

Abstract
We studied the effects of gut microbiome depletion by oral antibiotics on tumor growth in subcutaneous and liver metastases model of pancreatic cancer, colon cancer and melanoma. Gut microbiome depletion significantly reduced tumor burden in all the models tested. However, depletion of gut microbiome did not reduce tumor growth in Rag1-knockout mice, which lack mature T and B cells. Flowcytometry analyses demonstrated that gut microbiome depletion led to significant increase in interferon gamma-producing T cells with corresponding decrease in interleukin 17A and interleukin 10-producing T cells. Our results suggest that gut microbiome modulation could emerge as a novel immunotherapeutic strategy.
Keywords: gut bacteria, immune regulation, tumor promotion, metastases
There are more resident microbes in the human body than there are ‘human’ cells and most of these microbes occupy an ambiguous niche in the gut. The gut microbiota, forming a unique metagenome, is dynamic and changes with a person's nutrition state, geography and even age. A growing body of evidence hints towards a co-evolved relationship between gut microbes and our immune system1. In fact, some inflammatory diseases like colitis are characterized by a transition in the gut microbiome, which changes from a ‘eubiotic’ to a ‘dysbiotic’ state, with interesting therapeutic implications2. Although several epidemiological studies associate dysbiosis with cancer, the exact role of gut bacteria in the pathogenesis of cancer is still unclear.
We evaluated the impact of gut microbiome depletion on tumor growth in multiple mouse models. Gut microbiome was depleted in age and sex-matched C57BL/6J mice with a broad-spectrum cocktail of oral antibiotics (Vancomycin, Neomycin, Metronidazole, Ampicillin and Amphotericin B) using a well-established protocol3 (Figure 1A). Mice, with or without gut microbiome depletion, were used to establish cancer models by subcutaneous injection of i) KPC pancreatic cancer cells derived from tumors forming in KrasG12D/+; Trp53R172H/+; Pdx-1cre mice4; or ii) melanoma cells derived from tumors forming in Tyr-CreER; Braf V600E/+; Ptenfl/fl mice5 , and by splenic injection of i) KPC cells; or ii) B16-F10 melanoma cells; or iii) MC38 colon cancer cells to induce liver metastases.
Figure 1. Depletion of gut microbiome decreases tumor burden in multiple models of cancer.
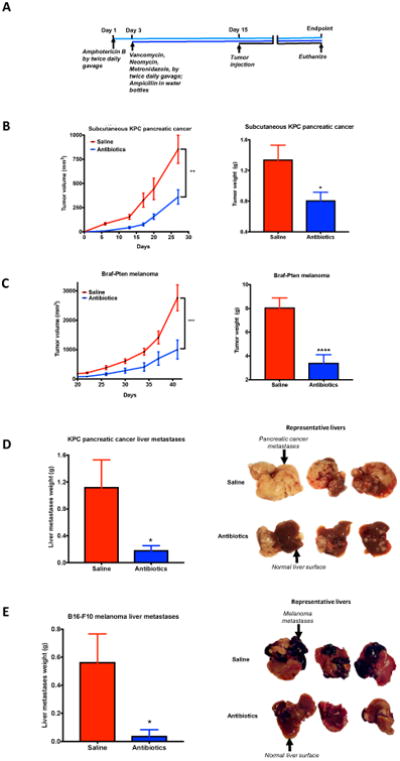
(A) schematic of the experiments; (B) & (C) saline and antibiotics-gavaged C57BL/6J mice were subcutaneously implanted with (B) KPC pancreatic cancer cells (n=13 for saline; 7 for antibiotics) or (C) Braf-Pten melanoma cells (n=14 for saline; 15 for antibiotics). Experiments were repeated four independent times with similar results. Results from one experiment are shown. X-axis label in (B) and (C) tumor kinetics represents days after tumor injection. (D) & (E) saline and antibiotics-gavaged mice were injected intrasplenically with (D) KPC cells (n=9 for saline; 7 for antibiotics) or (E) B16-F10 melanoma cells (n=10 for saline; 9 for antibiotics) (Unpaired Student's t-test with Welch's correction was used. Data is shown as mean±SEM; *, P<0.05; **, P<0.01; ***, P<0.005; ****, P<0.0005)
Our results show that gut microbiome depletion led to a significant decrease in subcutaneous tumor burden in pancreatic cancer and melanoma models. (Figure 1B, C). There was also a significant decrease in liver metastases burden in pancreatic cancer, colon cancer and melanoma models. (Figures 1D, E, S1A). Interestingly, the tumor-suppressing effect of gut microbiome depletion was abolished when the subcutaneous experiments were carried out in Rag1 knockout mice lacking mature T (and B) lymphocytes (Figures 2A; S2A). This suggests that the tumor-decreasing effect of antibiotics was not an off-target cytotoxic action on cancer cells but required active participation of adaptive immunity.
