

Abstract
Natural products have historically been a major source of antibiotics and therefore novel scaffolds are constantly of interest. The lipoxazolidinone family of marine natural products, with an unusual 4-oxazolidinone heterocycle at their core, represents a new scaffold for antimicrobial discovery; however, questions regarding their mechanism of action and high lipophilicity have likely slowed follow-up studies. Herein, we report the first synthesis of lipoxazolidinone A, 15 structural analogues to explore its active pharmacophore, and initial resistance and mechanism of action studies. These results suggest that 4-oxazolidinones are valuable scaffolds for antimicrobial development and reveal simplified lead compounds for further optimization.
Keywords: antibiotics, cyclization, heterocycles, medicinal chemistry, natural products
Cyclize for antibiotics
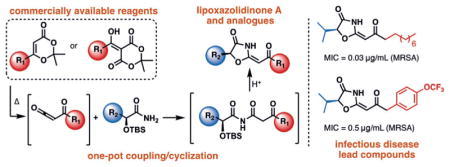
The first synthesis of lipoxazolidinone A and 15 structural analogues via a one-pot coupling/cyclization of β-keto-imides is reported. These studies revealed potent small molecules with improved properties, slow resistance development, and intriguing potential mechanisms of action. These results set the stage for the future development of 4-oxazolidinones as novel antibiotics.
Bacterial infections represent a significant threat to human and animal health and there is therefore a critical need for the development of antimicrobial agents.[1] With clinical resistance observed for all classes of antibiotics, there is a pressing need for the development of antibiotics with new, or multiple, mechanisms of action. As of 2013, the Center for Disease Control and Prevention (CDC) identified that antibiotic resistance contributed to more than two million illnesses and 23000 deaths in the United States alone.[2] Further, previously treatable infections are becoming a renewed concern with high resistance resulting in mortality rates for infections such as methicillin-resistant S. aureus (MRSA) of 14%.[2] Historically, natural products have held a unique place in infectious disease drug discovery, with the majority of antibiotics either being natural products or derivatives thereof.[3–10]
Lipoxazolidinone A (1) was isolated in 2002 from marine sediments collected off of the coast of Guam (Figure 1a).[11] This family of natural products contains a unique 4-oxazolidinone core with an exocyclic conjugated ketone moiety and two lipophilic carbon chains. The lipoxazolidinones are reported to be antimicrobial agents against Gram-positive pathogens (≈ 1 μgmL−1 MIC against MRSA) via an unknown mechanism of action. The synoxazolidinone family of natural products, highlighted by synoxazolidinone A (2) is the only other class of 4-oxazolidinone natural products; albeit, they possess diminished antimicrobial activity and instead possess potent anti-biofilm activity (Figure 1a).[12–16] The structurally related 2-oxazolidinone antibiotics, represented by linezolid (3) and tedizolid (4), are synthetic molecules that target bacterial protein synthesis (Figure 1b). Herein, we describe our efforts to synthesize the lipoxazolidinone family of natural products, a series of targeted analogues and provide insights into their susceptibility to resistance and their mechanism of antimicrobial action.
Figure 1.

4-Oxazolidinone-containing natural products and related antibiotics. a) Two families of 4-oxazolidinone natural products isolated to date; b) synthetic 2-oxazolidinones developed for the treatment of Gram-positive bacterial infections; c) a one-pot synthetic strategy to synthesize the 4-oxazolidinone core of lipoxazolidinone A.
Given the novelty of the lipoxazolidinone core, and the antimicrobial activity possessed by this family of natural products, we embarked on efforts to develop a concise synthesis of 1. We envisioned accessing the 4-oxazolidinone core (10) from a cascade consisting of intramolecular cyclization/dehydration of a β-keto imide, formed from coupling of α-hydroxy amide 8 and α-keto ketene derivative 7 (Figure 1c). The use of α-hydroxy amides as key building blocks was particularly desirable since various analogues can be readily prepared in enantiopure form from amino acids through a three-step sequence.[17] The α-keto ketene was envisioned to arise from dioxinone 5 or the acylation of Meldrum’s acid with a carboxylic acid derivative (6). Although ketenes are known to react with amides to form imides, there are limited reports for the synthesis of β-keto imides, and no previous reports of the coupling of amides with acyl ketenes possessing an α,β-unsaturated ketone moiety as would be required for the synthesis of lipoxazolidinone A (1) itself.[18]
Initially, simplified analogue 14 was targeted to test the proposed reaction sequence (Figure 2a). Employing the TBS-protected (S)-lactamide 12, the desired imide was obtained in 88% yield after reflux with dioxinone 11. Upon subsequent treatment with KF or TBAF, none of the desired cyclized product was obtained, with only loss of the TBS protecting group being observed; however, treatment of imide 13 with trifluoroacetic acid in dichloromethane (1:1) led to the desired 4-oxazolidinone 14 in 64% yield. The structure of 14 was confirmed by X-ray analysis. Following these initial studies, it was found that a two-step, one-pot procedure was more convenient and higher yielding, providing 14 in 62% yield from 11 and 12 (Figure 2b).
