

Abstract
Endometrial Cancer is the most common female genital tract malignancy, its pathogenesis is complex, not yet fully described. To identify key genes of Endometrial Cancer we downloaded the gene chip GSE17025 from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were identified through the GEO2R analysis tool. Functional and pathway enrichment analysis were performed for DEGs using DAVID database. The network of protein–protein-interaction (PPI) was established by STRING website and visualized by Cytoscape. Then, functional and pathway enrichment analysis of DEGS were performed by DAVID database. A total of 1000 significant differences genes were obtained, contain 362 up-regulated genes and 638 down-regulated genes. PCDH10, SLC6A2, OGN, SFRP4, TRH, ANGPTL, FOSB are down-regulated genes. The gene of IGH, CCL20, ELF5, LTF, ASPM expression level in tumor patients are up-regulated. Biological function of enrichment include metabolism of xenobiotics by cytochrome P450, MAPK signaling pathway, Serotonergic synapse, Protein digestion and absorption, IL-17 signaling pathway, Chemokine signaling pathway, HIF-1 signaling pathway, p53 signaling pathway. All in all, the current study to determine endometrial differentially expressed genes and biological function, comprehensive analysis of intrauterine membrane carcinoma pathogenesis mechanism, and might be used as molecular targets and diagnostic biomarkers for the treatment of endometrial cancer.
Keywords: Endometrial cancer, bioinformatical analysis, differentially expressed genes, functional enrichment
Introduction
In all the factors that affect women’s health, female genital tract tumors to their health poses a huge threat, include three major tumors, Endometrial Cancer (EC), Epithelial Ovarian Cancer (EOC) and Cervical Cancer (CC). Among EC is the most common in women. EC is the most widely recognized ynecological tumor in created nations, and its predominance is expanding. Most patients in the early vaginal bleeding, vaginal discharge abdominal discomfort and other symptoms, endometrial malignancy is frequently diagnosed at stage I (Amant et al., 2005). Endometrial growth emerges from the lining of the uterus. It is the fourth most regular danger among ladies in the United States, with an expected 60,050 new cases and 10,470 deaths in 2016 (Siegel et al., 2016). As per the study, 63,400 women in China were diagnosed have endometrial cancer, even more unfortunate, 21,800 people deaths because of endometrial tumor in 2015 (Chen et al., 2016). In recent years, Many researchers have verified a lot of genes associated with endometrial cancer, such as PTEN (Gao et al., 2017), ADRID1A and ARID1B (Espinosa et al., 2017), PDL, B7-H4 (Bregar et al., 2017), POLE (Kandoth C, et al., 2013), MLH1, MSH2 (Martin et al., 2010) and some of others. We have scholars bioinformatics analysis was carried out on the mechanism of EC, and obtained the results of the proud (Xue et al., 2015). Therefore, the mechanism of oncogenesis is extremely complicated and controlled by various factors, not a gene or a few genes that cause malignant tumors. In recent years, the microarray technology has been exetensively used to get general hereditary modification amid during tumorigenesis (Guo et al., 2017; Xu et al., 2016; Gao et al., 2017). Microarray technology has been broadly utilized for the examination of general genetic deviations engaged with different disease. In any case, there are few investigations coordinating these Bioinformatics on endometrial cancer. In this paper, bioinformatics technique is utilized to examine the qualities and instruments of endometrial carcinoma.
Materials and Methods
Microarray data
The Gene Expression Omnibus (GEO, http://www.ncbi. nlm.nih.gov/geo) is an open database for biological information stockpiling, for example, microarray and cutting edge sequencing, which is uninhibitedly accessible to clients. It’s based high-throughput microarray and next-generation sequence functional genomic datasets submitted by the research community (Barrett et al., 2009). The quality articulation profiles of GSE17025 were downloaded from GEO database. GSE17025, which depended on Affymetrix Human Genome U133 Plus 2.0 Array GPL570 plat-form, Analysis of stage I endometrial cancer. Results give understanding into sub-atomic systems fundamental early endometrial malignancy histological sorts. The GSE17025 dataset contained 103 specimens, contain 91 samples of pathologically reviewed stage I endometrial cancers with a heterogeneous distribution of grade and depth of myometrial invasion were examined in relation to 12 samples of atrophic endometrium from postmenopausal women. Specimens were analyzed using oligonucleotide array analysis.
