

Abstract
BACKGROUND AIMS
Chronic hepatitis B virus (HBV) infection remains incurable. While HBsAg-specific chimeric antigen receptor (HBsAg-CAR) T-cells have been generated, they have not been tested in animal models with authentic HBV infection.
METHODS
We generated a novel CAR targeting HBsAg, and evaluated its ability to recognize HBV+ cell lines and HBsAg particles in vitro. In vivo, we tested whether human HBsAg-CAR T-cells would have efficacy against HBV-infected hepatocytes in human liver chimeric mice.
RESULTS
HBsAg-CAR T-cells recognized HBV-positive cell lines and HBsAg particles in vitro as judged by cytokine production. However, HBsAg-CAR T-cells did not kill HBV-positive cell lines in cytoxicity assays. Adoptive transfer of HBsAg-CAR T-cells into HBV-infected humanized mice resulted in accumulation within the liver and a significant decrease in plasma HBsAg and HBV-DNA levels in comparison to control mice. Notably, the fraction of HBV core-positive hepatocytes among total human hepatocytes was greatly reduced after HBsAg-CAR T-cell treatment, pointing to non-cytopathic viral clearance. In agreement, changes in surrogate human plasma albumin levels were not significantly different between treatment and control groups.
DISCUSSION
HBsAg-CAR T-cells have anti-HBV activity in an authentic preclinical HBV infection model. Our results warrant further preclinical exploration of HBsAg-CAR T-cells as immunotherapy for HBV.
Keywords: adoptive immunotherapy, CAR T cells, hepatitis B virus
INTRODUCTION
Hepatitis B virus (HBV) is a global pandemic chronically infecting 300 million people across the world today [1]. In these chronic patients, HBV causes a lifelong infection that can lead to liver cirrhosis or cancer in 25% of patients [2]. HBV therapies currently remain limited to reverse transcriptase inhibitors (RTIs) and interferon (IFN)-α. RTIs only suppress HBV-DNA levels without significantly affecting the transcriptional template, covalently closed circular DNA (cccDNA) [3] while IFN-α causes significant side effects with little long-term therapeutic benefit [4]. Thus, new anti-HBV therapies are urgently needed in order to cure the virus.
During the HBV-specific immune response in acutely resolving patients, infiltrating T-cells rapidly purge the liver of HBV [5,6] In chronic HBV patients, however, HBV-specific T-cells are present in only low frequency and/or are anergic [7]. CD8-positive T-cells have been shown to be crucial in resolution of acute HBV infection [6]. They are able to clear HBV in both cytolytic and noncytolytic effector functions [8]. The cytokines, IFN-γ and tumor necrosis factor (TNF)-α, released by T-cells are important in driving noncytolytic suppression of virus [9,10]. Both IFN-γ and TNF-α can induce degradation of intracellular cccDNA [11] explaining part of this mechanism.
The adoptive transfer of T-cells genetically engineered to target hepatitis B surface antigen (HBsAg) on the surface of infected hepatocytes [12] with chimeric antigen receptors (HBsAg-CAR T-cells) is an attractive strategy to reconstitute HBV-specific T-cell immunity. Indeed, HBsAg-CAR T-cells have been shown to eliminate cccDNA from HBV-infected primary hepatocytes in vitro [13], and had transient anti-HBV activity in a transgenic HBV mouse model [14]. The HBV transgenic mouse model harbors an integrated copy of the HBV genome [15], is born with tolerance to viral antigens, and lacks cccDNA formation. Thus, transgenic HBV mice can only model viral suppression and not complete T-cell mediated cure. Given that HBV only naturally infects humans and chimpanzees at high levels [16], finding appropriate models to test cure strategies is challenging. With previous testing only in transgenic mice, it remains an open question whether HBsAg-CAR T-cells can induce a reduction of HBV levels in a model with authentic infection harboring episomal HBV cccDNA. Here we address this question by evaluating human HBsAg-CAR T-cells in HBV-infected human liver chimeric mice. These mice are immunodeficient and repopulated with human hepatocytes [17,18], allowing for spreading infection with HBV entry and cccDNA formation [19]. Thus, this model closely mimics HBV infection, and is ideal to test the ability of HBsAg-CAR T-cell to eradicate HBV genomes and/or infected hepatocytes.
