

Supplemental Digital Content is available in the text.
Keywords: fasting, leucine, lipoproteins, mass spectrometry, triglycerides
Abstract
Objective—
Inhibition of PCSK9 (proprotein convertase subtilisin/kexin type 9) and statins are known to lower plasma LDL (low-density lipoprotein)-cholesterol concentrations. However, the comparative effects of these treatments on the postprandial metabolism of TRLs (triglyceride-rich lipoproteins) remain to be investigated.
Approach and Results—
We performed a 2-by-2 factorial trial of the effects of 8 weeks of subcutaneous evolocumab (420 mg every 2 weeks) and atorvastatin (80 mg daily) on postprandial TRL metabolism in 80 healthy, normolipidemic men after ingestion of an oral fat load. We evaluated plasma total and incremental area under the curves for triglycerides, apo (apolipoprotein)B-48, and VLDL (very-LDL)-apoB-100. We also examined the kinetics of apoB-48 using intravenous D3-leucine administration, mass spectrometry, and multicompartmental modeling. Atorvastatin and evolocumab independently lowered postprandial VLDL-apoB-100 total area under the curves (P<0.001). Atorvastatin, but not evolocumab, reduced fasting plasma apoB-48, apoC-III, and angiopoietin-like 3 concentrations (P<0.01), as well as postprandial triglyceride and apoB-48 total area under the curves (P<0.001) and the incremental area under the curves for plasma triglycerides, apoB-48, and VLDL-apoB-100 (P<0.01). Atorvastatin also independently increased TRL apoB-48 fractional catabolic rate (P<0.001) and reduced the number of apoB-48–containing particles secreted in response to the fat load (P<0.01). In contrast, evolocumab did not significantly alter the kinetics of apoB-48.
Conclusions—
In healthy, normolipidemic men, atorvastatin decreased fasting and postprandial apoB-48 concentration by accelerating the catabolism of apoB-48 particles and reducing apoB-48 particle secretion in response to a fat load. Inhibition of PCSK9 with evolocumab had no significant effect on apoB-48 metabolism.
TRLs (triglyceride-rich lipoproteins) are comprised of both intestinally derived apo (apolipoprotein) B-48 containing chylomicrons and hepatically derived apoB-100 containing VLDL (very-low-density lipoproteins) and their remnants.1 Increasingly, evidence suggests that TRLs are implicated in the development of atherosclerotic cardiovascular disease (CVD) through their induction of endothelial dysfunction, inflammation, oxidative stress, and atherogenesis.2–4 The therapeutic regulation of TRL metabolism is therefore important to lower the risk of atherosclerotic CVD.
Chylomicrons are the predominant lipoproteins secreted by the intestine and are responsible for transporting absorbed dietary lipids to peripheral tissues. Because humans consume multiple meals during the day this system is continually in a dynamic state of postprandial lipid and lipoprotein metabolism. LPL (lipoprotein lipase) hydrolyzes chylomicron triglyceride and allows the delivery of free fatty acids to muscle and adipose tissue, resulting in the formation of smaller chylomicron remnants. Chylomicron remnants compete with VLDLs for hepatic uptake via the LDLR (LDL receptor) and the LRP (LDL receptor-related protein).1 ApoC-III and ANGPTL3 (angiopoietin-like protein 3) are emerging key regulators of TRL metabolism.5–8 ApoC-III is involved in chylomicron metabolism through the inhibition of LPL activity and chylomicron remnant uptake by hepatic lipoprotein receptors.5,6 In vitro studies have also demonstrated that expression of apoC-III increases apoB-48 secretion.9 Overexpression of ANGPTL3 decreases VLDL-triglyceride clearance by inhibition of LPL and inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion.8 Understanding the therapeutic regulation of postprandial TRLs is fundamental for effective treatment of dyslipidemias.
Statins decrease cholesterol synthesis and increase LDLR expression, leading to significantly lower concentrations of plasma apoB-100 and LDL-cholesterol.10 Statins may also lower plasma triglycerides by upregulating other hepatic receptors that in turn mediate the disposal of TRLs.11,12 We and others have shown that statins can lower fasting and postprandial plasma triglyceride and apoB-48 concentrations.13–18 Burnett et al19 first reported, in miniature pigs, that inhibition of statins with atorvastatin increased the catabolism of postprandial TRLs independent of LDLR. However, the precise effects of statins on the metabolism of TRLs in the postprandial state, particularly apoB-48 transport under normal physiological conditions, remains unclear in humans.
Recent research has focused on the impact of PCSK9 (proprotein convertase subtilisin/kexin type 9) on TRL metabolism.20,21 PCSK9 is a secretory protease that regulates cell surface receptors, principally the LDLR.22 Experimental evidence suggests that PCSK9 is also involved in the regulation and intracellular degradation of other lipoprotein receptors, including the VLDL receptor and LRP.23,24 In animal studies, PCSK9 deficiency is associated with reduced postprandial hypertriglyceridemia, in part because of an increase in hepatic clearance of chylomicrons.25 Patients carrying PCSK9 loss-of-function variants display attenuated levels of fasting and postprandial triglycerides and apoB-48.26 We previously reported that, in the absence of statin therapy, plasma PCSK9 concentration is directly associated with postprandial TRL metabolism, as reflected by the area under the curves (AUCs) of plasma triglycerides and apoB-48 in response to a fat load.27 Monoclonal antibodies to PCSK9 significantly lower LDL and TRL levels, as well as CVD events.28–31 However, in a small study of 10 individuals, Reyes-Soffer et al32 found that inhibition of PCSK9 did not alter postprandial TRL concentration. The impact of PCSK9 inhibition on postprandial TRL metabolism, including apoB-48 kinetics, remains to be formally investigated.
In the present study, we investigated the effects of atorvastatin and the PCSK9 inhibitor evolocumab (as monotherapy and combined with atorvastatin) on postprandial TRL kinetics in healthy men without significant dyslipidemia. Atorvastatin decreases intracellular cholesterol synthesis in the liver, resulting in upregulation of the LDLR expression. By contrast to atorvastatin, evolocumab inhibits extracellular binding between PCSK9 and LDLR, resulting in enhanced recycling of LDLR within hepatocytes. Hence, we hypothesized that atorvastatin and evolocumab would independently and additively improve TRL metabolism by accelerating the catabolism of apoB-48 particles via different primary mechanisms of action that increase LDLR function.
Materials and Methods
Qualified medical/scientific researchers may submit for consideration a data sharing request through the Amgen homepage.
Subjects and Study Design
The current study is a part of a large kinetic trial investigating the effect of atorvastatin and evolocumab on lipoprotein metabolism in healthy normolipidemic men (URL: http://www.clinicaltrials.gov. Unique identifier: NCT02189837). Full details of the recruitment of subjects and study design were published previously.29 Briefly, the main study recruited healthy nonobese men aged 18 to 65 years with fasting plasma LDL-cholesterol <4.9 mmol/L (79% had optimal LDL-cholesterol level of <3.4 mmol/L) and triglycerides of <1.7 mmol/L, that is, within a population reference range that excluded those with significant dyslipidemia. Exclusion criteria included factors known to influence lipoprotein metabolism, such as diabetes mellitus, chronic kidney disease, medications, and thyroid dysfunction. Eligible subjects were randomized into a double-blind, placebo-controlled, 2-by-2 factorial trial estimating the effects of oral atorvastatin 80 mg QD, SC evolocumab 420 mg Q2W, or oral atorvastatin 80 mg QD, and SC evolocumab 420 mg Q2W for 8 weeks. The study was approved by a National Ethics Committee (Bellberry Ltd, Eastwood, South Australia); all patients provided written informed consent.
Postprandial and Stable Isotope Protocol
The postprandial study was performed before and after intervention (ie, day 5 at baseline and week 8) after the completion of the first tracer postabsorptive study on apoB-100 kinetics.29 Briefly, all patients were admitted to the metabolic ward in the morning after a 14-hour fast. They were studied in a semirecumbent position and allowed to drink only water. After a baseline fasting blood sample, a bolus of d3-leucine (5 mg/kg of body weight) was administered intravenously, within a 2-minute period, into an antecubital vein via a 21g butterfly needle. A liquid formulated high-fat test meal consisting of 100 mL of full cream milk, 150 mL of pure cream, 70 mL of corn oil, 90 g of whole egg, and 10 g of sugar (a total of 4800 kJ, 130 g fat, 17 g protein, and 21 g carbohydrate) was consumed over several minutes. Additional blood samples were obtained after 30 minutes, and 1, 1.5, 2, 3, 4, 5, 6, 8, and 10 hours. Subjects were then given a snack and allowed to go home. All the procedures were repeated after the 8-week intervention period.