Data processing
The GEO database files an extensive number of high-throughput useful genomic thinks about that contain information that are handled and standardized utilizing different techniques. GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was connected to screen differentially communicated qualities between endometrial carcinoma and contrast examples. GEO2R provide a basic interface that enables clients to perform modern R-based investigation of GEO information to help distinguish and imagine differential quality articulation. GEO2R is an intelligent web instrument that looks at two gatherings of tests under the same trial conditions and can break down any GEO arrangement (Barrett et al., 2011). In this article, Using GEO2R was applied to screen differentially expressed between endometrial cancer and normal endometrial samples. The false positive result of microarray was then corrected by adjusted P value (adj.P). The adj. P were applied to correct for the occurrence of false positive results using Benjamini and Hochberg false discovery rate method by default. The littler the false positive rate of the littler value.Log FC is the distinction in quality articulation up - control, down - down logarithmic esteems, and diminishment in test measure. The DEG which Log FC<0 was the down-regulated gene, on the contrary, which gene was the up-regulated gene with Log FC>0. We set the adj. P <0.05 and |log FC| >2 were set as the cut-off criterion. This analysis of the meaningful differences in genes, the formation of volcanic charts, Blue point represents P ≥ 0.05, or | log FC | <2, red dot represents P <0.05 and | log FC |> 2, The blue dots in the figure are considered as genes that do not differ between patients with endometrial cancer and healthy individuals. The red dots in the picture are candidate genes for our analysis, and the genes with different significance in the study has clinical significance. analysis of the results will be shown in Figure 1.
Figure 1.
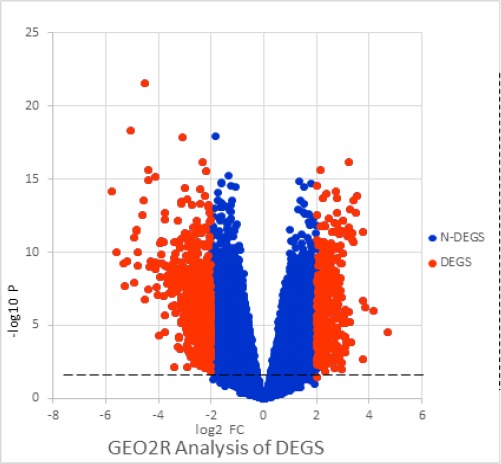
The Related Genes were Identified between Endometrial Cancer and Healthy Women. The blue points with P ≥ 0.05, or | log FC | <2. The red dots were meaningful with P <0.05 and | log FC |> 2.
String P-P-I
String Integration of protein– protein-interaction (PPI) organize Look Tool for the Search Tool for the Retrieval of Interacting Genes (STRING) database is online apparatus intended to assess the functional protein association networks. In the most recent variant 10.5 of STRING, the web frontend has been totally upgraded to lessen reliance on obsolete program innovations, and the database would now be able to likewise be questioned from inside the famous Cytoscape programming system (Szklarczyket al., 2017). Then, the molecular complex detection was performed to screen modules of PPI network with | log FC |> 2 and P <0.05. PPI interaction was performed on differentially expressed genes with significant differences in expression and visualized by Cytoscape.
Functional and pathway enrichment analysis
In present study, DAVID database was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) is a widely used public program that provides a comprehensive set of functional annotation tools for researchers to understand biological function behind abuntant of genes (Huang et al., 2009). The GO enrichment analysis were performed for identified DEGs using DAVID database. P < 0.05 was set as the cut-off criterion. It is constantly revised and expanded as biological knowledge accumulates. The GO describes function with respect to three aspects: molecular function (molecular-level activities performed by gene products), cellular component (the locations relative to cellular structures in which a gene product performs a function), and biological process (the larger processes, or ‘biological programs’ accomplished by multiple molecular activities) (S.Carbon et al., 2017). We uploaded all DEGs to the online software DAVID to identify overrepresented GO categories and KEGG pathways.
