
Fig. 1. GABA metabolism and intracellular interactions.
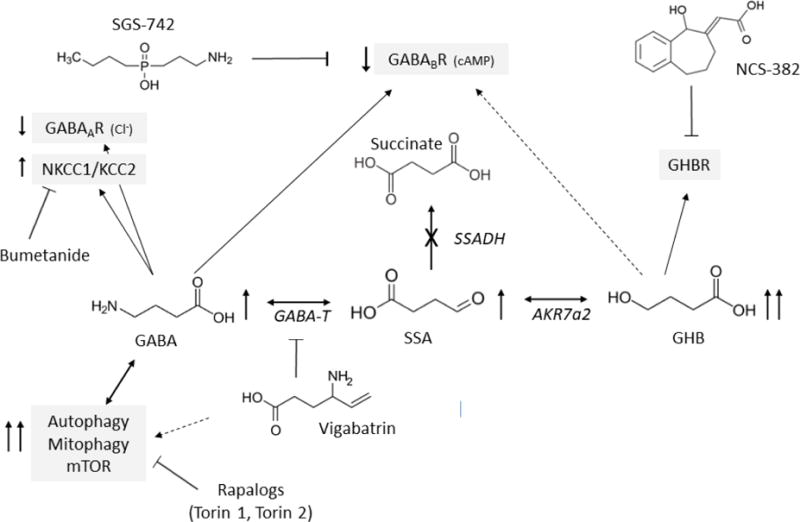
The site of the defect in patients with SSADHD is indicated by “X”. Abbreviations: GABA, γ-aminobutyric acid; GABAAR, ionotropic GABAA receptors; GABABR, metabotropic GABAB receptors. GABA-T, GABA-transaminase; SSA, succinic semialdehyde; AKR7a2, aldo-keto reductase 7a2; GHB, γ–hydroxybutyric acid; cAMP, cyclic AMP; NKCC1, sodium potassium chloride cotransporter 1; KCC2, neuronal potassium chloride cotransporter 2. In SSADHD, GABA, SSA and GHB accumulate (↑). Increased GABA and GHB activate GABAA and GABAB receptors and a putative GHB receptor (molecular nature unknown). However, a compensatory downregulation of GABA and GHB receptors (↓) has been reported in SSADHD suggesting excess GABA does not lead to increased inhibitory neurotransmission in vivo. NKCC1 and KCC2 control transmembrane chloride gradient and determine GABAA receptor directional transmembrane chloride flux. In experimental SSADHD, the expression ratio of NKCC1 and KCC2 (NKCC1/KCC2) is elevated (Vogel, 2017c), suggesting that activation of GABAA receptors causes chloride efflux from brain cells, membrane depolarization and activation of neurotransmission (see Fig. 3 for further details). NKCC1 inhibitors like bumetanide lower intracellular chloride concentration. In SSADHD, bumetanide may thus restore GABA inhibitory neurotransmission activity and efficiently suppress seizures. Finally, in SSADHD as well as in vigabatrin-treated animals, increased GABA activates the mTOR pathway with secondarily increased mitochondria number, oxidative stress, autophagy and mitophagy. mTOR inhibitors such as torin 1 and torin 2 improve GABA-induced, mTOR-pathway mediated intracellular defects and significantly prolong the lifespan of aldh5a1-deficient mice.