Biochemical Measurements
The quantification of analytes including lipids, lipoproteins, and apolipoproteins were detailed previously.29 VLDL-apoB-100 from the TRL fraction was measured using an ELISA kit (Mabtech, Nacka, Sweden). Plasma apoB-48 levels were measured with an enzyme immunoassay kit (Shibayagi, Shibukawa, Japan). Fasting plasma HL (hepatic lipase; MyBioSource, San Diego, CA), LPL (Cusabio, Wuhan, China), and ANGPTL3 (R&D Systems) levels were determined using enzyme immunoassay kits. Postprandial metabolism was quantified by calculating the incremental AUC for plasma triglycerides, VLDL-apoB-100, and apoB-48 (0–10 h), using the trapezium rule. The incremental AUC was estimated as the difference between the area defined below the baseline concentration and the area under the plasma curve between hours 0 and 10. These measures provide an integrated estimate of the total dynamic response of TRL particles to a fat load.33 Total AUC reflects exposure and potential impact of accumulation of TRLs on the artery and hence atherosclerotic CVD. By contrast, the incremental AUC reflects the acute change in TRLs after a fat load. Total AUC under TRL response curve has been shown to be a better predictor than fasting concentration or incremental AUC in predicting cardiovascular events.34
Isolation of ApoB-48 and Measurement of Isotopic Leucine Enrichment
The TRL fraction was isolated from 3.5 mL of plasma by ultracentrifugation (Optima XL-100K, Beckman Coulter, Australia) at density of 1.006 g/L (40 000 rpm, 16 h, 4°C).35,36 The TRL samples were then prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The apoB-48 band was excised from the membrane and hydrolyzed with 200 µL 6 mol/L HCl at 110°C for 16 hours. Derivatization of leucine to the oxazolinone derivative was described previously.37 Isotopic enrichment was determined using gas chromatography-mass spectrometry with selected ion monitoring of samples at a mass to charge ratio of 212 and 209, and negative ion chemical ionization. Tracer to tracee ratios were derived from isotopic ratios for each sample.
Model of ApoB-48
As previously reported,35,36 a nonsteady compartment model (SAAM II program, The Epsilon Group, VA) was used to account for changes in plasma apoB-48 concentration and tracer after consumption of the fat load (Figure I in the online-only Data Supplement). Briefly, the model includes 2 separate, but linked models; one to account for the leucine tracer data, including plasma leucine and apoB-48 leucine enrichment, and the other model for apoB-48 concentration data. The leucine compartment model consists of a 4-compartment subsystem that describes plasma leucine kinetics. This subsystem is connected to an intrahepatic delay compartment that accounts for the time required for leucine tracer to be incorporated into apoB-48 and subsequently secreted into plasma. The apoB-48 concentration compartment model consists of a delay compartment that represents 4 compartments in series and an additional compartment representing plasma apoB-48 particles. The model assumed that catabolism (the fractional catabolic rate [FCR], ie, the rate constants out of compartments 6 and 8) was time-invariant, similar to the kinetic study by Le et al,38 and that the increase in plasma apoB-48 concentration was because of an increase in apoB-48 secretion after consumption of the fat meal. The compartment model was fit to the plasma apoB-48 concentration and tracer curves to estimate model parameters: rate constants, including FCR, and production parameters. Because apoB-48 FCR is invariant across the postprandial study, apoB-48 secretion in the fasted state (basal production rate [PR]) was calculated as the product of the FCR and fasting apoB-48 concentration. The model calculated apoB-48 particle secretion in response to the fat meal as the product of the integral under the apoB-48 concentration curve (above fasting concentration) and the FCR.
Statistical Analyses
Statistical protocols have been described previously.29 Data were transformed to normalize distributions. Group characteristics were compared by 1-way ANOVA. Main effects (MEs) of treatment (ie, isolated effect of one treatment irrespective of effect of second treatment) and interactive effects of treatment (ie, effect of combination treatment not predicted by the MEs of each treatment) were assessed by maximum-likelihood random-effects regression models. The models contained 3-way interactions of time, atorvastatin, and evolocumab. If the 3-way interaction of atorvastatin, evolocumab, and time was not statistically significant (P>0.05), then only the MEs (time–atorvastatin and time–evolocumab) were included in the model. If the MEs of atorvastatin and evolocumab on a dependent variable (eg, total AUC of VLDL-apoB-100) are both significant and in the same direction, then the combination treatment has an additive effect on the outcome. If an interaction was identified, 6 comparisons were made among the 4 treatment groups using random-effects regression models; P values were expressed before and after adjustment for multiple intergroup comparisons using a Holm-Bonferroni test (step-down procedure). Changes in variables with interventions relative to baseline were described as a ratio of geometric means (post/preintervention). The association between the changes in apoB-48 kinetics and other metabolites was examined using a linear regression and Spearman/Pearson correlations. Statistical significance was defined at the 5% level.
Results
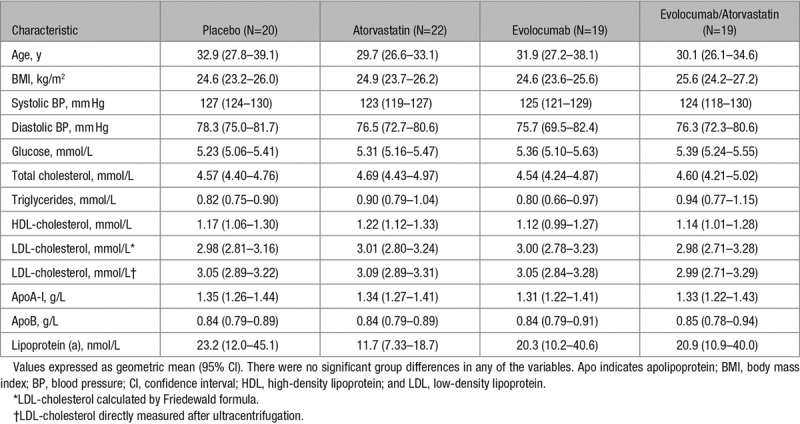
The selection of subjects and their disposition were summarized in a previous CONSORT (Consolidated Standards of Reporting Trials) diagram.29 Briefly, 81 subjects completed the 2×2 factorial study, with the exception of one subject from the evolocumab group who did not complete the fat load test. As shown in Table 1, the 80 subjects were on average 31 years old, nonobese, normotensive, nondiabetic and had overall normal plasma lipid and lipoprotein profiles. There were no significant group differences in any of the variables in Table 1. The group characteristics at baseline were similar to the parent cohort.29 Adherence to randomized treatments was 100%. The spectrum of adverse event after treatments was as reported previously.29
Table 1.
Clinical and Biochemical Characteristics of the 80 Subjects at Baseline
Fasting Plasma Lipid, Lipoprotein, and Apolipoprotein Concentration
Treatment effects on fasting plasma lipid, lipoprotein, and apolipoprotein concentration in 81 subjects were reported previously.29 The exclusion of the subject who did not undertake the fat load test did not alter the major findings when compared with the parent cohort. Except for total cholesterol, LDL-cholesterol, and apoB-100, there were no significant interactive effects between atorvastatin and evolocumab treatment on other variables (Table I in the online-only Data Supplement). Briefly, both atorvastatin and evolocumab independently decreased fasting plasma concentrations of total cholesterol, LDL-cholesterol, and apoB (P<0.001, both interventions), the reduction being significantly greater with combination therapy compared with monotherapy (P<0.001). As shown in Table I in the online-only Data Supplement, there were significant MEs (PME) of atorvastatin in lowering fasting triglycerides (−21%, PME <0.01), VLDL-apoB-100 (−30%, PME <0.001), apoB-48 (−31%, PME <0.01), apoC-III (−15%, PME <0.01), apoE (−38%, PME <0.001), ANGPTL3 (−25%, PME <0.001) concentrations, and lathosterol/campesterol ratio (−80%, PME <0.001) and of evolocumab in raising HDL (high-density lipoprotein)-cholesterol (+11%, PME <0.01) and lowering VLDL-apoB-100 (−31%, PME <0.001) and apoE (−39%, PME <0.001). There were no significant effects on fasting apo A-V, LPL, HL, or CETP (cholesteryl ester transfer protein) mass concentrations. Collectively, combination treatment with evolocumab and atorvastatin lowered LDL-cholesterol, apoB-100, VLDL-apoB-100, and apoE concentrations more than either agent did when administered alone.