Results
Identify the DEGS
The gene chip GSE17025 contains 103 samples, 91 endometrial cancer patients and 12 healthy people, The 54,333 gene was analyzed by GEO2R, A total of 1,000 genes with | log FC |> 2 and P <0.05, of which 362 differentially expressed genes were up-regulated genes and 638 were down-regulated genes. The PCDH10 gene of which was a down-regulation expressed gene with | log FC | is 5.78. It follows that PCDH10 maybe the most significant genes in tumor differentiation, SLC6A2, OGN, SFRP4, TRH, ANGPTL, FOSB are down-regulated genes. The gene of IGH, CCL20, ELF5, LTF, ASPM expression level in tumor patients are up-regulated. These differences in the expression of genes P values were less than 0.05, indicating that these genes in the tumor patients and the control group were significantly different, shows in Table 1.
Table 1.
By Analyzed with GEO2R, the Highest Difference Genes with Down-Regulated and up-Regulated
| Gene.symbol | logFC | |log FC| | P.Value | adj.P.Val |
|---|---|---|---|---|
| PCDH10 | -5.778583 | 5.778583 | 6.93E-15 | 1.58E-11 |
| SLC6A2 | -5.607417 | 5.607417 | 9.85E-11 | 2.42E-08 |
| OGN | -5.342162 | 5.342162 | 5.56E-10 | 9.28E-08 |
| SFRP4 | -5.269629 | 5.269629 | 2.14E-08 | 1.63E-06 |
| TRH | -5.032782 | 5.032782 | 4.70E-19 | 1.28E-14 |
| ANGPTL1 | -4.907703 | 4.907703 | 1.16E-08 | 1.01E-06 |
| FOSB | -4.836907 | 4.836907 | 3.19E-12 | 1.61E-09 |
| CECR2 | -4.800043 | 4.800043 | 7.73E-10 | 1.20E-07 |
| IGH | 4.70326 | 4.70326 | 2.73E-05 | 0.000512 |
| CCL20 | 4.16092 | 4.16092 | 9.83E-07 | 3.46E-05 |
| ELF5 | 3.83392 | 3.83392 | 5.87E-07 | 0.000023 |
| LTF | 3.76273 | 3.76273 | 0.0021 | 0.0157 |
| ASPM | 3.76096 | 3.76096 | 4.15E-12 | 1.94E-09 |
| SAA | 3.74062 | 3.74062 | 2.23E-07 | 1.06E-05 |
| TOP2A | 3.52639 | 3.52639 | 1.39E-14 | 2.79E-11 |
| MELK | 3.47246 | 3.47246 | 2.18E-13 | 1.99E-10 |
| RRM2 | 3.40456 | 3.40456 | 2.16E-11 | 7.16E-09 |
| RNFT2 | 3.37986 | 3.37986 | 2.93E-14 | 4.55E-11 |
| MKI67 | 3.35593 | 3.35593 | 5.13E-12 | 2.32E-09 |
P-P-I network
The genes involved in the STRING website were enriched by the interaction between genes. The key gene action network is shown below Figure 2. STRING, The main network includes 494 points, 3,023 sides, of which 246 down-regulated genes, 234 up-regulated genes and 14 genes have no differentially expressed but is closely related to the differentially expressed genes. The highest degree of the gene in the network is 99, that is, have 99 genes closely related with it. There are 65 DGES with the number of DGES with the value greater than or equal to 30, among which there are 8 down-regulated genes, 57 of the up-regulated genes and 36 of the genes had degree greater or equal than 50, of which 2 genes are down-regulated and 34 genes are up-regulated.
Figure 2.
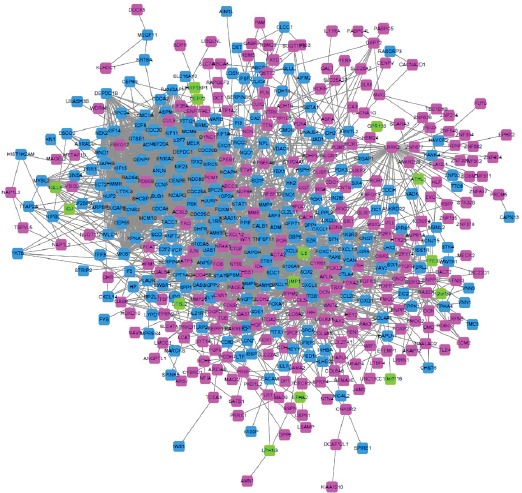
DEGS Protein-Protein-Interation Network. The pink points represent up-regulated genes, the blue points shows down-regulated genes, and the green points represent the genes which have no differentially expressed but is closely related to the differentially expressed genes.