RESULTS
Generation of a novel CAR targeting HBsAg
We first generated two HBsAg-CARs with a CD28.δ signaling domain and a single chain variable fragment (scFv) derived from the human monoclonal antibody (mAb) 19.79.5, which recognizes HBsAg from different serotypes [20], and has undergone successful Phase 1 testing [21]. Since the length of the spacer region of CARs is critical for their function [22], we first compared long and intermediate spacers. The IgG4 Fc domain with mutated Fc receptor binding sites (HBs-G4m-CAR) served a long [22] and the CH3 domain of IgG1 as an intermediate spacer (HBs-CH3-CAR; Figure 1A). As a control, we constructed a G4m-CAR with an scFv specific for an irrelevant antigen (EGFRvIII [23]; Ctrl-G4m-CAR; Figure 1A). CAR T-cells were generated by retroviral transduction, and the median transduction efficiency was 79.0% (range 60.5-89.9) as judged by FACS analysis with no significant differences between CAR constructs (Figure 1B).
Figure 1. Generation and functional characterization of HBsAg-CAR T-cells.
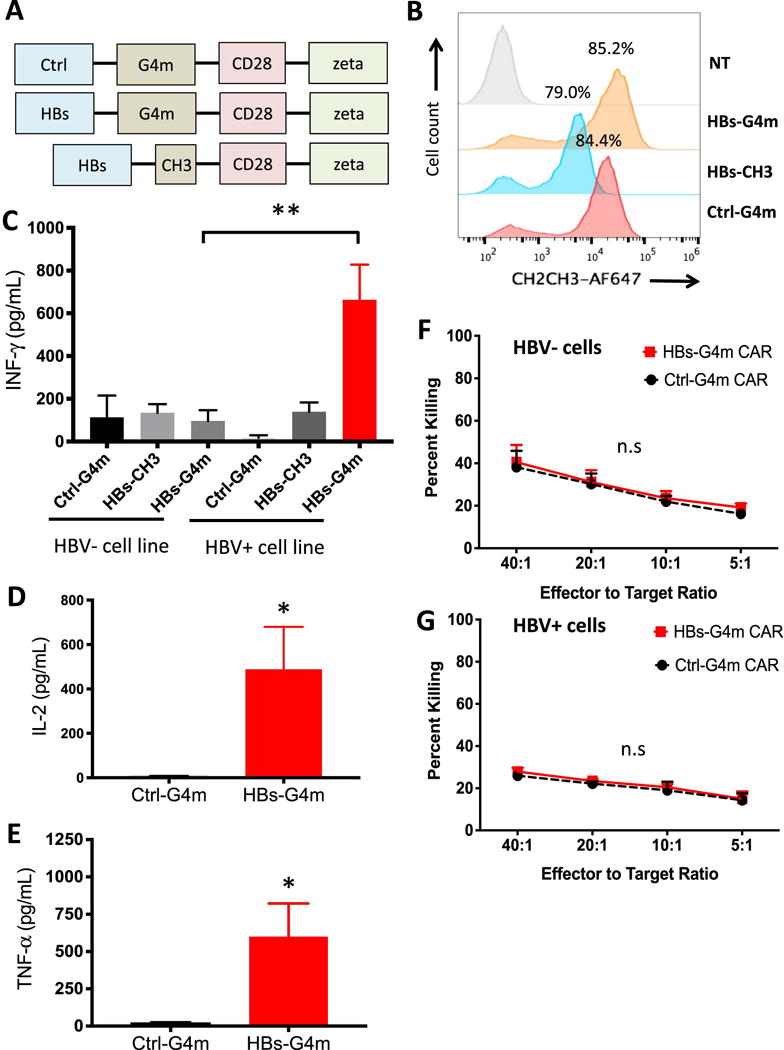
(A) Scheme of HBs-G4m, HBs-CH3, and Ctrl-G4m CAR constructs. (B) Representative FACS analysis of HBs-G4m-CAR (orange), HBs-CH3-CAR (blue), and Ctrl-G4m-CAR T-cells (red) confirming CAR expression (gray: non-transduced T-cells, NT). CAR-T cells were co-cultured with HBV+ or HBV-cell lines. Cytokine production, (C) IFN-γ, (D) IL-2, and (E) TNF-α, was measured by ELISA after 24 hours (for IFN-γ: **p<0.01, n=4; for IL-2, and TNF-α: *p<0.05, n=3). CAR-T cells were tested in a 5-hour chromium release assay against (F) HBV-or (G) HBV+ cell lines (n.s.: not significant, n=3). Error bars represent S.E.M. and significance is determined by unpaired, one-tailed t-tests.