Figure 2. Gut microbiome depletion modulates tumor immune-environment.
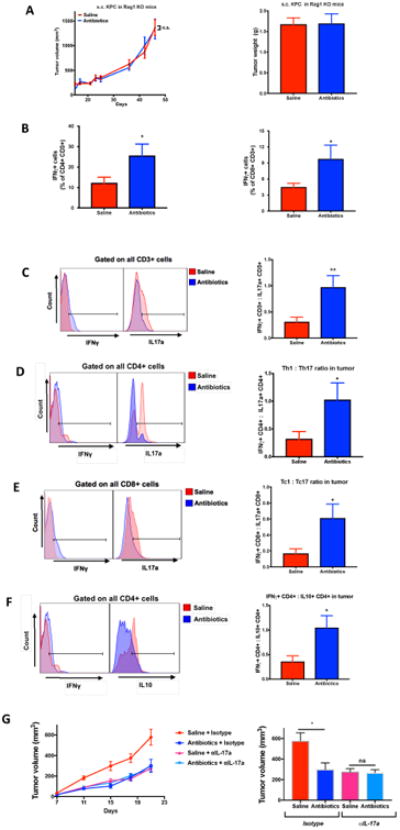
(A) C57BL/6J mice carrying a Rag1tm1Mom mutation were given saline or oral antibiotics and were subcutaneously implanted with KPC cells (n=10 for saline; 9 for antibiotics). X-axis label in tumor kinetics represents days after tumor injection; (B)–(F) KPC cells were subcutaneously implanted in wildtype mice and tumors were immunophenotyped by flowcytometry (n=9 for saline; 6 for antibiotics). Histograms depict individual representative samples. (G) Saline or antibiotics-treated KPC-bearing mice were either injected with anti-IL17a or isotype. (n=9 for saline+isotype, 8 for antibiotics+isotype, 10 for saline+anti-IL17a, 11 for antibiotics+anti-IL17a group) (Unpaired Student's t-test with Welch's correction was used. Data is shown as mean±SEM; *, P<0.05; ***, P<0.005).
We next evaluated the impact of gut microbiome depletion on the balance between pro- and anti-tumor T cells in tumor microenvironment (TME). It is known that naïve helper T cells (Th0), typically, mature into Th1, Th2, Treg or Th17 lineage. The classical Th1 cytokine-Interferon Gamma (IFNγ) plays an anti-tumorigenic role in TME whereas the Th2/Treg cytokines- IL4, IL5, IL10, etc., mediate a pro-tumorigenic role. As would be expected, a high Th1:Th2 ratio in TME correlates with improved survival in pancreatic cancer6. Moreover, Th17 cells are known to be pro-tumorigenic in pancreatic cancer7, melanoma8 and colorectal cancer9. IL17a is also intricately linked to the gut microbiome10 and plays a key role in defending against fungal and bacterial pathogens. Our results indicate that gut microbiome depletion caused a significant increase in Th1 (IFNγ+CD4+CD3+) and Tc1 (IFNγ+CD8+CD3+) cells in TME (Figure 2B). Furthermore, gut microbiome depletion caused a significant increase in numbers of anti-tumor IFNγ-secreting T cells (IFNγ+CD3+), with a corresponding decrease in numbers of pro-tumor IL17a(IL17a+CD3+) and IL10(IL10+CD4+CD3+) secreting immune populations (Figure 2C-F). Tumor-attenuating effect of antibiotics was abrogated in mice that were treated in vivo with IL17a neutralizing monoclonal antibody (Figure 2G), thereby confirming the essential role of IL17a in mediating this phenomenon.
Analysis of stool samples from subcutaneous KPC-bearing mice, given antibiotics, revealed an expected ablation of 16S ribosomal DNA (rDNA) and decrease in relative abundance of the two phyla majorly found in mouse (and human) gut: Bacteroidetes and Firmicutes (Figure S3A and E). Antibiotics also caused a significant decrease in α-diversity, a significant change in β-diversity, a reversed Bacteroidales:Clostridiales abundance ratio and colonization of gut by otherwise scarce (and likely antibiotic-resistant) Proteobacteria (mainly Alcaligenaceae and Enterobacteriaceae) and Tenericutes (mainly Mycoplasmataceae) (Figure S3 B-F). We also observed presence of 16S rDNA belonging to diverse microbial taxa in metastatic livers (Figure S4A-E).
The mechanism by which gut microbiome interacts with immune system and affects cancer progression is unclear but some inferences can be drawn from literature. Bacterial products recognized by toll-like receptors have been previously known to activate the IL23/IL17 axis and promote colon cancer development9. Thus, it is possible that gut microbes interact with immune system via pattern recognition receptors in pancreatic and other cancers too. The exact cell type which participates in this interaction and the potential site of this interaction (gut vs intra-tumoral), will be deciphered in future studies.