Biological function analysis
The differential gene function was enriched by the DAVID database, down-regulated DGES function categories include Polymorphism, Alternative splicing, Glycoprotein, Transmembrane helix, Transmembrane, therefore, the up-regulated DGES function categories contain Coiled coil Signal, and Nucleus, shows in Table 2.
Table 2.
The DEGS’ Function were Analysis by the DAVID
| Attributes | Term | Count | PValue |
|---|---|---|---|
| DOWN-DGES | Polymorphism | 266 | 2.51E-04 |
| Alternative splicing | 237 | 4.91E-04 | |
| Glycoprotein | 138 | 5.42E-09 | |
| Transmembrane helix | 125 | 0.041314 | |
| Transmembrane | 125 | 0.044761 | |
| Signal | 123 | 2.70E-07 | |
| Metal-binding | 100 | 1.28E-04 | |
| Disulfide bond | 95 | 1.59E-04 | |
| Secreted | 74 | 2.66E-08 | |
| UP-DEGS | Coiled coil | 59 | 8.41E-05 |
| Signal | 56 | 0.010438 | |
| Nucleus | 23 | 0.014648 | |
| DNA-binding | 17 | 0.013645 | |
| Secreted | 14 | 0.006977 |
Function Categories
The differential gene function was analyzed by DAVID, The functional orientation of genes is described, to determine the role of each gene play a place. We used the differential gene into DAVID to analyze the main function of the down-regulated gene Polymorphism, Alternative splicing, Glycoprotein, and the effect of the up-regulated gene is Coiled coil, Signal, Nucleus. Then we can DEGS GO Function for detailed analysis, the result shows in Table 2.
GO term enrichment analysis
GO biological function enrichment contain three functional groups: molecular function group (MF), biological process group (BP), and cellular component group (CC). The key genes of down-regulated GO enrichment analysis of CC were plasma membrane, extracellular region, extracellular space, integral component of plasma membrane; the MF of them were zinc ion binding, metal ion binding, transcription factor activity DNA binding, and calcium ion binding; throw the BP of regulation RNA polymerase II promoter. positive cell proliferation, cell adhesion, and negative transcription. The up-regulated DEGs’CC were extracellular space, membrane, nucleus cytoplasm and extracellular exposome;t here MF were chemokine activity, ATP binding, transcriptional activator activity, sequence-specific DNA binding, RNA polymerase II core promoter DNA binding, their BP were chemokine-mediated signaling pathway, cell division, immune response, regulation of cell proliferation, G-protein coupled receptor signaling pathway (Figure 3).
Figure 3.
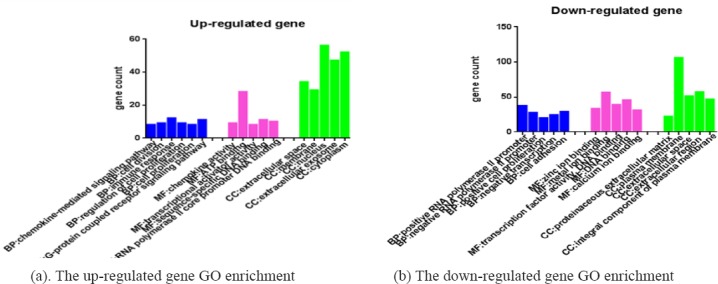
GO Biological Function Enrichment. Contain: Molecular Function Group(MF), Biological Process Group(BP), and Cellular Component Group(CC).
KEGG pathway analysis
As shown in Figure 4, contains the most significantly enriched pathways of the down-regulated DEGs and up-regulated DEGs analyzed by KEGG analysis. The down-regulated DEGs were enriched Metabolism of xenobiotics by cytochrome P450, MAPK signaling pathway, Rap1 signaling pathway, Focal adhesion pathways, while the down-regulated DEGs were enriched in IL-17 signaling pathway, Cell cycle, Toll-like receptor signaling pathway, p53 signaling pathway (Table 3).
Figure 4.