HBs-G4m-CAR T-cells recognize HBV-positive cells in vitro
To determine which HBs-CAR recognized HBV-positive cells, we performed 24-hour co-culture assays with HepG2 (HBV-negative) and HepG2.2.15 (HBV-positive) cell lines, washing the cells first before adding CAR-T cells. Only HBs-G4m-CAR T-cells produced significant amounts of IFN-γ in the presence of HepG2.2.15 in contrast to HBs-CH3-CAR and Ctrl-G4m-CAR T-cells (Figure 1C). HepG2 induced only background IFN-γ production confirming specificity. These results demonstrate that a long spacer is needed for CARs with a mAb 19.79.5-derived HBsAg binding domain. In addition to IFN-γ, HBs-G4m-CAR T-cells also produced IL-2 (Figure 1D) and TNF-α (Figure 1E) in the presence of HepG2.2.15 in contrast to Ctrl-G4m-CAR T-cells. Having established that HBs-G4m-CAR T-cells recognize HepG2.2.15 in an HBsAg-restricted fashion, we performed standard cytotoxicity assays with HepG2 and HepG2.2.15 (Figure 1F,G). Only background killing of HepG2.2.15 by HBs-G4m-CAR T-cells was observed.
HBs-G4m-CAR T-cells recognize HBsAg particles in vitro
To determine if HBs-G4m-CAR T-cells recognize HBsAg particles, 24-hour co-culture assays were performed with media supernatants derived from HepG2 and HepG2.2.15 cell lines, the latter containing 80 ng/mL HBsAg. Only HBs-G4m-CAR T-cells secreted significant amounts IFN-γ in the presence of HepG2.2.15-conditioned media in contrast to HBs-CH3-CAR or Ctrl-G4m-CAR T-cells (Supplementary Figure S1A). We confirmed T-cell recognition of HBsAg particles by performing FACS analysis for the T-cell activation markers CD25 and CD69 (Supplementary Figure S2). To acquire more evidence that HBsAg particles bind to HBs-G4m-CAR T-cells, electron microscopy of HepG2.2.15-conditioned media was performed 24 hours after exposure to HBs-G4m-CAR or Ctrl-G4m-CAR T-cells. While HepG2.2.15-conditioned media exposed to Ctrl-G4m-CAR T-cells contained abundant HBsAg particles, there was a significant reduction in the number of viral particles after exposure to HBs-G4m-CAR T-cells (Supplementary Figure S1B,C). Thus, HBsAg particles produced by HepG2.2.15 can bind to HBs-G4m-CAR T-cells and potentially inhibit CAR-T targeting or killing of infected cells.
HBs-G4m-CAR T-cells have anti-HBV activity in HBV-infected human liver chimeric mice
We next tested HBs-G4m-CAR T-cell therapy in human liver chimeric FRG mice [17,18], which can replicate HBV (Figure 2A) [19]. Mice received 2 × 107 HBs-G4m-CAR (n=4) or Ctrl-G4m-CAR T-cells (n=3) intraperitoneal, and HBV-DNA and HBsAg levels were measured for five weeks. There was an average 3.0-fold decrease in HBV-DNA levels after 36 days in mice treated with HBs-G4m-CAR T-cells (Figure 2B), whereas HBV-DNA levels increased an average 5.6-fold in mice that received Ctrl-G4m-CAR T-cells. The decrease in HBV-DNA levels was mirrored by an average 4.7-fold decrease in HBsAg levels in HBs-G4m-CAR T-cell-treated mice, and an average 4.6-fold increase in Ctrl-G4m-CAR T-cell-treated mice (Figure 2C, Supplementary Figure S3A,B). There was no significant difference in plasma human albumin levels between groups, indicating that the anti-HBV activity of HBs-G4m-CAR T-cells is noncytolytic toward infected hepatocytes (Figure 2D, Supplementary Figure S3C).
Figure 2. HBs-G4m-CAR T-cells have anti-HBV activity in vivo.