Postprandial Triglycerides, VLDL-ApoB-100, and ApoB-48 Responses
The postprandial responses for plasma triglycerides, VLDL-apoB-100, and apoB-48 to the fat load in 80 subjects are shown in Figure 1. There was no statistically significant interaction between atorvastatin and evolocumab on total and incremental AUCs for triglycerides, VLDL-apoB-100, and apoB-48 in response to the fat load. Atorvastatin significantly reduced postprandial triglyceride, VLDL-apoB-100, and apoB-48 total AUCs (−29%, −30%, and −30%, respectively; PME <0.001 for all) and incremental AUCs (−29%, −27%, and –37%, respectively; PME <0.01 for all) and of evolocumab in lowering postprandial VLDL-apoB-100 total AUCs (−27%, PME <0.001; Table 2). There were no significant changes to postprandial triglyceride and apoB-48 total AUCs and incremental triglyceride, VLDL-apoB-100, and apoB-48 AUCs with evolocumab (Figure 2). When analyzed by group, atorvastatin alone and atorvastatin plus evolocumab had comparable effects in lowering postprandial triglycerides (−31% versus −26%), VLDL-apoB-100 (−27% versus −27%), and apoB-48 (−34% versus −26%) total AUCs and postprandial triglyceride (−33% versus −24%) and apoB-48 (−35% versus −39%) incremental AUCs. The combination of atorvastatin and evolocumab, however, lowered VLDL-apoB-100 AUC more than either treatment did alone. These bygroup analyses were consistent with those ME analyses.
Figure 1.
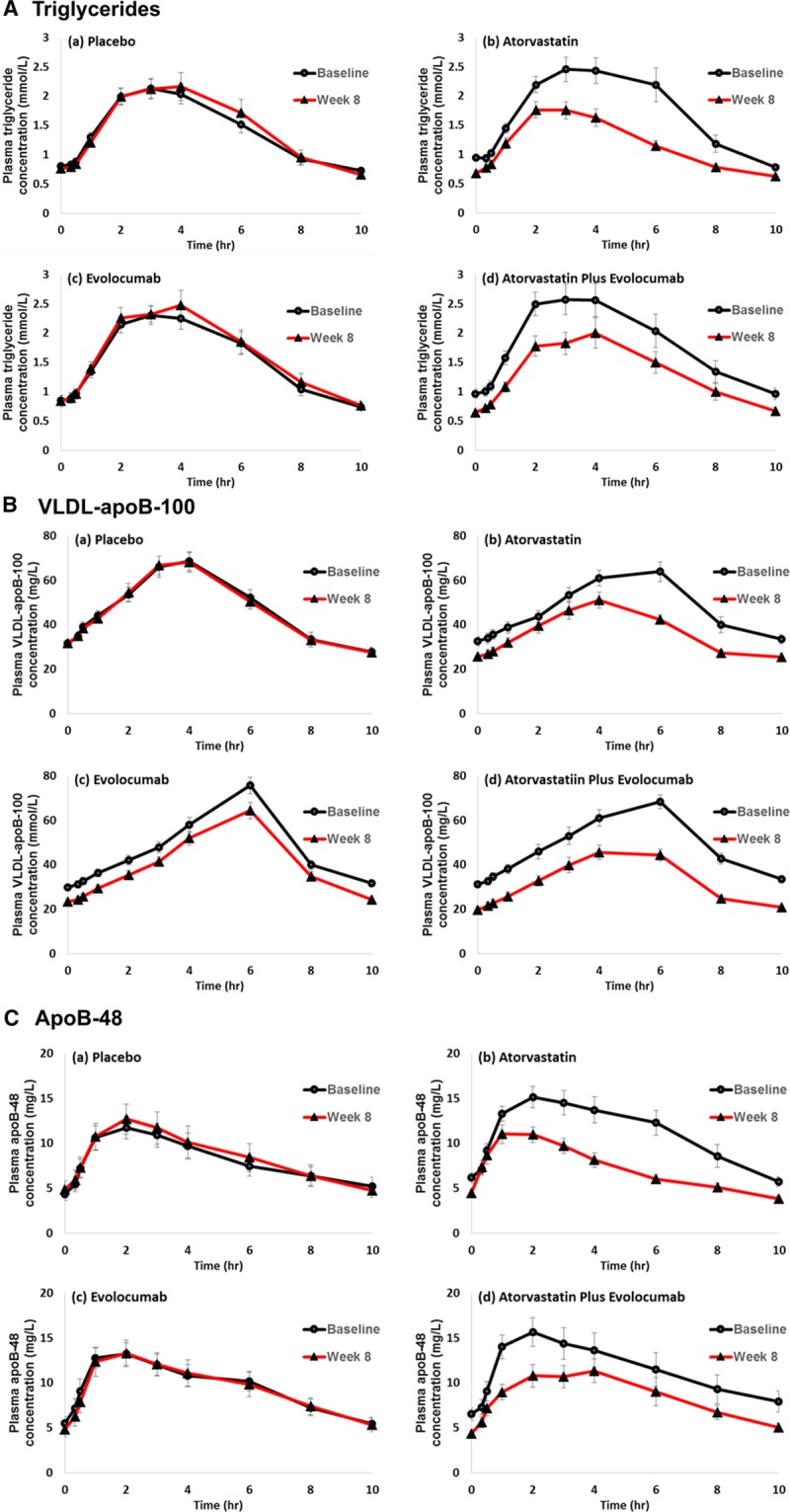
Plasma triglyceride (A), VLDL-apoB-100 (B), and apoB-48 (C) responses to the fat load in the placebo, atorvastatin, evolocumab, and atorvastatin plus evolocumab groups. Values are expressed as mean±SEM. Apo indicates apolipoprotein; and VLDL, very-low-density lipoprotein.
Table 2.
Effect of Interventions on Postprandial Changes for Plasma Triglyceride, VLDL-ApoB-100, and ApoB-48 After the Oral Fat Load
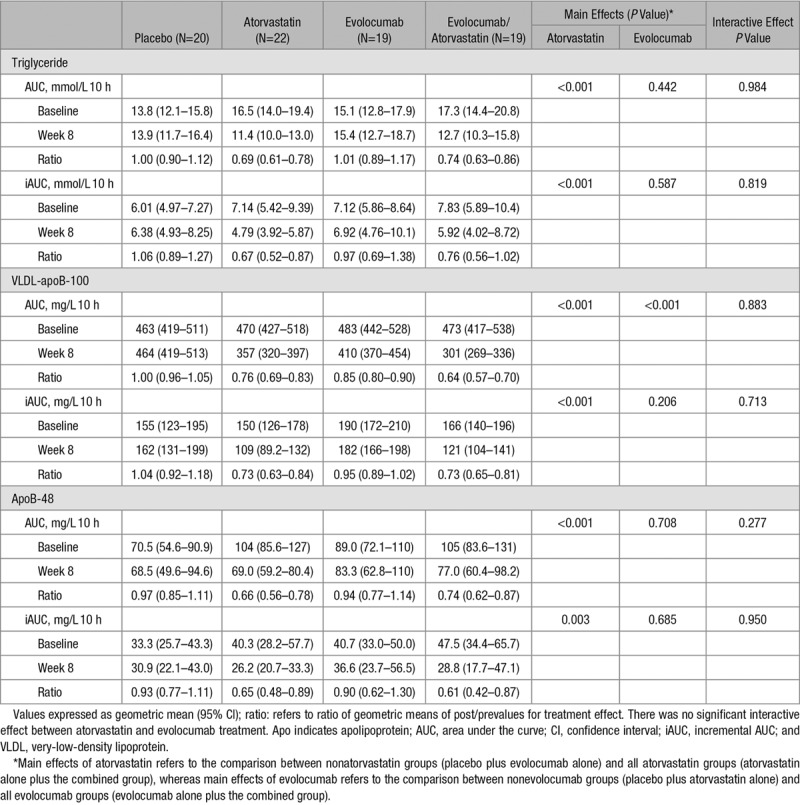
Figure 2.