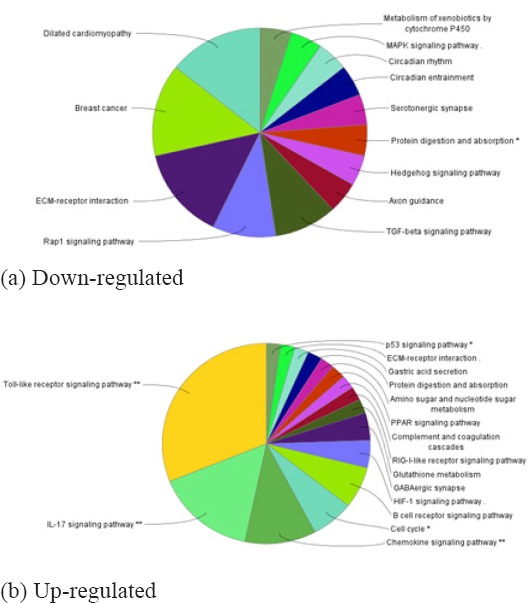
The DEGs Related Pathways were Analyzed by KEGG
Table 3.
The P-values and the Number of Genes in the Pathways by KEGG Analysis
| GOID | GO Term | Nr. Genes | Term P-Value | |
|---|---|---|---|---|
| DOWN | GO:0000980 | Metabolism of xenobiotics by cytochrome P450 | 3 | 0.16 |
| GO:0004010 | MAPK signaling pathway | 11 | 0.0082 | |
| GO:0004726 | Serotonergic synapse | 6 | 0.019 | |
| GO:0004974 | Protein digestion and absorption | 7 | 0.0014 | |
| GO:0004350 | TGF-beta signaling pathway | 5 | 0.02 | |
| GO:0004550 | Signaling pathways regulating pluripotency of stem cells | 6 | 0.046 | |
| GO:0004015 | Rap1 signaling pathway | 10 | 0.0059 | |
| GO:0005218 | Melanoma | 3 | 0.13 | |
| GO:0004512 | ECM-receptor interaction | 6 | 0.0042 | |
| GO:0005224 | Breast cancer | 6 | 0.053 | |
| GO:0005231 | Choline metabolism in cancer | 5 | 0.038 | |
| UP | GO:0004657 | IL-17 signaling pathway | 10 | 0.00001 |
| GO:0004062 | Chemokine signaling pathway | 13 | 0.000045 | |
| GO:0004066 | HIF-1 signaling pathway | 8 | 0.0007 | |
| GO:0004115 | p53 signaling pathway | 6 | 0.002 | |
| GO:0004620 | Toll-like receptor signaling pathway | 7 | 0.0038 | |
| GO:0004512 | ECM-receptor interaction | 6 | 0.0049 | |
| GO:0004068 | FoxO signaling pathway | 6 | 0.042 | |
| GO:0004933 | AGE-RAGE signaling pathway in diabetic complications | 5 | 0.042 | |
| GO:0004668 | TNF signaling pathway | 5 | 0.057 | |
| GO:0003320 | PPAR signaling pathway | 3 | 0.16 |
Disscusion
Endometrial carcinoma is the most common cancer of the female genital tract (Piulats et al., 2017). In the past decades, Numerous studies much has been learnt about the molecular mechanisms underlying EC disease from studies on human subjects, animals, or cell models to reveal the causes and underlying mechanisms of endometrial carcinoma and progression in the past several decades, but the incidence and mortality of EC is still very high in the world. However, the gene and mechanism of gene expression in endometrial cancer have not been systematically studied. In the present analysis, We analyzed GSE17025 gene chip obtained 1,000 different genes between EC and normal group, 362 up-regulated and 638 down-regulated genes. Large values of | log FC | are closely related to endometrial cancer. Protocadherin 10 (PCDH10) is differentially expressed in various human tumors. Previous studies have demonstrated that the expression of PCDH10 was noticeably downregulated in the tissue and cells of hepatocellular carcinoma (HCC), when compared to those in normal liver tissue throw inhibits cell proliferation and induces cell apoptosis by inhibiting the PI3K/Akt signaling pathway (Ye et al., 2017). In colorectal cancer, the progression of gastrointestinal stromal tumors or pancreatic cancer the PCDH10 gene all have down-regulated (Zhong et al., 2017; Lee et al., 2016; Qiu et al., 2016). And it is inactivated often by promoter hypermethylation in various human tumors. PCDH10 was down-regulated and a novel PCDH10-Wnt/beta-catenin-MALAT1 regulatory axis that contributes to EEC development suppressed cell growth and triggered apoptosis (Zhao et al., 2014). Transcriptional quieting by CpG island hypermethylation assumes a basic part though SFRP4 demonstrated demethylation in tumor through restrained WNT pathway and acted pathogenetic part in endometrial carcinogenesis (An et al., 2011). Experiments show that CCL20 contributed to invasion and EMT of RANK over-expressed EC cells (Liu et al., 2016). The differentially expressed genes STRING were enriched to form a differential gene interrelationship network. Its main network contain 494 points, 3,023 sides, of which 246 down-regulated genes, 234 up-regulated genes