

Abstract
The polyketide synthases found in a variety of plants and fungi provide a varied source of biologically active compounds of pharmacological and medicinal interest. Stilbene synthase and chalcone synthase catalyze the formation of a common tetraketide intermediate, but use different cyclization mechanisms to produce distinct and separate natural products. While key structural differences have been identified to explain this functional diversity, a fuller explication of the factors responsible for this mechanistic disparity is required. Based on the energetics of our models of the bound tetraketides, and our structural analysis of the active sites we propose that a key tautomeric conversion provides a mechanistic framework common to both cyclizations. A previously unidentified active water molecule facilitates cyclization in chalcone synthase through a Claisen mechanism. Such a “Claisen switch” is comparable to the previously characterized “aldol switch” mechanism proposed for the biosynthesis of resveratrol in stilbene synthase.
Keywords: stilbene synthase, chalcone synthase, resveratrol, polyketide biosynthesis, Claisen condensation, aldol condensation
Graphical Abstract
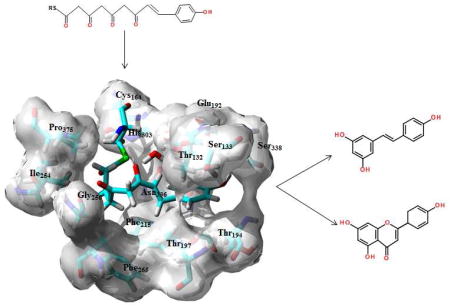
1. Introduction
Stilbene synthase (STS) and chalcone synthase (CHS) are plant polyketide synthases that catalyze the formation of a tetraketide intermediate (Fig. 1A) through successive additions of acetyl groups from malonyl-CoA to a p-coumaryl starter fragment. While STS and CHS share over 75% sequence identity STS cyclizes the tetraketide via an aldol condensation mechanism (Fig. 1B), yielding the hydroxystilbene resveratrol (RSVL, Fig. 1C), but CHS utilizes a Claisen condensation (Fig. 1D) of the same intermediate to generate chalcone (Fig. 1E), which can then be converted to the flavanone naringenin (Fig. 1F). In both enzymes the conserved residues Cys164, Phe 215, His303 and Asp336 define an active site where the cysteine functions as an attachment site for the polyketide intermediate, while the asparagine and histidine facilitate polyketide extension through nucleophilic attack by the malonyl fragment. The residues Ser133, Glu192, Thr194, Thr197 and Ser338 define a binding pocket that surrounds the p-coumaryl portion of the tetraketide. Finally Thr132, Phe215, Ile254, Gly256, Phe265 and Pro375 define the cyclization site, which facilitates the proper folding of the tetraketide either through the aldol (STS) or Claisen (CHS) condensation mechanisms. This site is also bounded by the Met137 residue from an adjoining monomer. Together these domains provide a cavity to facilitate and control chain elongation through successive acetylations of the bound coumaryl ligand. The growing polyketide is alternately attached and detached from the Cys164 residue by successive nucleophilic substitutions, and ultimately cyclized to the natural product.
Fig. 1.
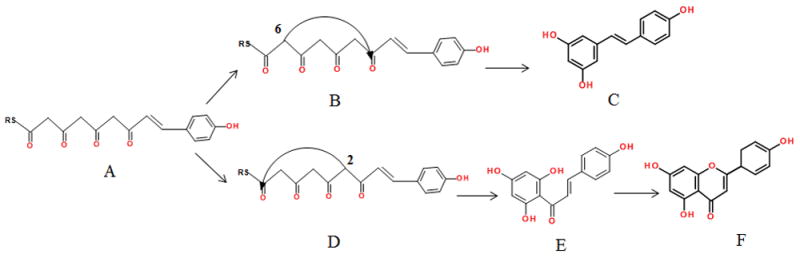
The common tetraketide intermediate (A) cyclized via an aldol condensation mechanism (B), yielding resveratrol (C), and cyclized via a Claisen condensation (D) to generate chalcone (E), which can then be converted to the flavanone naringenin (F).