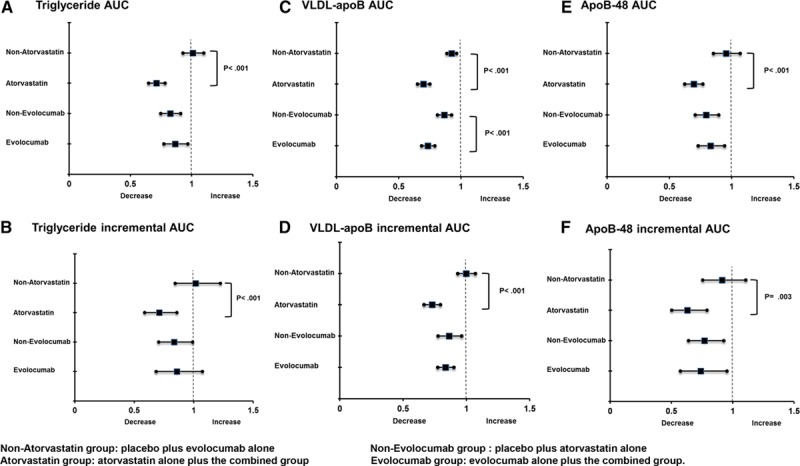
Ratio of geometric means (post/preintervention) for main effects of atorvastatin and evolocumab on the total and incremental area under the curves (AUCs) for plasma triglycerides (A and B), VLDL-apoB-100 (C and D), and apoB-48 (E and F). Apo indicates apolipoprotein; and VLDL, very-low-density lipoprotein.
ApoB-48 Kinetics
Figure 3 shows the isotopic enrichment of apoB-48 with the D3-leucine (expressed as tracer/tracee ratio over time), pre- and postintervention, for a representative subject from the placebo, atorvastatin, evolocumab, and evolocumab plus atorvastatin groups. In the atorvastatin and evolocumab plus atorvastatin groups, the peak enrichment values were higher. Enrichment curves in the placebo and evolocumab groups did not alter between baseline and week 8.
Figure 3.
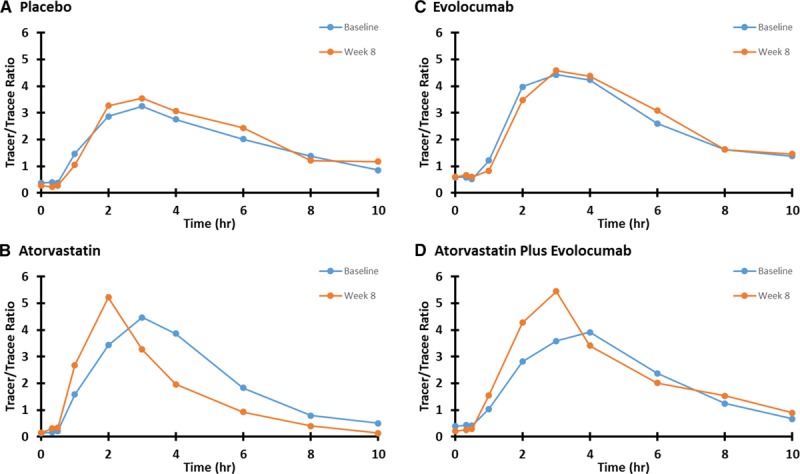
Tracer/tracee ratio of leucine in apoB-48 at preintervention and postintervention in a representative subject from the placebo (A), atorvastatin (B), evolocumab (C), and atorvastatin plus evolocumab (D) groups. Apo indicates apolipoprotein.
There was no statistically significant interaction between atorvastatin and evolocumab on the plasma pool size and kinetics of apoB-48 (Table 3; Figure 4). Atorvastatin had significant MEs in reducing plasma apoB-48 pool size (−30%, PME <0.01) and increasing apoB-48 FCR (+32%, PME <0.001). There was also a significant ME of atorvastatin in reducing the number of apoB-48-containing particles secreted in response to the fat load (−29%, PME <0.01). However, atorvastatin did not significantly alter the basal PR of apoB-48. Evolocumab had no significant ME (PME> 0.05 for all) on apoB-48 kinetics variables (Table 3). When analyzed by group, atorvastatin alone and atorvastatin plus evolocumab had comparable effects in lowering apoB-48 pool size (−26% versus −33%) and particle secretion (−29% versus −30%) and increasing apoB-48 FCR (+34% versus +30%).
Table 3.
Effect of the Interventions on ApoB-48 Kinetics in the Subjects
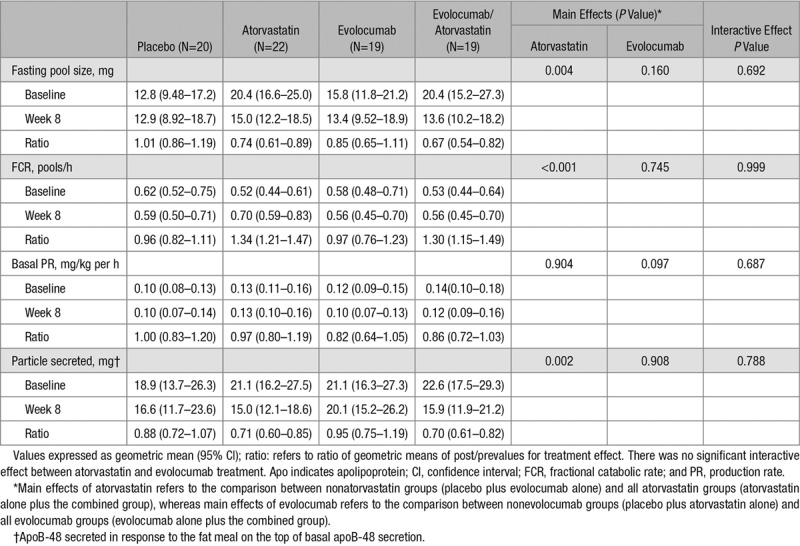
Figure 4.
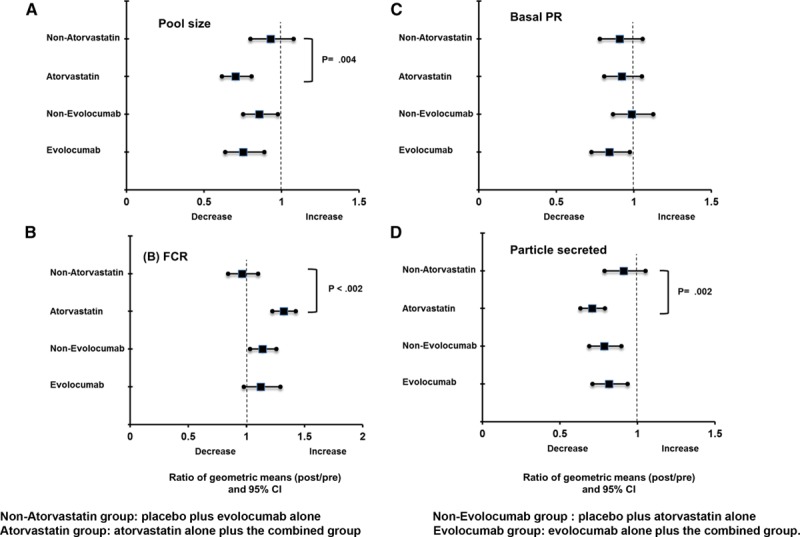
Ratio of geometric means (post/preintervention) for main effects of atorvastatin and evolocumab on the apoB-48 pool size (A), apoB-48 FCR (B), basal apoB-48 PR (C), and apoB-48 particle secreted on the top of basal apoB-48 secretion (D). Apo indicates apolipoprotein; CI, confidence interval; FCR, fractional catabolic rate; and PR, production rate.