and 14 genes have no significant difference in expression difference but closely related to the network. TOP2A has the highest degree, patients with TOP2A-positive tumors had significantly lower overall survival than did patients with TOP2A-negative tumors, and disease-free survival for patients with TOP2A-positive tumors tended to be shorter than for those with TOP2A-negative tumors. TOP2A will be anthor potential molecular markers (Ito et al., 2016), the gene of GAPDH (Bersinger et al., 2010), BIRC5 (Chuwa et al., 2016), CCNB2 (Gayyed et al., 2016), and so they have a close relationship with the development and prognosis of endometrial cancer. Increased expression of CYP1 family indicates the possibility of carcinogenesis by exposure of xenobiotics in endometrial and ovarian cancers. CYP1 as a downstream genes including cytochrome P450 (CYP) 1 family members, involved metabolism of xenobiotics by cytochrome P450 pathway (Go et al., 2015). IL-17 is emerging as an important cytokine in cancer promotion and progression by sustains a chronic inflammatory microenvironment that favors tumor formation (Ling et al., 2015). While IL-17 may regulate chemokines and cytokines in gynecologic cancers. Toll-like receptors may play an important role in the development of gynecologic cancers by trigger an inflammatory response and cell survival in the tumor micro-environment (Husseinzadeh and Davenport, 2014). Integrated the above enrichment analysis, Endometrial cancer is closely associated with Serotonergic synapse Protein digestion and absorption, p53 signaling pathway, Toll-like receptor signaling pathway, FoxO signaling pathway.
In conclusion, those malignancies have distinct biology and molecular features and differ from each other based on clinical behavior, nevertheless all them are associated with high mortality. The use of bioinformatics method to analyze the differences between patients and normal genes, biological function pathway analysis, a more comprehensive interpretation of pathogenic mechanisms for further exploration of EC mechanisms and treatment to provide direction and basis, and then lay the foundation for targeted therapy. We can according to the direction of the above analysis the experimental study on the EC, bioinformatics analysis pointed out the direction for the research of disease, which laid a foundation for design of experiment.
Funding Statement
Project supported by the Natural Science Foundation of Liaoning pvovince, China(Grant No. 20170540373).
Project supported by the Jinzhou Foundation for Science and Technology, China (Grant No. 16B1G35).
Laboratory
Liaoning Provincial Key Laboratory of Follicle Development and Reproductive Health (Office of Science and Technology).
Appendix
Schedule 1
| share name | character | degree | Average shortestpathlength |
|---|---|---|---|
| TOP2A | UP | 99 | 2.352941 |
| GAPDH | UP | 95 | 2.058824 |
| BIRC5 | UP | 75 | 2.389452 |
| CCNB2 | UP | 70 | 2.578093 |
| NDC80 | UP | 66 | 2.705882 |
| BUB1 | UP | 65 | 2.789047 |
| CDC20 | UP | 64 | 2.756592 |
| KIF11 | UP | 63 | 2.669371 |
| JUN | DOWN | 61 | 2.286004 |
| CDCA8 | UP | 61 | 2.782961 |
| FOXM1 | UP | 60 | 2.427992 |
| KIF20A | UP | 60 | 2.782961 |
| TTK | UP | 60 | 2.805274 |
| CENPE | UP | 59 | 2.782961 |
| CENPF | UP | 59 | 2.807302 |
| PBK | UP | 59 | 2.517241 |
| TPX2 | UP | 59 | 2.69574 |
| KIF23 | UP | 58 | 2.787018 |
| NCAPG | UP | 58 | 2.807302 |
| ASPM | UP | 57 | 2.803245 |
| MELK | UP | 57 | 2.703854 |
| DLGAP5 | UP | 56 | 2.813387 |
| KIF15 | UP | 56 | 2.801217 |
| NEK2 | UP | 56 | 2.730223 |
| RRM2 | UP | 56 | 2.592292 |
| CEP55 | UP | 54 | 2.730223 |
| HMMR | UP | 54 | 2.817444 |
| LRRK2 | DOWN | 53 | 2.537525 |
| KIAA0101 | UP | 53 | 2.667343 |