While the goal of using antibiotic cocktail was to deplete gut microbiome, Metronidazole has appreciable oral bioavailability and thus some systemic effects cannot be ruled out. However, the same antibiotic cocktail failed to reduce tumor size in Rag1 knockout mice or mice treated with IL17a neutralizing antibody, thereby, suggesting that a direct cytotoxic effect of antibiotics is not responsible for mediating the anti-tumor phenomenon. Additionally, our studies suggest that depleting the gut microbiome leads to infiltration of pancreatic tumors with effector-T cells. Conventional immunotherapeutic drugs like the modern checkpoint inhibitors have failed to show significant efficacy against pancreatic cancer, in part due to minimal effector-T cell infiltration in this cancer. Inducing T cell immunity has been previously shown to overcome pancreatic cancer's resistance to immune checkpoint inhibitors11 and therefore, future studies should evaluate if a gut microbial modulation strategy can potentiate the efficacy of checkpoint inhibitors or cytotoxic drugs in pancreatic and other cancers with minimal adverse effects.
In summary, our studies suggest that the gut microbiome modulates tumor progression and that manipulation of gut bacteria could emerge as a novel immunotherapeutic strategy, either alone or in combination with conventional immunotherapy.
Supplementary Material
Supplementary Figure S1: (A) Saline and antibiotics-gavaged mice were given intrasplenic injections of 200,000 MC-38 colon cancer cells tagged with red fluorescent protein to induce hepatic metastases (n=9 for saline and 6 for antibiotics group) (Data is shown as mean±SEM; ***, p<0.005. Mann Whitney test was used for comparisons. Representative images show metastatic burden from tumor-bearing livers imaged ex vivo using IVIS. Weight of the liver metastases was calculated as the difference between experimental metastatic liver weight and mean liver weight of same-aged cancer-naïve mice given saline or antibiotics.)
Supplementary Figure S2: (A) C57BL/6J mice carrying a Rag1tm1Mom mutation were either given oral antibiotics or saline and were subcutaneously implanted with Braf-Pten melanoma cells and tumor progression was serially followed (n=10 per group). X-axis label in tumor kinetics represents days after tumor injection (Unpaired Student's t-test with Welch's correction was used for comparisons. Data is shown as mean±SEM; ns, non-significant)
Supplementary Figure S3: Oral antibiotics lead to widespread gut microbial changes. (A)– (F) Saline and antibiotics-gavaged mice were implanted with KPC cancer cells subcutaneously and stool samples were collected from the distal rectum after euthanasia. These samples were then probed for bacterial 16S rDNA through PCR and gel electrophoresis (representative samples shown in (A) and also passed through a standardized 16S rRNA gene sequencing pipeline. For simplicity, only families with >1% relative abundance are depicted in (E)). (For 16S rRNA gene sequencing analysis, n=8 for saline and 6 for antibiotics). (One-way ANOVA with Tukey's multiple comparison test was used for UniFrac distance comparisons in (D). Unpaired Student's t-test with Welch's correction was used for all other comparisons. Data is shown as mean ± SEM; *, P<0.05; *****, P<0.0005, ******, P<0.0001)
Supplementary Figure S4: Oral antibiotics significantly change metastatic liver microbiome (A)–(E) saline and antibiotics-gavaged mice were given KPC hepatic metastases and metastatic livers were collected at endpoint. Livers were probed for relative abundance of 16S rDNA/mouse genomic DNA through RT-PCR and also passed through a 16S rRNA gene sequencing pipeline. For simplicity, only families with >1% relative abundance are depicted in (E). (n=8 for saline and 7 for antibiotics) (One-way ANOVA with Tukey's multiple comparison test was used for UniFrac distance comparisons in (D). Unpaired Student's t-test with Welch's correction was used for all other comparisons. Data is shown as mean ± SEM; **, P<0.01, ***, P<0.005, ******, P<0.0001)
Acknowledgments
Grant Support: This work was supported by National Institutes of Health awards CA124723 (Ashok Saluja), CA170946 (Ashok Saluja) and CA184274 (Sulagna Banerjee), DK058694 (Ashok Saluja), DK093047 (Ashok Saluja), DK111834 (Vikas Dudeja) and DA044582 (Sabita Roy)
We thank the University of Miami's Sylvester Comprehensive Cancer Center's IVIS Facility and Flow Cytometry Core Facility as well as the University of Minnesota's Genomic Core for valuable assistance in data acquiring. We also thank Ms. Patricia L. Guevara, BS, MA and Ms. Sydney Paige Kopen, BSB for important assistance during flowcytometry sample preparation and data acquiring. We also thank Yuguang James Ban, PhD and Xi Steven Chen, Ph.D. from the Department of Public Health Sciences at Sylvester Comprehensive Cancer Center, University of Miami for assistance in statistical and microbiome analysis.