Discussion
We demonstrated that inhibition of statins with atorvastatin, but not inhibition of PCSK9 with evolocumab, improved TRL metabolism after an oral fat load in healthy subjects without dyslipidemia. These effects of atorvastatin were chiefly attributed to a significant increase in apoB-48 catabolism and the concomitant reduction of apoB-48 particle secretion during the postprandial period. The enhancement in the catabolism of apoB-48 with atorvastatin may be mediated by its effect in lowering plasma apoC-III and ANGPTL3 concentrations.
Previous ApoB-48 Kinetic Studies With Statins and PCSK9 Inhibitors
The effects of statins on fasting and postprandial apoB-48 have been previously examined, mostly in patients with dyslipidemia.13–18 In an uncontrolled study of 10 normolipidemic men, Parhofer et al15 found that atorvastatin (10 mg/d) reduced the total and incremental AUCs for chylomicron remnant-triglycerides and apoB-48 in response to a fat load, suggesting increased clearance of chylomicron remnants with atorvastatin. Lamon-Fava et al39 reported that atorvastatin significantly reduced TRL-apoB-48 concentration under constant feeding conditions in 9 patients with combined hyperlipidemia by increasing the FCR and reducing the PR of TRL-apoB-48, but these kinetic parameters failed to reach statistical significance. Similar nonsignificant changes on apoB-48 FCR and PR were also observed with simvastatin in 16 hyperlipidemic men.40 Hogue et al41 reported that under constant feeding conditions low dose atorvastatin significantly reduced TRL-apoB-48 chiefly because of decreased production in 12 hypertriglyceridemic patients with type 2 diabetes mellitus. Discrepancies among the various kinetic studies might have been because of small sample size and different patient characteristics. Only one study has reported on the effect of PCSK9 monoclonal antibodies on apoB-48 metabolism. Reyes-Soffer et al32 found that treatment with alirocumab had no effect on postprandial TRL metabolism in 10 healthy individuals, but no isotope tracer data on apoB-48 kinetics were reported. We have extended previous studies by comprehensively examining a larger number of patients and investigated the comparative effect of atorvastatin and evolocumab on postprandial apoB-48 metabolism under normal physiological conditions.
Atorvastatin
Inhibition of de novo cholesterol synthesis by statins is well recognized to upregulate LDLR activity, thereby increasing hepatic removal of apoB-containing lipoproteins.10 We confirmed this in an earlier report by showing that atorvastatin treatment increased the FCRs of apoB-100 in the VLDL, IDL (intermediate-density lipoprotein), and LDL fractions in these patients.29 We now provide evidence that atorvastatin decreases plasma apoC-III and ANGPTL3 concentrations in individuals without significant dyslipidemia, thereby contributing to increased lipolysis of VLDL. This may explain why atorvastatin, compared with evolocumab, does not lower the production of IDL-apoB-100. The clearance of chylomicrons and its remnants would be expected to be increased with atorvastatin because this agent increases LDLR activity and decreases the competition of chylomicron remnants with apoB-100 containing lipoproteins for the receptor-mediated clearance pathway. Accordingly, we found that atorvastatin increased the clearance of triglyceride-rich chylomicron particles as reflected by the increase in FCR of apoB-48. The reduction in postprandial total and incremental AUCs for both triglycerides and apoB-48 is also in agreement with an effect of atorvastatin increasing the catabolism of apoB-48 containing lipoprotein particles.
Both apoC-III and ANGPTL3 are important regulators of TRL removal by inhibiting LPL activity for chylomicron and VLDL lipolysis.5–9 The hepatic removal of apoB-48 particles requires initial lipolysis of chylomicrons with formation of chylomicron remnant particles.42 We have previously reported that atorvastatin significantly reduces plasma apoC-III concentrations by increasing its FCR.43 Gene expression data also suggest that atorvastatin treatment is associated with reduced hepatic ANGPTL3 mRNA expression.44 Consistent with these, we found that atorvastatin but not evolocumab reduced plasma apoC-III and ANGPTL3 concentrations. The significant reductions in plasma apoC-III and ANGPTL3 with atorvastatin may, in part, account for the associated increase in conversion of chylomicron into chylomicron remnant particles and subsequent catabolism of apoB-48 in our study. The significant effect of atorvastatin, but not evolocumab, in accelerating the catabolism of apoB-48 particles highlights the importance of apoC-III and ANGPTL3 in regulating chylomicron catabolism under normal physiological conditions. Whether reductions in apoC-III and ANGPTL3 with atorvastatin contributes to the reduction in the number of apoB-48 particles secreted in response to a fat load merits further investigation.
Several in vitro studies have suggested that the availability of cholesterol substrate regulates the hepatic and intestinal synthesis of apoB-100 and apoB-48.45–47 Hence, inhibition of cholesterol synthesis with atorvastatin would be anticipated to limit apoB-100 and apoB-48 synthesis in liver and intestine, respectively. However, atorvastatin did not decrease the PRs of VLDL, IDL-, and LDL-apoB-100 in our previous report.29 In the present study, we also failed to show a significant effect of atorvastatin on basal apoB-48 PR. Consistent with this, suppression of cholesterol synthesis with atorvastatin did not attenuate the production of apoB-48 from human epithelial Caco2 cells.47
Another important finding is that atorvastatin significantly reduced the number of apoB-48 containing particles secreted in response to a fat load. This is consistent with some of the intervention studies we referred to earlier.39–41 In vitro cellular data suggest that statins can enhance the intracellular degradation of apoB-48 in the presence of exogenous lipids.46 Animal and gene expression studies also suggest that statin treatment is associated with reduced intestinal microsomal triglyceride transfer protein activity for apoB-48 assembly and secretion.48,49 By contrast to evolocumab, atorvastatin may increase GLP-1 (glucagon-like peptide 1) activity by inhibiting DPP-IV (dipeptidyl peptidase-4) activity and this may contribute to reduce postprandial lipemia,50,51 but this speculation requires further investigation.
Evolocumab
The role of PCSK9 inhibition in the upregulation of LDLR activity is well recognized.22 Accordingly, we previously reported in the present group of subjects under postabsorptive conditions that evolocumab significantly decreases the pool sizes of VLDL-, IDL-, and LDL-apoB-100 by increasing the corresponding FCRs.29 The reduction in IDL-apoB-100 production with evolocumab in that study could be exclusively related to greater enhancement in the direct hepatic uptake of VLDL, leaving less to be converted to IDL by delipidation. Consistent with this, we now show that evolocumab significantly decreases the total AUC of VLDL-apoB-100 in the postprandial state. This observation is likely to be a consequence of reduction in the fasting VLDL-apoB-100 pool size given that the total AUC of VLDL-apoB-100 is a function of the fasting and postprandial VLDL-apoB-100 pool sizes in response to a fat load. In contrast to atorvastatin, evolocumab had no significant effect on the total and incremental AUCs for plasma triglycerides and apoB-48. The FCR and PR of apoB-48 was also not significantly altered by evolocumab. We have previously reported that evolocumab significantly increased the FCRs of VLDL- and IDL-apoB-100. The lack of effect of evolocumab on apoB-48 FCR might seem paradoxical. However, evolocumab did not lower apoC-III and ANGPTL3 concentrations, implying that the increased clearance of VLDL and IDLs with this agent may relate exclusively to a direct receptor-mediated mechanism. There is also evidence showing that certain hepatic receptors (eg, VLDL and LRP receptors) are less sensitive to the effects of PCSK9 than the LDLR.20,23 These explanation could be attributed to the apparently less potent effect of evolocumab (ME +23%) than atorvastatin (+40%) in accelerating the catabolism of VLDL particles and lowering plasma triglyceride concentrations in the postabsorptive state.29 Given that the present study was performed under postprandial conditions, the flux of VLDL and chylomicron particles into the circulation system would be significantly increased. Hence, it is possible that evolocumab, unlike atorvastatin, was unable to accelerate the catabolism of apoB-48 particles because of less effective lipolysis of nascent chylomicron particles, and less effective hepatic uptake by the VLDL (or LRP) receptors in the postprandial condition.