Given that STS and CHS share substantial sequence identity without any significant insertions or deletions it is not unexpected that their mechanisms of product formation should be very similar. But while they do share a common method for ketide elongation, cyclization of the final tetraketide occurs using different mechanisms, namely the aldol condensation for the production of RSVL (Fig 1B) and a Claisen condensation in CHS (Fig 1D) that results in the production of either chalcone or nargenin. Several structural features have been identified that correlate with the observed selectivities. The most notable of these is what is termed an “aldol switch”, a modulation of the cyclization mechanism mediated by an active water molecule unique to STS crystal structures [1]. Through hydrolysis of the thioesterase this active water facilitates the base catalysis necessary for the aldol mechanism. However no equivalent base capable of catalyzing the Claisen condensation has been identified, and in the absence of a discrete proton abstraction step the current mechanism for cyclization in CHS requires that “the intermediate itself provide the driving force for carbanion formation” [2]. The Phe256 residue, positioned between the binding pocket and the cyclization site, is thought to function as a “steric gate” [2], mediating polyketide elongation and cyclization. This residue is disorderd in the ligand-free STS crystal structure, but adopts a well-defined conformation when RSVL is bound, a conformation however distinct from that characterized in CHS structures [3]. Most dramatically, whereas in CHS the cyclization mechanism results in cleavage of the Cys16-tetraketide thioester linkage, in STS the biosynthesis of RSVL involves separate thioester cleavage followed by cyclization and subsequent decarboxylation [1].
Because of its antioxidant and biomedical properties resveratrol has been co-crystallized with a variety of enzymes including transthyretin [4], quinone reductase [5], leukotriene-A4-hyrolyase [6], the cardiac regulatory protein troponin C [7] and, not surprisingly, stilbene synthase [3]. In all of these structures RSVL is planar or just slightly distorted from planarity. The notable exception is STS itself, where the dihedral angle between the π-bond and the aromatic rings is 60° (Fig. 2A), despite the fact that both NMR data and theoretical calculation confirm that the planar conformation is an energy minimum. Even a mutant chalcone synthase crystallized with a bound RSVL yields a conformation for RSVL with only a marginal, less than 3°, deviation from planarity [1]. Using the STS-bound RSVL ligand as a template we have developed a model for the STS-bound tetraketide intermediate. Quantum mechanical (QM) and quantum mechanical-molecular mechanical (QM-MM) calculations on this, and an analogously derived CHS-Bound tetraketide model has allowed us to develop a common mechanistic framework for the production of both the hydroxystilbenes and flavanones. Our analysis points to an energetically favored enol-keto tautomerism as a common element in both cyclizations, with the isomerism catalyzed by the previously characterized active water in the case of STS, and by a previously overlooked active water in the case of CHS. For the Claisen condensation in CHS the reaction enthalpy indicates a late transition, that is one most closely resembling the ketone. This in turn favors a concerted mechanism, one where formation of the enolate coincides with nucleophilic attack by the nascent carbanion at C2 on the thioester carbonyl, Fig 1D. Such a mechanism obviates the need for proton abstraction as the carbanion is formed by general base catalyzed tautomerization to the enolate.
Fig. 2.