Figure 2.

4-Oxazolidinone method development and synthesis of lipoxazolidinone A. a) initial reaction development for the preparation of 4-oxazolidinones; b) optimized one-pot protocol; c) synthesis of ketene precursor and subsequent conversion to lipoxazolidinone A.
At this stage we moved to extend this approach to the synthesis of lipoxazolidinone A (1). Dioxinone 16 was prepared in 73% yield from β-keto tert-butyl ester 15,[19,20] and was allowed to react with α-hydroxy amide 17 (prepared from L-norleucine in 3 steps, see Supporting Information)[21–24] under our optimized protocol for 4-oxazolidinone formation, providing 1 in 52% yield. The absolute configuration of the natural product, previously unreported, was established to be (S)-1 by comparison of its optical rotation with that reported by the isolation group ([α]D −34° vs. [α]D −31° (lit)).[11] We further confirmed this by preparing the enantiomer of 17 and carrying this through the synthetic route to provide 18 (Figure 3a). This enantiomeric compound demonstrated an equal, but opposite rotation as expected ([α]D + 30°). Additionally, lipoxazolidinone A (1) was evaluated in antimicrobial assays against S. aureus[25a] and methicillin-resistant S. aureus (MRSA)[25b] and was found to possess activity of 1 μgmL−1 against S. aureus and 0.5 μgmL−1 against MRSA, in line with what was reported by the isolation group.[11]
Figure 3.

Structure–activity profile of lipoxazolidinone A (1). a) Key structural modifications were performed around the molecule to probe the role of various changes on antimicrobial activity; b) initial evaluation of antimicrobial activity against two S. aureus strains with red highlighting compounds with significant activity or structural modifications. 1 ATCC 29213; 2 ATCC 33591.
With the natural product (1) and its enantiomer (18) in hand, we sought to ask fundamental questions regarding the structure–activity profile of these lipophilic natural products. At the outset of our studies it was unclear if this family of natural products was antimicrobial due to their insertion into bacterial membranes, or if instead, there was a key pharmacophore present in this scaffold that warranted further investigation. To begin to address this question, a series of derivatives were prepared to define the structural elements required for antimicrobial activity and to set the stage for more in-depth mechanism of action studies (Figure 3a, see Supporting Information for synthetic protocols). Initially, we utilized MIC assays on the same strains of S. aureus and MRSA as was used for the parent natural product to determine the relative activity of our targeted analogues (Figure 3b).
Stereochemical and alkene substitution changes were first explored to evaluate the significance of varying the heterocyclic core (vs. lipophilic side chains) and the impact of the Michael acceptor on antimicrobial activity. We first explored the impact of stereochemical changes with the enantiomer of the natural product (18) being 2–4 times less active than 1 (Figure 2b). Although this is not a significant difference given the error associated with microdilution MIC assays, it provided an initial glimpse that the core structure of the natural products may indeed drive their activity, at least in part. Deletion of the alkenyl methyl group (19) resulted in an increase of activity; however, deletion of the alkene (20) provided a compound with equipotent activity to that of 1 highlighting that the potential Michael acceptor is not an important factor in antimicrobial activity (Figure 2b).
Another key question for the structure–activity profile study was the role of the lipophilic side chain. To this end, truncated (21, 22) and extended (23) analogues were prepared and, in all cases, a significant decrease in activity was observed. These results were initially discouraging; however, it was our next set of compounds that replaced the N—H of the oxazolidinone with an N—Me group (24), and replaced the oxygen of the exocyclic carbonyl group with sulfur (25)[26] that provided key insights. In both cases, significant decreases in activity were observed, highlighting that single-atom changes and/or removal of the N—H group had profound impacts on the antimicrobial activity (Figure 3b). This level of structure–activity detail would not be expected for molecules that function mainly by physical effects or membrane permeabilization, and in fact, both 24 and 25 are more nonpolar than 1, highlighting that there is not a correlation with polarity and antimicrobial activity.
