013e3182805c72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Sue LI, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta neuropathologica. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, White CL, 3rd, Hladik CL, et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta neuropathologica. 2009;117:169–174. doi: 10.1007/s00401-008-0450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besong-Agbo D, Wolf E, Jessen F, et al. Naturally occurring alpha-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology. 2013;80:169–175. doi: 10.1212/WNL.0b013e31827b90d1. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brenowitz WD, Hubbard RA, Keene CD, Hawes SE, Longstreth WT, Jr, Woltjer RL, Kukull WA. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2017;13:654–662. doi: 10.1016/j.jalz.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan T, Luo X, Forsgren L, Morozova-Roche LA, Davis JJ. The robust electrochemical detection of a Parkinson's disease marker in whole blood sera. Chemical Science. 2012;3:3468–3473. [Google Scholar]
- Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical Features of Alzheimer Disease With and Without Lewy Bodies. JAMA neurology. 2015;72:789–796. doi: 10.1001/jamaneurol.2015.0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compta Y, Pereira JB, Rios J, et al. Combined dementia-risk biomarkers in Parkinson's disease: a prospective longitudinal study. Parkinsonism & related disorders. 2013;19:717–724. doi: 10.1016/j.parkreldis.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Doty RL, Stern MB, Pfeiffer C, Gollomp SM, Hurtig HI. Bilateral olfactory dysfunction in early stage treated and untreated idiopathic Parkinson's disease. Journal of neurology, neurosurgery, and psychiatry. 1992;55:138–142. doi: 10.1136/jnnp.55.2.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer's disease: Definition, natural history, and diagnostic criteria. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2016;12:292–323. doi: 10.1016/j.jalz.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunmire SK, Hogquist KA, Balfour HH. Infectious Mononucleosis. Current topics in microbiology and immunology. 2015;390:211–240. doi: 10.1007/978-3-319-22822-8_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology. 1992;42:726–732. doi: 10.1212/wnl.42.4.726. [DOI] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain : a journal of neurology. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Gaig C, Tolosa E. When does Parkinson's disease begin? Movement disorders : official journal of the Movement Disorder Society. 2009;24(Suppl 2):S656–664. doi: 10.1002/mds.22672. [DOI] [PubMed] [Google Scholar]
- Games D, Valera E, Spencer B, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease-like models. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. Journal of neuroscience research. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. The Journal of biological chemistry. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Gruden MA, Sewell RD, Yanamandra K, et al. Immunoprotection against toxic biomarkers is retained during Parkinson's disease progression. Journal of neuroimmunology. 2011;233:221–227. doi: 10.1016/j.jneuroim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nature medicine. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawa M, Beyan H, Leslie RD. Principles of autoantibodies as disease-specific markers. Autoimmunity. 2004;37:253–256. doi: 10.1080/08916930410001710668. [DOI] [PubMed] [Google Scholar]
- Horvath I, Iashchishyn IA, Forsgren L, Morozova-Roche LA. Immunochemical Detection of alpha-Synuclein Autoantibodies in Parkinson's Disease: Correlation between Plasma and Cerebrospinal Fluid Levels. ACS chemical neuroscience. 2017 doi: 10.1021/acschemneuro.7b00063. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Annals of neurology. 2014;76:168–184. doi: 10.1002/ana.24200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Annals of neurology. 2012;72:587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii R, Tokuda T, Tatebe H, Ohmichi T, Kasai T, Nakagawa M, Mizuno T, El-Agnaf OM. Decrease in plasma levels of alpha-synuclein is evident in patients with Parkinson's disease after elimination of heterophilic antibody interference. PloS one. 2015;10:e0123162. doi: 10.1371/journal.pone.0123162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Irwin DJ, Chen-Plotkin AS, et al. Association of cerebrospinal fluid beta-amyloid 1–42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA neurology. 2013;70:1277–1287. doi: 10.1001/jamaneurol.2013.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K, Nishizawa M, Ikeuchi T. alpha-Synuclein as CSF and Blood Biomarker of Dementia with Lewy Bodies. International journal of Alzheimer's disease. 2012;2012:437025. doi: 10.1155/2012/437025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EJ, Jr, Creighton ET. Venereal Disease Research Laboratory (VDRL) Slide Test. In: Larsen SA, Pope V, Johnson RE, Kennedy EJ Jr, editors. Manual of Tests for Syphilis. American Public Health Association; Washington, D.C: 1998. [Google Scholar]