PCSK9 is active in the small intestine,52 and phase 2 and 3 studies of evolocumab with or without combination statin therapy have shown that evolocumab treatment leads to modest triglyceride reductions of 6% to 18% versus placebo.53,54 However, evolocumab did not reduce fasting and postprandial triglyceride concentration in our study. The observed differences might be because of different subject characteristics given that this study examined fasting and postprandial triglyceride levels in subjects with normolipidemia. The lack of effect of evolocumab on postprandial triglyceride and apoB-48 metabolism is consistent with another report by Reyes-Soffer et al32 showing that inhibition of PCSK9 by alirocumab had no effects on fasting and postprandial triglyceride and apoB-48 AUCs in 10 healthy normolipidemic individuals.
Strengths and Limitations
The strengths of our study include the large sample size. We also used a well-validated compartment model to describe the nonsteady kinetics of apoB-48 based on previous studies.35,36,38 We also used a single fat load test for postprandial investigation, which permits measurements of postprandial triglyceride and apoB-48 AUCs,33 2 common end points used in these types of studies.
We only studied white men. Whether our findings apply to pre- and postmenopausal women or non-whites remains to be tested. The selection of healthy, nonobese, insulin sensitive, overall normolipidemic subjects ensured that the LDLR (or other related hepatic TRL receptor) pathway was fully functional and plasma TRL transport physiological. However, the effect of atorvastatin on basal PR of apoB-48, or evolocumab on postprandial triglyceride and apoB-48 response nor apoB-48 kinetics might not been observed under normal physiological conditions. Whether our findings of our study apply to patients with dyslipidemia and diabetes mellitus merits further investigation. This is being addressed in an ongoing tracer study to evaluate the effect of evolocumab on postprandial lipidemia in type 2 diabetes mellitus (URL: http://www.clinicaltrials.gov. Unique identifier: NCT02948777). Although there are multiple mechanisms that can contribute to the removal of chylomicron remnants, both LPL and HL are critical for the hydrolysis of triglycerides in CM and VLDL and play key roles in postprandial TRL metabolism.55,56 The lack of effect of atorvastatin and evolocumab on LPL or HL concentration should be interpreted with caution because we only measured fasting LPL and HL concentrations, which may not fully reflect postheparin mass and activity. We cannot exclude an effect of atorvastatin on the activities of LPL and HL, as suggested elsewhere.57,58 The lipid and apolipoprotein composition (eg, triglyceride, cholesterol, phospholipids, apoC-III, and apoE) of apoB-48 particles could also affect the conformation of apoB-48 particles for binding to the LDLR and possibly LRP receptors.42 Atorvastatin, but not evolocumab, has been shown to raise intestinal cholesterol absorption and this could also alter the composition and induce conformational changes of apoB-48 particles. We did not measure the composition of apoB-48 particles. Further studies should therefore investigate the effects of atorvastatin and evolocumab on the composition of apoB-48 particles. Measurements of postprandial apoC-III and ANGPTL3 levels may help to further elucidate their roles in postprandial TRL metabolism.
Clinical Implications
Statins are currently the most common medications for reducing LDL-cholesterol levels. Our findings suggest that atorvastatin also regulates TRL metabolism beyond LDL-cholesterol. Whether the reduction in postprandial TRLs with statins contributes to reductions in cardiovascular events in primary and secondary prevention setting remains to be demonstrated.
That evolocumab did not improve TRL metabolism suggests that the improvement in clinical outcomes, in FOURIER trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk), was principally related to reduction in plasma concentration of LDL particles.31 The marked reduction in LDL particle numbers after evolocumab treatment has been previously reported.59 Our findings furthermore suggest that because evolocumab treatment does not impact apoB-48 concentrations, PCSK9 inhibition may not impact intestinal fat and attendant nutrient absorption. These results are consistent with a post hoc analysis demonstrating a relatively modest effect of evolocumab on cholesterol absorption.60 That atorvastatin and evolocumab have primary intracellular and extracellular mechanisms of action, respectively, may account for the divergent effects on TRL metabolism observed in the present study. The effect of PCSK9 inhibition on postprandial TRL metabolism using modalities other than extracellular acting monoclonal antibodies requires further investigation.
Conclusions
Our data suggest that in healthy, normolipidemic men, inhibition of cholesterol synthesis with atorvastatin lowers fasting and postprandial apoB-48 concentrations by accelerating the catabolism of apoB-48 particles and by reducing the number of apoB-48 particles secreted in response to the fat load. Inhibition of PCSK9 with evolocumab did not impact apoB-48 metabolism. Although a study in healthy individuals, from a clinical perspective, our findings support the combined use of high-intensity statin and a PCSK9 inhibitor to reduce atherosclerotic CVD events in high-risk patients.31
Acknowledgments
We would like to thank Annalise Nawrocki, PhD (of Amgen Inc) for editorial support. All authors had full access to all the data in the study and take responsibility for its integrity and the data analysis.
Sources of Funding
This study was funded by Amgen Inc.
Disclosures
G. Watts has received honoraria for advisory boards and speakers bureau or research grants from Amgen Inc, Sanofi, Regeneron, Kowa, and Genfit. R. Somaratne, S.M. Wasserman, and R. Scott are current or former employees of Amgen Inc and own Amgen stock/stock options. R. Somaratne is an inventor on at least one pending patent involving evolocumab. S.M. Wasserman appears on a number of pending patents owned by Amgen relating to evolocumab and PCSK9 inhibition. The other authors report no conflicts of interest.
Nonstandard Abbreviations and Acronyms
- ANGPTL3
- angiopoietin-like protein 3
- Apo
- apolipoprotein
- AUC
- area under the curve
- CETP
- cholesteryl ester transfer protein
- CVD
- cardiovascular disease
- DPP-IV
- dipeptidyl peptidase-4
- FCR
- fractional catabolic rate
- GLP-1
- glucagon-like peptide 1
- HDL
- high-density lipoprotein
- HL
- hepatic lipase
- IDL
- intermediate-density lipoprotein
- LDL
- low-density lipoprotein
- LDLR
- LDL receptor
- LPL
- lipoprotein lipase
- LRP
- LDL receptor-related protein
- ME
- main effect
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- PR
- production rate
- TRL
- triglyceride-rich lipoprotein
- VLDL
- very-low-density lipoprotein
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.310882/-/DC1.
Highlights
The comparative effects of evolocumab and atorvastatin on the postprandial metabolism of TRLs (triglyceride-rich lipoproteins) in humans are unclear.
We demonstrated that atorvastatin improved TRL metabolism by a significant increase in apo (apolipoprotein)B-48 catabolism and the concomitant reduction of apoB-48 particle secretion during the postprandial period.
Evolocumab had no significant effect on apoB-48 metabolism.