We next explored the sidechain of the amino acid-derived portion of the molecule, varying the linear butyl group with benzyl (26), isobutyl (27), hydrogen (28), and isopropyl (29) substituents (Figure 3a). To our surprise, whereas 26 and 28 had slightly reduced activity overall, the branched alkyl derivatives 27 and 29 had improved activity (0.0338–0.5 μgmL−1), particularly against the MRSA strain ATCC 33591 (Figure 3b). Although this is only a narrow snapshot of the potential utility of these branched derivatives, we sought to explore whether coupling the isopropyl substitution with shorter, or more polar, side chains on the ketene-derived portion could provide potent antimicrobial compounds. Indeed, installation of benzyl (30), p-bromobenzyl (31), p-trifluoromethoxybenzyl (32) and n-propylbenzene (33) groups provided compounds with potent activity, highlighted by 31 and 32 which possess potency equivalent to that of lipoxazolidinone A (1), but with significantly lowered cLogP values (≈ 3.8 vs. ≈ 6.6). Current efforts are aimed at continuing to increase the polarity of the molecules without sacrificing potency.
To further define the activity of our compounds against clinically relevant strains of Gram-positive bacteria, 1 and 29 were tested against other resistant strains of MRSA and S. epidermidis. Compound 29 exhibited significant potency against linezolid-, tetracycline-, and erythromycin-resistant S. aureus and several S. epidermidis strains, including vancomycin-intermediate S. epidermidis (NR45860) (Table 1). We have also evaluated 1 and several derivatives against Gram-negative pathogens such as A. baumannii (ATCC 19606); however, we have not achieved sufficient activity to date.[27] To demonstrate that the lack of activity in Gram-negative pathogens is due to cellular uptake and not lack of target or other resistance mechanisms we prepared an LPS-deficient A. baumannii (ATCC 19606) and subjected it to an MIC assay with 29. Gratifyingly, an MIC of 2 μgmL−1 was obtained and this result highlights the potential to chemically evolve this class of compounds to achieve Gram-negative activity going forward.
Table 1.
Antimicrobial activity against additional strains of MRSA, S. epidermidis, and A. baumannii.
| Strain | 1 MIC [μgmL−1] | 29 MIC [μg mL−1] |
|---|---|---|
| MRSA NR45924 (Lin resistant)[a] | 0.5 | 0.125 |
| MRSA NR45926 (Lin resistant)[a] | 0.25 | 0.0675 |
| MRSA NR45930 (Lin resistant)[a] | 0.5 | 0.25 |
| MRSA NR46062 (Lin resistant)[a] | 2 | 0.5 |
| MRSA NR45906 (Tet resistant)[b] | 0.25 | 0.25 |
| MRSA NR45898 (Ery resistant)[c] | 1 | 1 |
| S. epidermidis NR46376 | 2 | 0.25 |
| S. epidermidis NR46379 | 1 | 0.5 |
| S. epidermidis NR45860 (VISE)[d] | 1 | 1 |
| A. baumannii ATCC 19606 | >128 | 128 |
| A. baumannii ATCC 19606R[e] | n/d | 2 |
Lin =linezolid.
Tet =tetracycline.
Ery =erythromycin.
VISE =vancomycin intermediate.
LPS deficient A. baumannii generated via treatment with colistin.
The next aspect of our initial evaluation of 29 was focused on resistance studies. To explore the frequency of resistant mutant generation we conducted single-pass resistance studies by plating bacteria in their exponential growth phase on agar containing 2–10 × MIC of 29. We were unable to obtain mutants of S. aureus resistant to 29 even when plating on media with a low dose (4 × MIC) of the compound, providing a calculated frequency of resistance of <10−10.[7] Given this surprising lack of resistance development we sought to attempt to generate a resistant mutant through serial passage of S. aureus with sub-inhibitory concentrations of 29. Utilizing this approach we were able to slowly generate a mutant under forcing conditions (≈ 2 × MIC at 30 days, ≈ 64 × MIC at 50 days) (Figure 4, see Supporting Information for complete protocol).[7] Current efforts are focused on understanding the mechanisms by which resistance is achieved, although the lack of mutants via single exposure and the extremely slow generation of mutants upon serial passage (relative to most commonly employed antibiotics) is an encouraging starting point for this class of compounds going forward.