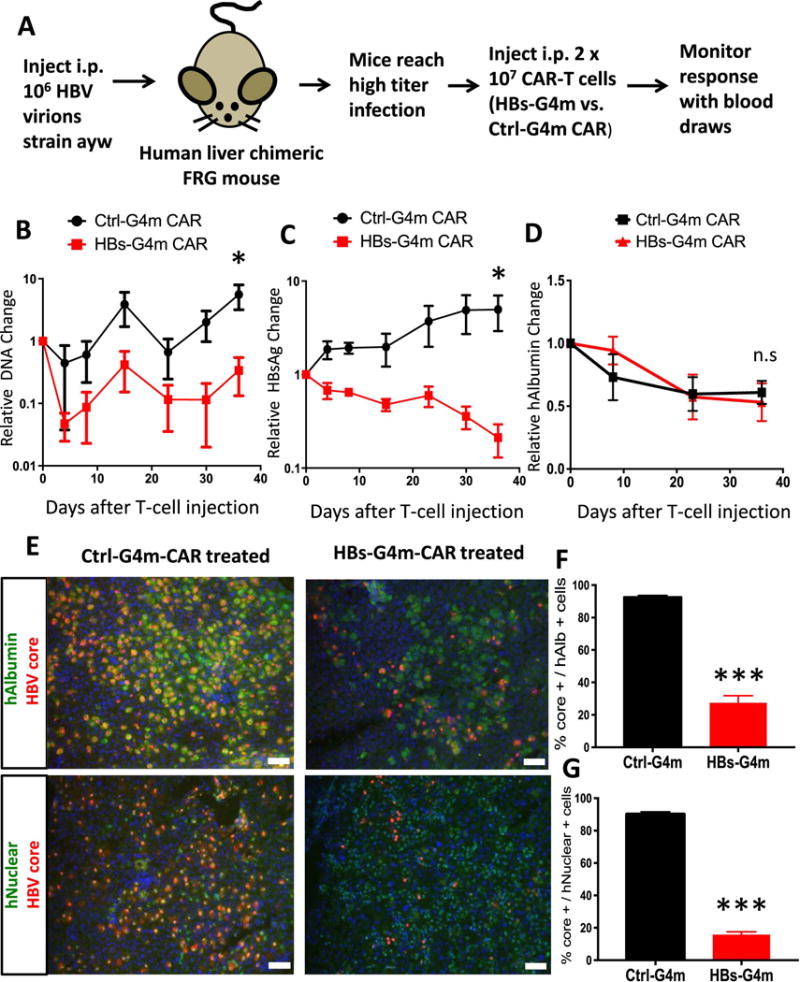
(A) Scheme of animal experiment. Median pre-treatment values were 17,221 ng/mL HBsAg, and 6.04×107 copies/ml HBV DNA. Serial monitoring of (B) HBsAg, (C) HBV-DNA, and (D) human albumin (hAlbumin) in plasma of infused mice. Data points were normalized to values pre T-cell injection in individual mice and averaged together. Average value +/− SEM is depicted; n=4 for HBs-G4m-CAR T-cell group; n=3 for Ctrl-G4m-CAR T-cell group; *p<0.05, n.s.: not significant). (E) Immunofluorescence of liver sections for hAlbumin (green), HBV-Core (red), and DAPI (blue) or human nucleus (green), HBV-Core (red), and DAPI (blue), scale bar=50 μm. (F,G) HBV-Core+, hAlbumin+, and human nuclear+ hepatocytes were quantified. Ratio of HBV-Core+ to hAlbumin+, and HBV-Core+ to human nuclear+ cells is shown (***p<0.0001, n=4 fields). Unpaired, two-tailed t-tests determined significance.
To examine liver sections for the presence of T-cells and HBV directly after T-cell infusion, mice received a second dose of 2 × 107 HBs-G4m-CAR or Ctrl-G4m-CAR T-cells, and were euthanized 10 days post injection. There was a 70.4% decrease in HBV-core staining among human albumin-positive hepatocytes in HBs-G4m-CAR T-cell-treated mice in comparison to control mice (Figure 2E,F), indicating clearance of HBV through noncytopathic mechanisms, similar to studies in chimpanzees [24]. This finding was confirmed by staining for human nuclei (Figure 2E,G). Human T-cells could only be detected in livers of mice treated with HBs-G4m-CAR T-cells in contrast to livers of Ctrl-G4m-CAR T-cell-treated mice (Figure 3), indicating that HBs-G4m-CAR T-cells traffic to livers to exert their anti-HBV activity. At the conclusion of the experiment, we did not detect long-term persistence of CAR T-cells in the liver by immunohistochemistry for CD3 (data not shown).
Figure 3. HBs-G4m-CAR T-cells redirected to HBsAg accumulate in the liver of HBV-infected humanized mice.