References
- 1.Havel RJ. Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler Thromb Vasc Biol. 2019;30:9–19. doi: 10.1161/ATVBAHA.108.178756. doi: 10.1161/ATVBAHA.108.178756. [DOI] [PubMed] [Google Scholar]
- 2.Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60:473–485. doi: 10.1161/01.cir.60.3.473. [DOI] [PubMed] [Google Scholar]
- 3.Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:2525–2540. doi: 10.1016/j.jacc.2014.09.042. doi: 10.1016/j.jacc.2014.09.042. [DOI] [PubMed] [Google Scholar]
- 4.Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118:547–563. doi: 10.1161/CIRCRESAHA.115.306249. doi: 10.1161/CIRCRESAHA.115.306249. [DOI] [PubMed] [Google Scholar]
- 5.Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R, Brown WV. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–1295. doi: 10.1172/JCI112713. doi: 10.1172/JCI112713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL, Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 2016;126:2855–2866. doi: 10.1172/JCI86610. doi: 10.1172/JCI86610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52:187–193. doi: 10.1007/s12020-015-0838-9. doi: 10.1007/s12020-015-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, Ueda K, Inaba T, Minekura H, Kohama T, Furukawa H. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–33748. doi: 10.1074/jbc.M203215200. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- 9.Sundaram M, Zhong SM, Khalil MB, Links PH, Zhao Y, Iqbal J, Hussain MM, Parks RJ, Wang YW, Yao Z. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51:150–161. doi: 10.1194/jlr.M900346-JLR200. doi: 10.1194/M900346-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundy SM. Statins: definitive translational research. Mol Med. 2014;20(suppl 1):S20–S23. doi: 10.2119/molmed.2014.00194. doi: 10.2119/molmed.2014.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim S, Kim CH, Vaziri ND. Upregulation of hepatic LDL receptor-related protein in nephrotic syndrome: response to statin therapy. Am J Physiol Endocrinol Metab. 2005;288:E813–E817. doi: 10.1152/ajpendo.00266.2004. doi: 10.1152/ajpendo.00266.2004. [DOI] [PubMed] [Google Scholar]
- 12.Imagawa M, Takahashi S, Zenimaru Y, Kimura T, Suzuki J, Miyamori I, Iwasaki T, Hattori H, Yamamoto TT, Nakano T, Nakajima K. Comparative reactivity of remnant-like lipoprotein particles (RLP) and low-density lipoprotein (LDL) to LDL receptor and VLDL receptor: effect of a high-dose statin on VLDL receptor expression. Clin Chim Acta. 2012;413:441–447. doi: 10.1016/j.cca.2011.10.033. doi: 10.1016/j.cca.2011.10.033. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC, Watts GF, Barrett PH, Martins IJ, James AP, Mamo JC, Mori TA, Redgrave TG. Effect of atorvastatin on chylomicron remnant metabolism in visceral obesity: a study employing a new stable isotope breath test. J Lipid Res. 2002;43:706–712. [PubMed] [Google Scholar]
- 14.Ng TW, Watts GF, Stuckey BG, Ching HL, Chan DC, Uchida Y, Sakai N, Yamashita S, Martins IJ, Redgrave TG, Barrett PH. Does pravastatin increase chylomicron remnant catabolism in postmenopausal women with type 2 diabetes mellitus? Clin Endocrinol (Oxf) 2005;63:650–656. doi: 10.1111/j.1365-2265.2005.02396.x. doi: 10.1111/j.1365-2265.2005.02396.x. [DOI] [PubMed] [Google Scholar]
- 15.Parhofer KG, Barrett PH, Schwandt P. Atorvastatin improves postprandial lipoprotein metabolism in normolipidemlic subjects. J Clin Endocrinol Metab. 2000;85:4224–4230. doi: 10.1210/jcem.85.11.6978. doi: 10.1210/jcem.85.11.6978. [DOI] [PubMed] [Google Scholar]
- 16.Parhofer KG, Laubach E, Barrett PH. Effect of atorvastatin on postprandial lipoprotein metabolism in hypertriglyceridemic patients. J Lipid Res. 2003;44:1192–1198. doi: 10.1194/jlr.M300011-JLR200. doi: 10.1194/jlr.M300011-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Battula SB, Fitzsimons O, Moreno S, Owens D, Collins P, Johnson A, Tomkin GH. Postprandial apolipoprotein B48-and B100-containing lipoproteins in type 2 diabetes: do statins have a specific effect on triglyceride metabolism? Metabolism. 2000;49:1049–1054. doi: 10.1053/meta.2000.7744. doi: 10.1053/meta.2000.7744. [DOI] [PubMed] [Google Scholar]
- 18.Otokozawa S, Ai M, Van Himbergen T, Asztalos BF, Tanaka A, Stein EA, Jones PH, Schaefer EJ. Effects of intensive atorvastatin and rosuvastatin treatment on apolipoprotein B-48 and remnant lipoprotein cholesterol levels. Atherosclerosis. 2009;205:197–201. doi: 10.1016/j.atherosclerosis.2008.11.001. doi: 10.1016/j.atherosclerosis.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnett JR, Barrett PH, Vicini P, Miller DB, Telford DE, Kleinstiver SJ, Huff MW. The HMG-CoA reductase inhibitor atorvastatin increases the fractional clearance rate of postprandial triglyceride-rich lipoproteins in miniature pigs. Arterioscler Thromb Vasc Biol. 1998;18:1906–1914. doi: 10.1161/01.atv.18.12.1906. [DOI] [PubMed] [Google Scholar]
- 20.Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114:1022–1036. doi: 10.1161/CIRCRESAHA.114.301621. doi: 10.1161/CIRCRESAHA.114.301621. [DOI] [PubMed] [Google Scholar]
- 21.Druce I, Abujrad H, Ooi TC. PCSK9 and triglyceride-rich lipoprotein metabolism. J Biomed Res. 2015;29:429–436. doi: 10.7555/JBR.29.20150052. doi: 10.7555/JBR.29.20150052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(suppl):S172–S177. doi: 10.1194/jlr.R800091-JLR200. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363–2372. doi: 10.1074/jbc.M708098200. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 24.Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. 2013;8:e64145. doi: 10.1371/journal.pone.0064145. doi: 10.1371/journal.pone.0064145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le May C, Kourimate S, Langhi C, Chétiveaux M, Jarry A, Comera C, Collet X, Kuipers F, Krempf M, Cariou B, Costet P. Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol. 2009;29:684–690. doi: 10.1161/ATVBAHA.108.181586. doi: 10.1161/ATVBAHA.108.181586. [DOI] [PubMed] [Google Scholar]
- 26.Ooi TC, Krysa JA, Chaker S, Abujrad H, Mayne J, Henry K, Cousins M, Raymond A, Favreau C, Taljaard M, Chrétien M, Mbikay M, Proctor SD, Vine DF. The effect of PCSK9 loss-of-function variants on the postprandial lipid and ApoB-lipoprotein response. J Clin Endocrinol Metab. 2017;102:3452–3460. doi: 10.1210/jc.2017-00684. doi: 10.1210/jc.2017-00684. [DOI] [PubMed] [Google Scholar]
- 27.Chan DC, Wong AT, Pang J, Barrett PH, Watts GF. Inter-relationships between proprotein convertase subtilisin/kexin type 9, apolipoprotein C-III and plasma apolipoprotein B-48 transport in obese subjects: a stable isotope study in the postprandial state. Clin Sci (Lond) 2015;128:379–385. doi: 10.1042/CS20140559. doi: 10.1042/CS20140559. [DOI] [PubMed] [Google Scholar]
- 28.Koren MJ, Sabatine MS, Giugliano RP, Langslet G, Wiviott SD, Kassahun H, Ruzza A, Ma Y, Somaratne R, Raal FJ. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 Extension Study. JAMA Cardiol. 2017;2:598–607. doi: 10.1001/jamacardio.2017.0747. doi: 10.1001/jamacardio.2017.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watts GF, Chan DC, Dent R, Somaratne R, Wasserman SM, Scott R, Burrows S, R Barrett PH. Factorial effects of evolocumab and atorvastatin on lipoprotein metabolism. Circulation. 2017;135:338–351. doi: 10.1161/CIRCULATIONAHA.116.025080. doi: 10.1161/CIRCULATIONAHA.116.025080. [DOI] [PubMed] [Google Scholar]
- 30.Robinson JG, Farnier M, Krempf M, et al. ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. doi: 10.1056/NEJMoa1501031. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 31.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 32.Reyes-Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352–362. doi: 10.1161/CIRCULATIONAHA.116.025253. doi: 10.1161/CIRCULATIONAHA.116.025253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith D, Watts GF, Dane-Stewart C, Mamo JC. Post-prandial chylomicron response may be predicted by a single measurement of plasma apolipoprotein B48 in the fasting state. Eur J Clin Invest. 1999;29:204–209. doi: 10.1046/j.1365-2362.1999.00431.x. [DOI] [PubMed] [Google Scholar]