Figure 4.
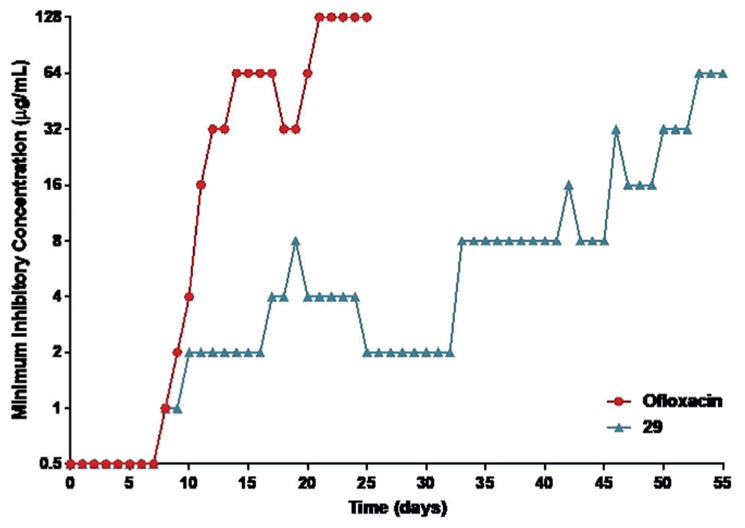
Serial passage resistance studies of 29 and ofloxacin on S. aureus ATCC 29213. Resistant mutants were generated for both ofloxacin and 29 through the daily sequential passage of the bacterial with a range of concentrations of antibiotic.
In addition to the structure–activity profile and resistance studies, we have begun efforts to define the mechanism of action for this class of antibiotics. Initially, given the highly lipophilic nature of the parent compounds and their structural similarities with antibiotics such as the reutericyclins, we sought to explore the effect of these compounds on membrane integrity.[28] We subjected the compounds to a red blood cell hemolysis assay (defibrinated sheep blood),[29] revealing less than 1% hemolysis at 40 μM for both natural product 1 and compound 29. Furthermore, a BacLight™ assay was utilized with S. aureus ATCC 29213 to evaluate the membrane permeabilization.[30] It was determined that both 1 and 29 do not significantly alter the bacterial membrane up to 8 × MIC (highest concentration tested, see Supporting Information for experimental details). Finally, 29 does not have broadly toxic effects on mammalian HepG2 cells up to 50 μgmL−1 (highest concentration tested).[31] Together these data demonstrate that the primary mechanism of antimicrobial action does not originate from membrane activity or broad toxicity and inspired further studies to explore how these molecules function.
To this end, we sought to investigate the effect of 29 on macromolecular synthesis pathways and utilized a radiolabeled macromolecular synthesis assay to explore the impact upon treatment with 29 at various concentrations.[32] The percentage of incorporation of radiolabeled precursors into macromolecules was determined upon treatment with 29 and was compared with results obtained after treatment with antibiotics of known mechanism (Figure 5). These studies reveal strong, dose-dependent inhibition of cell wall synthesis and protein synthesis at concentrations as low as 0.125 × MIC, whereas limited inhibition of DNA, RNA, or lipid synthesis was observed until >2 × MIC. This dual pathway inhibition, and lack of membrane permeabilization, provides exciting preliminary data warranting an array of follow-up studies. As an initial confirmation of protein synthesis, we have conducted an in vitro transcription/translation assay and demonstrated that 29 indeed inhibits protein synthesis at similar potency to that of Linezolid (see Supporting Information for data and experimental details). It is intriguing to consider whether the observed dual cell wall/protein synthesis activities are due to interrelated macromolecular synthesis pathways or independent molecular targets.[33]
Figure 5.