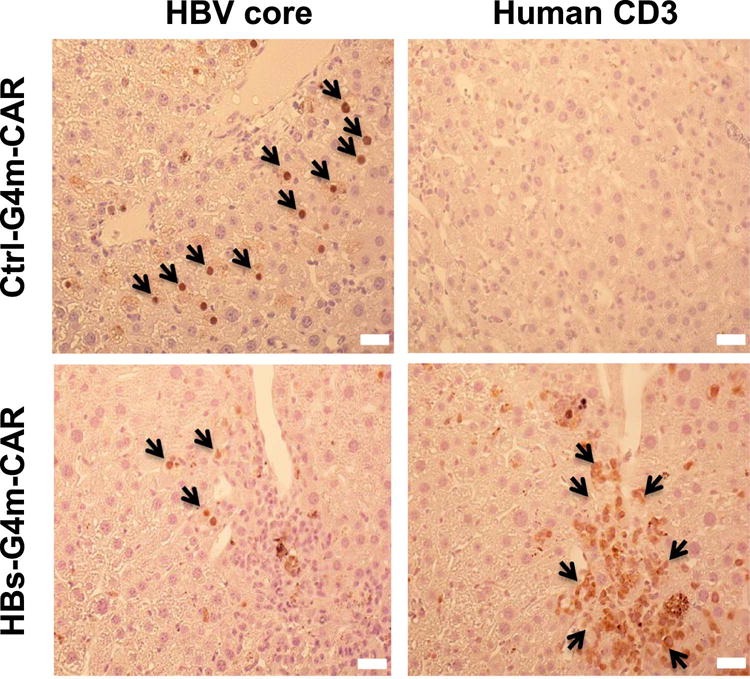
Localization of Ctrl-G4m-CAR and HBs-G4m-CAR T-cells in human liver chimeric mice 10 days post injection was studied using serial sections of paraformaldehyde fixed, paraffin embedded liver tissue, stained for core protein to detect HBV-infected cells (left panels; brown, nuclear stain delineates HBV core protein) and human CD3 to detect human T-cells (right panels; brown, membranous stain delineates CD3 protein; scale bar=20 μm).
DISCUSSION
In this study, we demonstrate that HBs-G4m-CAR T-cells are effective in reducing HBV-DNA and HBsAg levels in HBV-infected human liver chimeric mice [19], which possess cccDNA transcriptional templates. After CAR T-cell therapy, a portion of human hepatocytes was also found to have histologically absent HBV core expression, demonstrating clearance without destruction. However, we did not observe complete elimination of HBV, which is most likely due to the limited persistence of human CAR T-cells in immunodeficient mouse models [25], possibly facilitated in immunodeficient mice on the FRG background by lack of murine SIRPα engagement [26], as opposed to NOD background models [27].
Despite this limitation, HBs-G4m-CAR T cells had superior anti-HBV activity than HBV entry inhibitors [28] that did not reduce the number of HBV-infected hepatocytes in a humanized mouse model of established HBV infection. This finding also extends the work of the previous HBV-CAR publication in transgenic mice, which was unable to assess if HBV could be cleared from hepatocytes, since every cell had integrated virus allowing HBV core expression to quickly return [14]. Our results with HBsAg-CAR T cells were similar to HLA-restricted TCR-redirected T-cells in HBV-infected human liver chimeric mice [29], which observed transient reduction in serum viral markers. In contrast, their study found increases in liver enzymes resembling the administration of PBMCs from HBV-seropositive donors [30]. This suggests that T-cells targeting an extracellular and secreted HBsAg have different effector profiles versus targeting viral peptides on HLA proteins.
Since HBsAg-CAR T-cell therapy employs a different mechanism to eradicate HBV than currently approved therapies, there is an opportunity to explore combinatorial approaches. For example, combining CAR T-cells with RTIs may prevent rebound of viral DNA levels post CAR T-cell therapy observed in our study. Furthermore, hepatitis B immunoglobulin administration prior to CAR T-cell infusion may be useful to reduce serum HBsAg levels. Intriguingly, we found the mouse with the lowest initial HBsAg levels had the greatest HBsAg and DNA knockdown (−1.65 log and −3.67 log, respectively), suggesting some inhibitory role of HBsAg, or that higher disease burden may be too great for CAR T-cells. Future work testing mice with different HBsAg levels will be needed to examine this question. In addition, future studies are needed to characterize the phenotype of HBs-G4m-CAR T cells, and the expression of exhaustion markers (PD-1, LAG3, TIM3) pre-and post-stimulation with an extended panel of HBsAg-positive target cells.
In conclusion, HBs-G4m-CAR T cells are effective in reducing HBV levels in plasma and tissue in HBV-infected human liver chimeric mice. Thus, further preclinical exploration of our approach to HBV-targeted CAR T-cell therapy is warranted.