Footnotes
- Conception and design: Vrishketan Sethi, Ashok Saluja and Vikas Dudeja
- Acquisition and analyses of data: Vrishketan Sethi, Saba Kurtom, Mohammad Tarique, Shweta Lavania, Zoe Malchiodi, Leonor Hellmund, Li Zhang, Umakant Sharma, Bhuwan Giri, Bharti Garg, Anthony Ferantella, Sabita Roy, Sundaram Ramakrishnan, Selwyn Vickers, Ashok Saluja and Vikas Dudeja
- Microbiome data analyses: Vrishketan Sethi, Umakant Sharma and Li Zhang
- Writing, review and/or revision of the manuscript: Vrishketan Sethi, Saba Kurtom, Bhuwan Giri, Shweta Lavania, Rajinder Dawra, Selwyn Vickers, Sundaram Ramakrishnan, Sabita Roy, Ashok Saluja and Vikas Dudeja.
Disclosures: Ashok Saluja: The University of Minnesota has a patent for Minnelide, which has been licensed to Minneamrita Therapeutics, LLC. A.S. is the co-founder and the Chief Scientific Officer of this company. This relationship is managed by the University of Miami.
Sulagna Banerjee: S.B. is a compensated consultant with Minneamrita Therapeutics LLC, the licensee of Minnelide, and this relationship is managed by the University of Miami.
Other authors do not have any disclosures to make.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hooper LV, et al. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sartor RB. Gastroenterology. 2004;126:1620–1633. doi: 10.1053/j.gastro.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 3.Reikvam DH, Erofeev A, et al. PLOS ONE. 2011;6:e17996. doi: 10.1371/journal.pone.0017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hingorani SR, et al. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Dankort D, et al. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Monte L, et al. The Journal of Experimental Medicine. 2011;208:469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAllister F, et al. Cancer Cell. 2014;25:621–37. doi: 10.1016/j.ccr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, et al. The Journal of Experimental Medicine. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grivennikov SI, et al. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanov II, et al. Cell Host & Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winograd R, et al. Cancer Immunol Res. 2015;3:399–411. doi: 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: (A) Saline and antibiotics-gavaged mice were given intrasplenic injections of 200,000 MC-38 colon cancer cells tagged with red fluorescent protein to induce hepatic metastases (n=9 for saline and 6 for antibiotics group) (Data is shown as mean±SEM; ***, p<0.005. Mann Whitney test was used for comparisons. Representative images show metastatic burden from tumor-bearing livers imaged ex vivo using IVIS. Weight of the liver metastases was calculated as the difference between experimental metastatic liver weight and mean liver weight of same-aged cancer-naïve mice given saline or antibiotics.)
Supplementary Figure S2: (A) C57BL/6J mice carrying a Rag1tm1Mom mutation were either given oral antibiotics or saline and were subcutaneously implanted with Braf-Pten melanoma cells and tumor progression was serially followed (n=10 per group). X-axis label in tumor kinetics represents days after tumor injection (Unpaired Student's t-test with Welch's correction was used for comparisons. Data is shown as mean±SEM; ns, non-significant)
Supplementary Figure S3: Oral antibiotics lead to widespread gut microbial changes. (A)– (F) Saline and antibiotics-gavaged mice were implanted with KPC cancer cells subcutaneously and stool samples were collected from the distal rectum after euthanasia. These samples were then probed for bacterial 16S rDNA through PCR and gel electrophoresis (representative samples shown in (A) and also passed through a standardized 16S rRNA gene sequencing pipeline. For simplicity, only families with >1% relative abundance are depicted in (E)). (For 16S rRNA gene sequencing analysis, n=8 for saline and 6 for antibiotics). (One-way ANOVA with Tukey's multiple comparison test was used for UniFrac distance comparisons in (D). Unpaired Student's t-test with Welch's correction was used for all other comparisons. Data is shown as mean ± SEM; *, P<0.05; *****, P<0.0005, ******, P<0.0001)
Supplementary Figure S4: Oral antibiotics significantly change metastatic liver microbiome (A)–(E) saline and antibiotics-gavaged mice were given KPC hepatic metastases and metastatic livers were collected at endpoint. Livers were probed for relative abundance of 16S rDNA/mouse genomic DNA through RT-PCR and also passed through a 16S rRNA gene sequencing pipeline. For simplicity, only families with >1% relative abundance are depicted in (E). (n=8 for saline and 7 for antibiotics) (One-way ANOVA with Tukey's multiple comparison test was used for UniFrac distance comparisons in (D). Unpaired Student's t-test with Welch's correction was used for all other comparisons. Data is shown as mean ± SEM; **, P<0.01, ***, P<0.005, ******, P<0.0001)