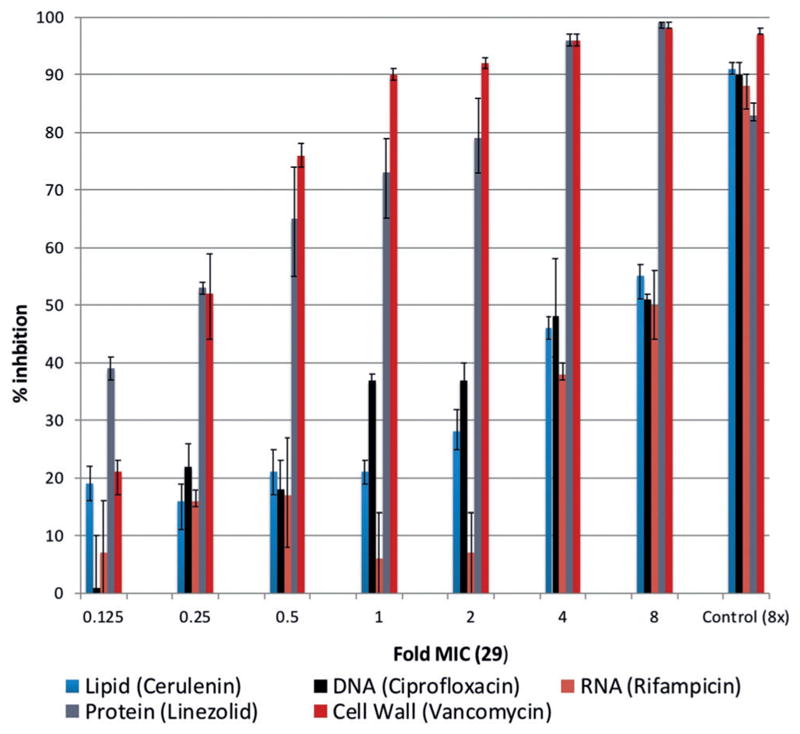
Macromolecular synthesis assay. Radiolabeled precursors (color coded by biosynthetic pathway) were employed with ascending concentrations of 29 to measure the relative impact on macromolecular synthesis pathways. Control antibiotics shown at right.
In conclusion, we have developed a rapid synthetic approach to prepare lipoxazolidinone A and a series of designed analogues to explore the structure–activity profile of this novel antibiotic scaffold. Not only have simplified, more polar derivatives proven active across a range of resistant S. aureus and S. epidermidis strains, they exert their activity via the inhibition of both cell-wall and protein synthesis. This exciting finding further reveals itself in the slow development of resistance during prolonged exposure. Together, these findings set the groundwork for continued study and development of the 4-oxazolidinone scaffold and these studies will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful to the NIH (1R01GM110154) for generous support of this work and to North Carolina State University for support of our program. Partial funding for this work was also obtained through the North Carolina State University Chancellor’s Innovation Fund. Mass spectrometry data, NMR data, and X-ray data were obtained at the North Carolina State Molecular, Education, Technology, and Research Innovation Center (METRIC). Dr. Roger Sommer is acknowledged for obtaining an X-ray of 14. The authors thank Prof. Christian Melander and Dr. Roberta Melander for helpful advice throughout the progression of this project. We also acknowledge the technical expertise of Dean Shinabarger and Bev Murray of Micromyx (Kalamazoo, MI, USA), who performed the macromolecular synthesis inhibition studies. The bacterial reagents in Table 1 were provided by the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for distribution by BEI Resources, NIAID, NIH.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201805078.