METHODS
Generation of retroviral vectors encoding CARs
To generate pSFG-HBs-G4m-28-zeta, a scFv encoding the amino acid sequence VH and VL domains of the XTL-19 antibody (mAb 19.79.5) [20] termed HBs was synthesized (IDTDNA, Coralville, IA), and cloned into an SFG retroviral vector using 5′ NcoI and 3′ BamHI, replacing the antigen binding domain from a second generation CAR vector, IL13Rα2-hIgG1-CD28-zeta CAR [31]. Next, the hIgG1 hinge was replaced with a mini-gene (synthesized by IDTDNA, Coralville, IA) encoding the CH2-CH3 domain from human IgG4 with mutated Fc receptor binding sites [22] (G4m) to generate pSFG-HBs-G4m-28-zeta. To generate pSFG-Ctrl-G4m-28-zeta, the HBs-specific scFv in pSFG-HBs-G4m-28-zeta was replaced by PCR cloning with an scFv specific for EGFRvIII [23]. pSFG-HBs-CH3-28-zeta was generated by cloning the HBs-specific scFv into the 5′ NcoI and 3′ BamHI sites of a pSFG vector with a CAR.IgG1 CH3. 28-zeta expression cassette (gift of Dr. Maksim Mamonkin, Baylor College of Medicine, Houston, TX). Cloning was verified by sequencing (Lone Star Labs, Houston TX). RD114-pseudotyped retroviral particles were generated by transient transfection of 293T cells as previously described [32].
Generation of CAR-T cells
To generate CAR-T cells, PBMCs were isolated by Lymphoprep (Greiner Bio-One, Monroe, NC) gradient centrifugation and then stimulated on treated non-tissue culture 24-well plates, which were pre-coated with OKT3 (CRL-8001, ATCC) and CD28 (BD Bioscience, Mountain View, CA) antibodies. Recombinant human interleukin-7 and interleukin-15 (IL-7, 10 ng/mL; IL-15, 5ng/mL; PeproTech, Rocky Hill, NJ) were added to cultures on day 2. On day 3, OKT3/CD28 stimulated T-cells (2.5 × 105 cells/well) were transduced on RetroNectin® (Clontech, Mountainview, CA) coated plates in the presence of IL-7 and IL-15. On day 5 or 6, T-cells were transferred into tissue culture plates and subsequently expanded with IL-7 and IL-15. CAR expression was determined 4 to 5 days post transduction by FACS analysis using a human IgG (H+L) antibody (Jackson Immunoresearch, West Grove, PA), and appropriate isotype control.
Cell culture assays
HepG2.2.15 cells (gift of Dr. Betty Slagle, Baylor College of Medicine, Houston, TX) and HepG2 cells (ATCC, Manassas, Virginia) were cultured in Dulbecco’s minimal essential media (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (Gibco ThermoFisher, Waltham, MA). For co-culture assays, 1 million HepG2 or HepG2.2.15 cells were plated; 24-hours later cells were washed and 2 million T-cells were added in one well of a 24-well plate in duplicate without exogenous cytokines. After 24 hours, supernatant was removed in order to assess cytokine release. For cytokine ELISAs, concentrations of INF-γ were assessed using manufacturer’s protocol (R&D Systems, Minneapolis, MN). For IL-2 and TNF-α, cytokine levels were determined using a multiplex assay (Millipore, St. Charles, MO). For cytotoxicity assays, standard chromium release protocols were followed as previously described [31] utilizing the same 2:1 ratio of T-cell to target cell, wherein target cells were plated on the same day as T-cell addition preventing any HBsAg particle accumulation. Cell culture assays were repeated with at least 3 different donors.
For cytokine assays based on co-culture with HBV particles, the HepG2.2.15 supernatant was collected (measured to be 80 ng/mL using HBsAg ELISA protocol below). Culture media contained 50% HepG2.2.15 supernatant with 1 million CAR-T cells. At 24 hours, supernatant was collected for use in cytokine ELISA assays and electron microscopy particle counts. T-cells were also collected at this time point for analysis by flow cytometry using established protocols [31] and staining cells with anti-CD25-PE (BD Biosciences, San Jose, CA) and anti-CD69-APC (BD Biosciences, San Jose, CA). Flow cytometric data were acquired by Gallios (Beckman Coulter, Brea, CA) and analyzed using FlowJo ver.10 (FlowJo, Ashland, Oregon).