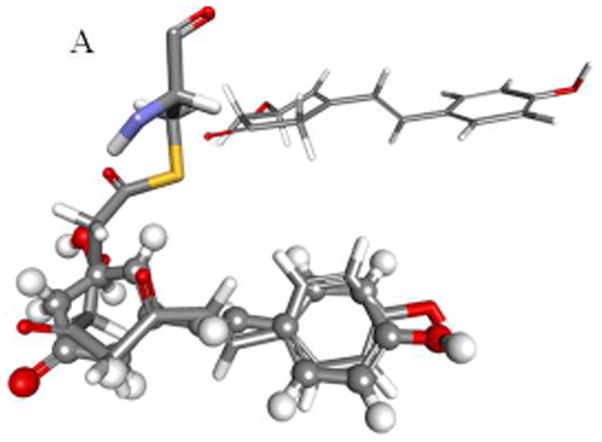
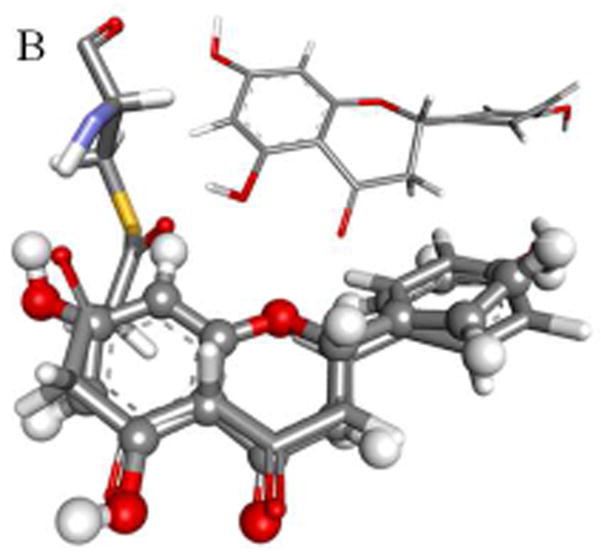
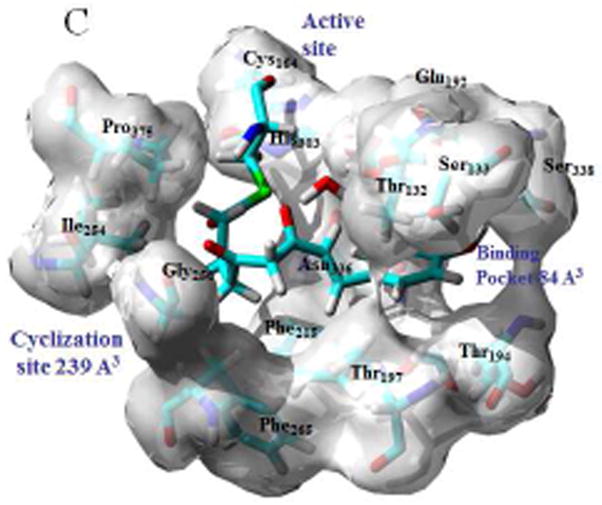
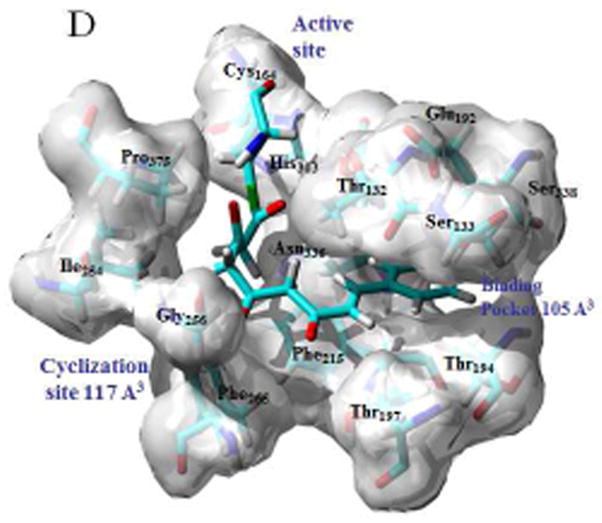
Modeled structures for the bound tetraketide in STS (A) and CHS (B) are shown overlaid with the bound resveratrol and naringenin ligands, with the crystal structures for the ligands shown in inset; binding pockets, active sites and cyclization sites in STS (C) and CHS (C), showing the calculated cavity volumes for the cyclization sites and binding pockets.