| NUF2 | UP | 53 | 2.825558 |
| EXO1 | UP | 52 | 2.634888 |
| MCM10 | UP | 52 | 2.831643 |
| PTTG1 | UP | 52 | 2.718053 |
| ANLN | UP | 50 | 2.831643 |
| MCM4 | UP | 50 | 2.811359 |
| MMP9 | UP | 50 | 2.37931 |
| CDC25C | UP | 49 | 2.578093 |
| HJURP | UP | 48 | 2.841785 |
| MKI67 | UP | 47 | 2.801217 |
| NCAPH | UP | 47 | 2.837728 |
| SHCBP1 | UP | 47 | 2.84787 |
| DEPDC1 | UP | 46 | 2.849899 |
| KIF14 | UP | 46 | 2.851927 |
| MND1 | UP | 45 | 2.890467 |
| SPC25 | UP | 45 | 2.853955 |
| STAT3 | UP | 45 | 2.448276 |
| FOS | DOWN | 44 | 2.484787 |
| ECT2 | UP | 44 | 2.839757 |
| RAD54L | UP | 44 | 2.845842 |
| GTSE1 | UP | 43 | 2.853955 |
| ZWILCH | UP | 41 | 2.922921 |
| share name | character | degree | Average shortestpathlength |
| CXCL12 | DOWN | 40 | 2.557809 |
| CCNF | UP | 39 | 2.884381 |
| ACTA2 | DOWN | 37 | 2.356998 |
| CDCA2 | UP | 36 | 2.929006 |
| KPNA2 | UP | 36 | 2.864097 |
| DCN | DOWN | 32 | 2.651116 |
| THBS1 | DOWN | 32 | 2.567951 |
| CDC25A | UP | 32 | 2.634888 |
| ITGB1 | UP | 32 | 2.527383 |
| HGF | DOWN | 31 | 2.598377 |
| FAM83D | UP | 31 | 2.93712 |
| E2F8 | UP | 30 | 3.040568 |
| SPP1 | UP | 30 | 2.498986 |
| TACC3 | UP | 30 | 2.8357 |
References
- 1.Amant F, Moerman P, Neven P, et al. Endometrial cancer. Lancet. 2005;366:491–505. doi: 10.1016/S0140-6736(05)67063-8. [DOI] [PubMed] [Google Scholar]
- 2.An SJ, Pandeya D, Park SW, et al. Epigenetic fingerprint in endometrial carcinogenesis:the hypothesis of a uterine field cancerization. Cancer Biol Ther. 2011;12:447–57. doi: 10.4161/cbt.12.5.15963. [DOI] [PubMed] [Google Scholar]
- 3.Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO:archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:885–90. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO:archive for functional genomics data sets--update. Nucleic Acids Res. 2011;41:991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bersinger NA, Schneider B, Vorburger SA, et al. Prognostic value of tumour endothelial markers in patients with endometrial cancer. Oncol Lett. 2010;1:203–7. doi: 10.3892/ol_00000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bregar A, Deshpande A, Grange C, et al. Characterization of immune regulatory molecules B7-H4 and PD-L1 in low and high grade endometrial tumors. Gynecol Oncol. 2017;145:446–52. doi: 10.1016/j.ygyno.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China 2015. CA Cancer J Clin. 2016;66:115. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 8.Chuwa AH, Sone K, Oda K, et al. Significance of survivin as a prognostic factor and a therapeutic target in endometrial cancer. Gynecol Oncol. 2016;141:564–9. doi: 10.1016/j.ygyno.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Espinosa I, Lee CH, Angelo ED, Palacios J, Prat J. Undifferentiated and dedifferentiated endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Am J Surg Pathol. 2017;41:1121–8. doi: 10.1097/PAS.0000000000000873. [DOI] [PubMed] [Google Scholar]
- 10.Gao H, Wang H, Wei Y. Identification of key genes and construction of microRNA–mRNA regulatory networks in multiple myeloma by integrated multiple GEO datasets using bioinformatics analysis. IJHOBMT. 2017;106:1–9. doi: 10.1007/s12185-017-2216-2. [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Lin P, Lydon JP, Li Q. Conditional abrogation of Transforming Growth Factor Beta Receptor 1 in PTEN-inactivated endometrium promotes endometrial cancer progression in mice. J Pathol. 2017;243:89–99. doi: 10.1002/path.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gayyed MF, Elmaqsoud NM, Tawfiek ER, et al. A comprehensive analysis of CDC20 overexpression in common malignant tumors from multiple organs:its correlation with tumor grade and stage. Tumour Biol. 2016;37:749–62. doi: 10.1007/s13277-015-3808-1. [DOI] [PubMed] [Google Scholar]