Animal experiments
Fah−/−Rag2−/−Il2-rγ−/−(FRG) mice were repopulated with human cadaveric hepatocytes, as described previously [17,19]. For the current experiment, all human liver chimeric mice were repopulated with hepatocytes from the same hepatocyte donor and lot in order to minimize differences. Furthermore, at the initiation of cell injection, mice were kept on 100% NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione) in order to stabilize the levels of human hepatocytes, and prevent ongoing destruction of murine hepatocytes and proliferation of human hepatocytes, that otherwise might contribute to either increasing HBV levels, or the loss of cccDNA after hepatocyte mitosis.
One million genome equivalents of HBV genotype D (serotype ayw) were inoculated intraperitoneal into humanized FRG mice as previously described [19]. The infection was allowed to spread over 1-2 months until mice reached high levels of HBV infection, monitored by qPCR and HBsAg ELISA. HBsAg-CAR or Ctrl-CAR T-cells were injected intraperitoneal into humanized mice. Mice were monitored with retro-orbital bleeds collected into EDTA containing tubes, and plasma collected after centrifugation for 30 minutes at 2.3G. Collected plasma was frozen until further use for HBV-DNA, HBsAg, and human albumin analysis.
Plasma analysis
Plasma HBsAg levels was quantified using commercially available ELISA reagents (International Immuno Diagnostics, Foster City, CA) and HBsAg standards (Alpha Diagnostic International, San Antonio, TX). Plasma HBV DNA levels were determined by quantitative PCR as previously described [33]. Human plasma albumin levels were assessed by ELISA (Bethyl laboratories, Montgomery, TX) and was performed according to manufacturer’s protocol.
Supplementary Material
Acknowledgments
We thank James Broughman for assistance with electron microscopy, Stefan Wieland and Catherine Gillespie for critical comments on the manuscript. This work was supported by the National Institutes of Heath (NIH) grant R01AI094409, R01HL134510, T32DK060445, 5T32HL092332, the Texas Hepatocellular Carcinoma Consortium (CPRIT #RP150587), and the Diana Helis Henry and Adrienne Helis Malvin Medical Research Foundations. The Dan L. Duncan Cancer Center is supported by P30-CA125123. The Integrated Microscopy Core at the Texas Medical Center Digestive Disease Center is supported by P30-DK56338.
ABBREVIATIONS
- HBV
Hepatitis B virus
- RTIs
reverse transcriptase inhibitors
- IFN
interferon
- TNF
tumor necrosis factor
- cccDNA
covalently closed circular DNA
- HBsAg
hepatitis B surface antigen
- HBsAg-CAR T-cells
T-cells genetically engineered to target HBsAg with chimeric antigen receptors
- HBs-G4m-CAR
CAR-T cell targeted HBsAg with an IgG4 Fc domain with mutated Fc receptor binding sites as the spacer domain
- HBs-CH3-CAR
CAR-T cell targeted HBsAg with the CH3 domain of IgG1 as an spacer domain
- Ctrl-G4m-CAR
CAR-T cell target to EGFRvIII with an IgG4 Fc domain with mutated Fc receptor binding sites as the spacer domain
- FRG
Fah−/−Rag2−/−Il2-rγ−/−mouse strain used for human liver repopulation
- NOD
non-obese diabetic mouse background
- TCR
T-cell receptor
- HLA
human leukocyte antigen, presenting antigens on the major histocompatibility complex in human cells
- VL
variable light domain of antibody
- VH
variable heavy domain of antibody
- DMEM
Dulbecco’s minimal essential media
- NTBC
2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional methods are provided in the supplementary materials.
Disclosure of Interest:
RLK, TS, SG and KDB have filed a patent application based on this work with Baylor College of Medicine. All other authors declare no conflict of interest.