References
- 1.Infectious Diseases Society of America (IDSA) Clin Infect Dis. 2011;52:S397. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. Antibiotic Resistance Threats in the United States. 2013. [Google Scholar]
- 3.Xie J, Pierce JG, James RC, Okano A, Boger DL. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh CT, Wencewicz TA. J Antibiot. 2014;67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 5.O’Connell KMG, Hodgkinson JT, Sore HF, Welch M, Salmond GPC, Spring DR. Angew Chem Int Ed. 2013;52:10706–10733. doi: 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:10904–10932. [Google Scholar]
- 6.Brown DG, Lister T, May-Dracka TL. Bioorg Med Chem Lett. 2014;24:413–418. doi: 10.1016/j.bmcl.2013.12.059. [DOI] [PubMed] [Google Scholar]
- 7.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patridge E, Gareiss P, Kinch MS, Hoyer D. Drug Discovery Today. 2016;21:204–207. doi: 10.1016/j.drudis.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Wright GD. Nat Prod Rep. 2017;34:694–701. doi: 10.1039/c7np00019g. [DOI] [PubMed] [Google Scholar]
- 10.Rossiter SE, Fletcher MH, Wuest WM. Chem Rev. 2017;117:12415–12474. doi: 10.1021/acs.chemrev.7b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macherla VR, Liu J, Sunga M, White DJ, Grodberg J, Teisan S, Lam KS, Potts BCM. J Nat Prod. 2007;70:1454–1457. doi: 10.1021/np0702032. [DOI] [PubMed] [Google Scholar]
- 12.Tadesse M, Strøm MB, Svenson J, Jaspars M, Milne BF, Tørfoss V, Andersen JH, Hansen E, Stensvåg K, Haug T. Org Lett. 2010;12:4752–4755. doi: 10.1021/ol101707u. [DOI] [PubMed] [Google Scholar]
- 13.Tadesse M, Svenson J, Jaspars M, Strøm MB, Abdelrahman MH, Andersen JH, Hansen E, Kristiansen PE, Stensvåg K, Haug T. Tetrahedron Lett. 2011;52:1804–1806. [Google Scholar]
- 14.Edwards GA, Shymanska NV, Pierce JG. Chem Commun. 2017;53:7353–7356. doi: 10.1039/c7cc03626d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H, Ling T, Wei S, Zhang C. Rec Nat Prod. 2015;9:247. [Google Scholar]
- 16.Shymanska NV, An IH, Guevara-Zuluaga S, Pierce JG. Bioorg Med Chem Lett. 2015;25:4887–4889. doi: 10.1016/j.bmcl.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Menche D, Hassfeld J, Li J, Mayer K, Rudolph S. J Org Chem. 2009;74:7220–7229. doi: 10.1021/jo901565n. [DOI] [PubMed] [Google Scholar]
- 18.Guo C. Tetrahedron Lett. 2010;51:548–549. [Google Scholar]
- 19.Carroll MF, Bader AR. J Am Chem Soc. 1953;75:5400–5402. [Google Scholar]
- 20.Reber KP, Tilley SD, Sorensen EJ. Chem Soc Rev. 2009;38:3022–3034. doi: 10.1039/b912599j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalafi-Nezhad A, Parhami A, Soltani Rad MN, Zarea A. Tetrahedron Lett. 2005;46:6879–6882. [Google Scholar]
- 22.Kapferer T, Brückner R, Herzig A, Kçnig WA. Chem Eur J. 2005;11:2154–2162. doi: 10.1002/chem.200401135. [DOI] [PubMed] [Google Scholar]
- 23.Ivanov AS. Chem Soc Rev. 2008;37:789–811. doi: 10.1039/b716020h. [DOI] [PubMed] [Google Scholar]
- 24.Sørensen US, Falch E, Krogsgaard-Larsen P. J Org Chem. 2000;65:1003–1007. doi: 10.1021/jo991409d. [DOI] [PubMed] [Google Scholar]
- 25.a) ATCC 29213; b) ATCC 33591.
- 26.Compound 25 is prepared by the thionation of 20 with Lawesson’s reagent. This reaction provides complete selectivity for the external carbonyl, highlighting the unique properties of these vinylogous imide-like species. See Supporting Information for complete experimental details.
- 27.For recent approaches toward overcoming lack of Gram-negative activity and discussion of Gram-negative compound development, see: Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ. Nature. 2017;545:299–304. doi: 10.1038/nature22308.Silver LL. Bioorg Med Chem. 2016;24:6379–6389. doi: 10.1016/j.bmc.2016.06.044.
- 28.Cherian PT, Wu X, Maddox MM, Singh AP, Lee RE, Hurdle JG. Sci Rep. 2014;4:4721. doi: 10.1038/srep04721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Brady A, Young A, Rasimick B, Chen K, Zhou C, Kallenbach NR. Antimicrob Agents Chemother. 2007;51:597–603. doi: 10.1128/AAC.00828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, Sandvik E, Hubbard JM, Kaniga K, Schmidt DE, Gao Q, Cass RT, Karr DE, Benton BM, Humphrey PP. Antimicrob Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Additional studies to more broadly evaluate the toxicity of the 4-oxazolidinones in vitro and in vivo are in progress.
- 32.Cotsonas King A, Wu L. Curr Protoc Pharmacol. 2009;chap 13:Unit 13A.7–13A.7.23. doi: 10.1002/0471141755.ph13a07s47. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham ML, Kwan BP, Nelson KJ, Bensen DC, Shaw KJ. J Biomol Screening. 2013;18:1018–1026. doi: 10.1177/1087057113487208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.