- 34.Patsch JR, Miesenböck G, Hopferwieser T, Mühlberger V, Knapp E, Dunn JK, Gotto AM, Jr, Patsch W. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler Thromb. 1992;12:1336–1345. doi: 10.1161/01.atv.12.11.1336. [DOI] [PubMed] [Google Scholar]
- 35.Pang J, Chan DC, Hamilton SJ, Tenneti VS, Watts GF, Barrett PH. Effect of niacin on triglyceride-rich lipoprotein apolipoprotein B-48 kinetics in statin-treated patients with type 2 diabetes. Diabetes Obes Metab. 2016;18:384–391. doi: 10.1111/dom.12622. doi: 10.1111/dom.12622. [DOI] [PubMed] [Google Scholar]
- 36.Wong AT, Chan DC, Barrett PH, Adams LA, Watts GF. Effect of ω-3 fatty acid ethyl esters on apolipoprotein B-48 kinetics in obese subjects on a weight-loss diet: a new tracer kinetic study in the postprandial state. J Clin Endocrinol Metab. 2014;99:E1427–E1435. doi: 10.1210/jc.2013-4037. doi: 10.1210/jc.2013-4037. [DOI] [PubMed] [Google Scholar]
- 37.Dwyer KP, Barrett PH, Chan D, Foo JI, Watts GF, Croft KD. Oxazolinone derivative of leucine for GC-MS: a sensitive and robust method for stable isotope kinetic studies of lipoproteins. J Lipid Res. 2002;43:344–349. [PubMed] [Google Scholar]
- 38.Le NA, Coates PM, Gallagher PR, Cortner JA. Kinetics of retinyl esters during postprandial lipemia in man: a compartmental model. Metabolism. 1997;46:584–594. doi: 10.1016/s0026-0495(97)90198-0. [DOI] [PubMed] [Google Scholar]
- 39.Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Matthan NR, Lichtenstein AH, Dolnikowski GG, Horvath K, Asztalos BF, Zago V, Schaefer EJ. Effects of different doses of atorvastatin on human apolipoprotein B-100, B-48, and A-I metabolism. J Lipid Res. 2007;48:1746–1753. doi: 10.1194/jlr.M700067-JLR200. doi: 10.1194/jlr.M700067-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay AJ, Lamarche B, Hogue JC, Couture P. Effects of ezetimibe and simvastatin on apolipoprotein B metabolism in males with mixed hyperlipidemia. J Lipid Res. 2009;50:1463–1471. doi: 10.1194/jlr.P800061-JLR200. doi: 10.1194/jlr.P800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hogue JC, Lamarche B, Deshaies Y, Tremblay AJ, Bergeron J, Gagné C, Couture P. Differential effect of fenofibrate and atorvastatin on in vivo kinetics of apolipoproteins B-100 and B-48 in subjects with type 2 diabetes mellitus with marked hypertriglyceridemia. Metabolism. 2008;57:246–254. doi: 10.1016/j.metabol.2007.09.008. doi: 10.1016/j.metabol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38:2173–2192. [PubMed] [Google Scholar]
- 43.Chan DC, Nguyen MN, Watts GF, Ooi EM, Barrett PH. Effects of atorvastatin and n-3 fatty acid supplementation on VLDL apolipoprotein C-III kinetics in men with abdominal obesity. Am J Clin Nutr. 2010;91:900–906. doi: 10.3945/ajcn.2009.28422. doi: 10.3945/ajcn.2009.28422. [DOI] [PubMed] [Google Scholar]
- 44.Pramfalk C, Parini P, Gustafsson U, Sahlin S, Eriksson M. Effects of high-dose statin on the human hepatic expression of genes involved in carbohydrate and triglyceride metabolism. J Intern Med. 2011;269:333–339. doi: 10.1111/j.1365-2796.2010.02305.x. doi: 10.1111/j.1365-2796.2010.02305.x. [DOI] [PubMed] [Google Scholar]
- 45.Thompson GR, Naoumova RP, Watts GF. Role of cholesterol in regulating apolipoprotein B secretion by the liver. J Lipid Res. 1996;37:439–447. [PubMed] [Google Scholar]
- 46.Pal S, Allister E, Thomson A, Mamo JC. Cholesterol esters regulate apoB48 secretion in CaCo2 cells. Atherosclerosis. 2002;161:55–63. doi: 10.1016/s0021-9150(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 47.Pau E, He Y, Lougheed M, Steinbrecher UP. Inhibition of hydroxymethylglutaryl coenzyme A reductase activity does not affect the secretion rate of apolipoproteins B and AI by CaCo-2 cells. Biochem Cell Biol. 1995;73:81–90. doi: 10.1139/o95-010. [DOI] [PubMed] [Google Scholar]
- 48.Aoki T, Yoshinaka Y, Yamazaki H, Suzuki H, Tamaki T, Sato F, Kitahara M, Saito Y. Triglyceride-lowering effect of pitavastatin in a rat model of postprandial lipemia. Eur J Pharmacol. 2002;444:107–113. doi: 10.1016/s0014-2999(02)01547-9. doi: 10.1016/S0014-2999(02)01547-9. [DOI] [PubMed] [Google Scholar]
- 49.Phillips C, Mullan K, Owens D, Tomkin GH. Intestinal microsomal triglyceride transfer protein in type 2 diabetic and non-diabetic subjects: the relationship to triglyceride-rich postprandial lipoprotein composition. Atherosclerosis. 2006;187:57–64. doi: 10.1016/j.atherosclerosis.2005.08.020. doi: 10.1016/j.atherosclerosis.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 50.Xiao C, Dash S, Morgantini C, Adeli K, Lewis GF. Gut peptides are novel regulators of intestinal lipoprotein secretion: experimental and pharmacological manipulation of lipoprotein metabolism. Diabetes. 2015;64:2310–2318. doi: 10.2337/db14-1706. doi: 10.2337/db14-1706. [DOI] [PubMed] [Google Scholar]
- 51.Taldone T, Zito SW, Talele TT. Inhibition of dipeptidyl peptidase-IV (DPP-IV) by atorvastatin. Bioorg Med Chem Lett. 2008;18:479–484. doi: 10.1016/j.bmcl.2007.11.107. doi: 10.1016/j.bmcl.2007.11.107. [DOI] [PubMed] [Google Scholar]
- 52.Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, Fazio S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. 2014;130:431–441. doi: 10.1161/CIRCULATIONAHA.113.006720. doi: 10.1161/CIRCULATIONAHA.113.006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, Bolognese M, Wasserman SM. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006. doi: 10.1016/S0140-6736(12)61771-1. doi: 10.1016/S0140-6736(12)61771-1. [DOI] [PubMed] [Google Scholar]
- 54.Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H MENDEL-2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–2540. doi: 10.1016/j.jacc.2014.03.018. doi: 10.1016/j.jacc.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Olivecrona G. Role of lipoprotein lipase in lipid metabolism. Curr Opin Lipidol. 2016;27:233–241. doi: 10.1097/MOL.0000000000000297. doi: 10.1097/MOL.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi J, Miyashita K, Nakajima K, Mabuchi H. Hepatic Lipase: a Comprehensive view of its role on plasma lipid and lipoprotein metabolism. J Atheroscler Thromb. 2015;22:1001–1011. doi: 10.5551/jat.31617. doi: 10.5551/jat.31617. [DOI] [PubMed] [Google Scholar]
- 57.Schneider JG, von Eynatten M, Parhofer KG, Volkmer JE, Schiekofer S, Hamann A, Nawroth PP, Dugi KA. Atorvastatin improves diabetic dyslipidemia and increases lipoprotein lipase activity in vivo. Atherosclerosis. 2004;175:325–331. doi: 10.1016/j.atherosclerosis.2004.04.003. doi: 10.1016/j.atherosclerosis.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 58.Berk-Planken II, Hoogerbrugge N, Stolk RP, Bootsma AH, Jansen H DALI Study Group. Atorvastatin dose-dependently decreases hepatic lipase activity in type 2 diabetes: effect of sex and the LIPC promoter variant. Diabetes Care. 2003;26:427–432. doi: 10.2337/diacare.26.2.427. [DOI] [PubMed] [Google Scholar]
- 59.Toth PP, Sattar N, Blom DJ, Martin SS, Jones SR, Monsalvo ML, Elliott M, Davis M, Somaratne R, Preiss D. Effect of evolocumab on lipoprotein particles. Am J Cardiol. 2018;121:308–314. doi: 10.1016/j.amjcard.2017.10.028. doi: 10.1016/j.amjcard.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 60.Blom DJ, Djedjos CS, Monsalvo ML, Bridges I, Wasserman SM, Scott R, Roth E. Effects of evolocumab on vitamin E and steroid hormone levels: results from the 52-week, phase 3, double-blind, randomized, placebo-controlled DESCARTES Study. Circ Res. 2015;117:731–741. doi: 10.1161/CIRCRESAHA.115.307071. doi: 10.1161/CIRCRESAHA.115.307071. [DOI] [PubMed] [Google Scholar]