References
- 1.Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev. 2006;28:112–25. doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- 2.Perz JF, Armstrong GL, Farrington LA, Hutin YJF, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. Journal of Hepatology. 2006;45:529–38. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Liaw Y-F. HBeAg seroconversion as an important end point in the treatment of chronic hepatitis B. Hepatol Int. 2009;3:425–33. doi: 10.1007/s12072-009-9140-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ning Q, Han M, Sun Y, Jiang J, Tan D, Hou J, et al. Switching from entecavir to PegIFN alfa-2a in patients with HBeAg-positive chronic hepatitis B: a randomised open-label trial (OSST trial) Journal of Hepatology. 2014;61:777–84. doi: 10.1016/j.jhep.2014.05.044. [DOI] [PubMed] [Google Scholar]
- 5.Wieland SF, Spangenberg HC, Thimme R, Purcell RH, Chisari FV. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci USA. 2004;101:2129–34. doi: 10.1073/pnas.0308478100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. Journal of Virology. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. Journal of Virology. 2007;81:4215–25. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoh A, Heeg M, Ni Y, Schuch A, Binder B, Hennecke N, et al. Hepatitis B Virus-Infected HepG2hNTCP Cells Serve as a Novel Immunological Tool To Analyze the Antiviral Efficacy of CD8+ T Cells In Vitro. Journal of Virology. 2015;89:7433–8. doi: 10.1128/JVI.00605-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 10.Phillips S, Chokshi S, Riva A, Evans A, Williams R, Naoumov NV. CD8(+) T cell control of hepatitis B virus replication: direct comparison between cytolytic and noncytolytic functions. J Immunol. 2010;184:287–95. doi: 10.4049/jimmunol.0902761. [DOI] [PubMed] [Google Scholar]
- 11.Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hösel M, et al. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology. 2016;150:194–205. doi: 10.1053/j.gastro.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 12.Chu CM, Liaw YF. Membrane staining for hepatitis B surface antigen on hepatocytes: a sensitive and specific marker of active viral replication in hepatitis B. J Clin Pathol. 1995;48:470–3. doi: 10.1136/jcp.48.5.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohne F, Chmielewski M, Ebert G, Wiegmann K, Kürschner T, Schulze A, et al. T Cells Redirected Against Hepatitis B Virus Surface Proteins Eliminate Infected Hepatocytes. Gastroenterology. 2008;134:239–47. doi: 10.1053/j.gastro.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Krebs K, Böttinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–65. doi: 10.1053/j.gastro.2013.04.047. [DOI] [PubMed] [Google Scholar]
- 15.Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. Journal of Virology. 1995;69:6158–69. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wieland SF. The chimpanzee model for hepatitis B virus infection. Cold Spring Harb Perspect Med. 2015;5:a021469–9. doi: 10.1101/cshperspect.a021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissig K-D, Le TT, Woods N-B, Verma IM. Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc Natl Acad Sci USa. 2007;104:20507–11. doi: 10.1073/pnas.0710528105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol. 2007;25:903–10. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bissig K-D, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, et al. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest. 2010;120:924–30. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eren R, Ilan E, Nussbaum O, Lubin I, Terkieltaub D, Arazi Y, et al. Preclinical Evaluation of Two Human Anti–Hepatitis B Virus(HBV) Monoclonal Antibodies in the HBV-Trimera Mouse Model and in HBV Chronic Carrier Chimpanzees. Hepatology. 2000;32:588–96. doi: 10.1053/jhep.2000.9632. [DOI] [PubMed] [Google Scholar]
- 21.Galun E, Eren R, Safadi R, Ashour Y, Terrault N, Keeffe EB, et al. Clinical evaluation (phase I) of a combination of two human monoclonal antibodies to HBV: safety and antiviral properties. Hepatology. 2002;35:673–9. doi: 10.1053/jhep.2002.31867. [DOI] [PubMed] [Google Scholar]
- 22.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–35. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human Gene Therapy. 2012;23:1043–53. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–9. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 25.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 26.Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer-(NK) cell homeostasis in vivo. Proc Natl Acad Sci USa. 2011;108:13224–9. doi: 10.1073/pnas.1101398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takenaka K, Prasolava TK, Wang JCY, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313–23. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 28.Nakabori T, Hikita H, Murai K, Nozaki Y, Kai Y, Makino Y, et al. Sodium taurocholate cotransporting polypeptide inhibition efficiently blocks hepatitis B virus spread in mice with a humanized liver. Nautre. 2016;6:27782. doi: 10.1038/srep27782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kah J, Koh S, Volz T, Ceccarello E, Allweiss L, Lütgehetmann M, et al. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J Clin Invest. 2017;127:3177–88. doi: 10.1172/JCI93024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida T, Hiraga N, Imamura M, Tsuge M, Abe H, Hayes CN, et al. Human Cytotoxic T Lymphocyte-Mediated Acute Liver Failure and Rescue by Immunoglobulin in Human Hepatocyte Transplant TK-NOG Mice. Journal of Virology. 2015;89:10087–96. doi: 10.1128/JVI.01126-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krebs S, Chow KKH, Yi Z, Rodriguez-Cruz T, Hegde M, Gerken C, et al. T cells redirected to interleukin-13Rα2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy. 2014;16:1121–31. doi: 10.1016/j.jcyt.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–85. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Billioud G, Kruse RL, Carrillo M, Whitten-Bauer C, Gao D, Kim A, et al. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. Journal of Hepatology. 2016;64:781–9. doi: 10.1016/j.jhep.2015.11.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.