- Kotzbauer PT, Trojanowsk JQ, Lee VM. Lewy body pathology in Alzheimer's disease. Journal of molecular neuroscience : MN. 2001;17:225–232. doi: 10.1385/jmn:17:2:225. [DOI] [PubMed] [Google Scholar]
- Lebouvier T, Neunlist M, Bruley des Varannes S, et al. Colonic biopsies to assess the neuropathology of Parkinson's disease and its relationship with symptoms. PloS one. 2010;5:e12728. doi: 10.1371/journal.pone.0012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Maetzler W, Apel A, Langkamp M, et al. Comparable autoantibody serum levels against amyloid- and inflammation-associated proteins in Parkinson's disease patients and controls. PloS one. 2014;9:e88604. doi: 10.1371/journal.pone.0088604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. Journal of neuropathology and experimental neurology. 2009;68:816–822. doi: 10.1097/NEN.0b013e3181ac10a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. Journal of the American Geriatrics Society. 2005;53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Obeso JA. The significance of defining preclinical or prodromal Parkinson's disease. Movement disorders : official journal of the Movement Disorder Society. 2012;27:666–669. doi: 10.1002/mds.25019. [DOI] [PubMed] [Google Scholar]
- Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain : a journal of neurology. 2005;128:2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- Papachroni KK, Ninkina N, Papapanagiotou A, Hadjigeorgiou GM, Xiromerisiou G, Papadimitriou A, Kalofoutis A, Buchman VL. Autoantibodies to alpha-synuclein in inherited Parkinson's disease. Journal of neurochemistry. 2007;101:749–756. doi: 10.1111/j.1471-4159.2006.04365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, Kordower JH. Alpha-synuclein in colonic submucosa in early untreated Parkinson's disease. Movement disorders : official journal of the Movement Disorder Society. 2012;27:709–715. doi: 10.1002/mds.23838. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Annals of neurology. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta neuropathologica. 2011;121:597–609. doi: 10.1007/s00401-011-0808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid {beta} 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins GT, Goetz CG, Burn DJ, Jankovic J, Khoo TK, Tilley BC. How to identify tremor dominant and postural instability/gait difficulty groups with the movement disorder society unified Parkinson's disease rating scale: comparison with the unified Parkinson's disease rating scale. Movement disorders : official journal of the Movement Disorder Society. 2013;28:668–670. doi: 10.1002/mds.25383. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature. 2017;546:656–661. doi: 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2014;10:477–484. e471. doi: 10.1016/j.jalz.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Movement disorders : official journal of the Movement Disorder Society. 2010;25:2649–2653. doi: 10.1002/mds.23429. [DOI] [PubMed] [Google Scholar]
- Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell reports. 2014;7:2054–2065. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, et al. Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2010;6:230–238. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nature protocols. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J. Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta neuropathologica. 2015;129:729–748. doi: 10.1007/s00401-015-1406-3. [DOI] [PubMed] [Google Scholar]
- Woulfe J, Gray MT, Ganesh MS, Middeldorp JM. Human serum antibodies against EBV latent membrane protein 1 cross-react with alpha-synuclein. Neurology(R) neuroimmunology & neuroinflammation. 2016;3:e239. doi: 10.1212/NXI.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woulfe J, Hoogendoorn H, Tarnopolsky M, Munoz DG. Monoclonal antibodies against Epstein-Barr virus cross-react with alpha-synuclein in human brain. Neurology. 2000;55:1398–1401. doi: 10.1212/wnl.55.9.1398. [DOI] [PubMed] [Google Scholar]
- Woulfe JM, Duke R, Middeldorp JM, et al. Absence of elevated anti-alpha-synuclein and anti-EBV latent membrane protein antibodies in PD. Neurology. 2002;58:1435–1436. doi: 10.1212/wnl.58.9.1435. [DOI] [PubMed] [Google Scholar]
- Woulfe JM, Gray MT, Gray DA, Munoz DG, Middeldorp JM. Hypothesis: a role for EBV-induced molecular mimicry in Parkinson's disease. Parkinsonism & related disorders. 2014;20:685–694. doi: 10.1016/j.parkreldis.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Yanamandra K, Gruden MA, Casaite V, Meskys R, Forsgren L, Morozova-Roche LA. alpha-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson's disease patients. PloS one. 2011;6:e18513. doi: 10.1371/journal.pone.0018513. [DOI] [PMC free article] [PubMed] [Google Scholar]