- 13.Go RE, Hwang KA, Choi KC. Cytochrome P450 1 family and cancers. J Steroid Biochem Mol Biol. 2015;147:24–30. doi: 10.1016/j.jsbmb.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Bao Y, Ma M, Yang W. Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci. 2017;18:834–40. doi: 10.3390/ijms18040722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 16.Husseinzadeh N, Davenport SM. Role of Toll-like receptors in cervical, endometrial and ovarian cancers:A review. Gynecol Oncol. 2014;135:359–63. doi: 10.1016/j.ygyno.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Ito F, Furukawa N, Nakai T. Evaluation of TOP2A as a predictive marker for endometrial cancer with taxane-containing adjuvant chemotherapy. Int J Gynecol Cancer. 2016;26:325–30. doi: 10.1097/IGC.0000000000000607. [DOI] [PubMed] [Google Scholar]
- 18.Kandoth C, Schultz N, Cherniack A D, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee NK, Lee JH, Kim WK, et al. Promoter methylation of PCDH10 by HOTAIR regulates the progression of gastrointestinal stromal tumors. Oncotarget. 2016;7:75307–18. doi: 10.18632/oncotarget.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ling W, Xing C, Zhao J, et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4–ERK5 axis. J Exp Med. 2015;212:1571–87. doi: 10.1084/jem.20150204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Wang J, Ni T, et al. CCL20 mediates RANK/RANKL-induced epithelial-mesenchymal transition in endometrial cancer cells. Oncotarget. 2016;7:25328–39. doi: 10.18632/oncotarget.8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin SA, Mccabe N, Mullarkey M, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17:235–48. doi: 10.1016/j.ccr.2009.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piulats JM, Guerra E, Gilmartín M, et al. Molecular approaches for classifying endometrial carcinoma. Gynecol Oncol. 2017;145:200–7. doi: 10.1016/j.ygyno.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Qiu C, Bu X, Jiang Z. Protocadherin-10 acts as a tumor suppressor gene, and is frequently downregulated by promoter methylation in pancreatic cancer cells. Oncol Rep. 2016;36:383–9. doi: 10.3892/or.2016.4793. [DOI] [PubMed] [Google Scholar]
- 25.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;56:106. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 26.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:362–8. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:331–8. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Z, Yu Z, Cao Y, et al. Identification of candidate biomarkers and analysis of prognostic values in ovarian cancer by integrated bioinformatics analysis. Med Oncol. 2016;33:130. doi: 10.1007/s12032-016-0840-y. [DOI] [PubMed] [Google Scholar]
- 29.Xue LC, Xu ZW, Wang KH, et al. Regulatory network of microRNAs, target genes, transcription factors and host genes in endometrial cancer. Asian Pac J Cancer Prev. 2015;16:475–83. doi: 10.7314/apjcp.2015.16.2.475. [DOI] [PubMed] [Google Scholar]
- 30.Ye M, Li J, Gong J. PCDH10 gene inhibits cell proliferation and induces cell apoptosis by inhibiting the PI3K/Akt signaling pathway in hepatocellular carcinoma cells. Oncol Rep. 2017;37:3167–74. doi: 10.3892/or.2017.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Yang Y, Trovik J, et al. A novel wnt regulatory axis in endometrioid endometrial cancer. Cancer Res. 2014;74:5103–17. doi: 10.1158/0008-5472.CAN-14-0427. [DOI] [PubMed] [Google Scholar]
- 32.Zhong X, Shen H, Mao J, Zhang J, Han W. Epigenetic silencing of protocadherin 10 in colorectal cancer. Oncol Lett. 2017;13:2449–53. doi: 10.3892/ol.2017.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]