2. Methods
The crystal structures for ligand free STS [1Z1E], STS with RSVL bound [1Z1F], 18xCHS mutant with RSVL [1U0W], transthyretin with RSVL [1DVS], quinone reductase with RSVL [1SG0], leukotriene-A4-hyrolyase with RSVL [3FTS], troponin C with RSVL [2L98], and CHS bound to naringenin [1CGK] are available from the RCSB (www.rcsb.org). After adding hydrogens all proteins were subjected to a short energy minimization using the CHARMm force field [8]. Protein alignments and superimposition were done using the MODELER protocol as implemented in the Discovery Studio program suite. Using the carbon backbone of the RSVL ligand co-crystallized with STS as a template, a model for the STS-bound tetraketide intermediate was constructed. After applying harmonic constraints to the ketide backbone and fixed atom constraints to the Cys164 side-chain the protein and bound ligand were subjected to energy minimization using the the CHARMm force field (Fig 2A). A similar process for CHS complexed with nargenin yielded a model for the CHS-bound tetraketide intermediate (Fig 2B).
Quantum Mechanical-Molecular-mechanical (QM/MM) calculations on the bound tetraketide were carried out at the DFT (B3LYP/DNP)/CHARMm level, using the Becke, three-parameter, Lee-Yang-Parr (B3LYP) exchange-correlation functional, the double numerical polarized (DNP) basis set, and the CHARMm force field, where the QM region was defined as the Cys164 side chain, as well as the bound ligand. QM calculations on the free tetraketide, where the SCoA-linkage is modelled as CH3S-, were performed as B3LYP/6-31G+(d,p) single point calculations for both tautomers of the tetraketide-bound conformations, and both tautomers of the CH3S-tetraketide optimized using the AM1 semiempirical Hamiltonian [9]. These calculations yielded the relative tautomeric stabilities reported in Table 1. Finally highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals were generated for the C6 anion responsible for Aldol cyclization (Fig 3A and 3B), and the C2 anion leading to the Claisen condensation (Fig 3C and 3D).
Table 1.
Relative keto-enol tautomeric stabilities calculated by QM/MM for the CHS and STS-bound ligand; as well as the relative keto-enol stability for the free tetraketide, calculated by DFT after geometry optimization using the AM1 semiempirical Hamiltonian.
| QM-MM ΔEketo-enol (kcal mol−1) |
DFT//AM1 ΔEketo-enol (kcal mol−1) |
|
|---|---|---|
| CHS-tetraketide | −16.4 | |
| STS-tetraketide | −36.2 | |
| CH3S-tetraketide | 4.6 |
Fig. 3.
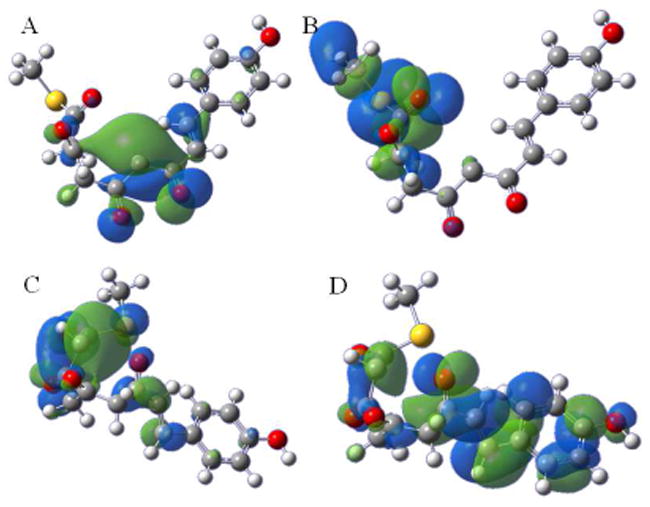
Frontier molecular Orbital (FMO) analysis of the C6 anion of the STS-bound tetraketide showing the highest occupied molecular orbital (HOMO) (A) and the lowest unoccupied molecular orbital (LUMO) (B); FMO analysis of the C2 anion of the CHS-bound tetraketide showing the HOMO (C) and the LUMO (D).
Cavity volumes for the cyclization sites and binding pockets of both STS and CHS were calculated using the CASTP utility available at the http://cast.engr.uic.edu/castp portal [10]. Because of its proximate position as an overhang to the bound ligand the Thr132 residue was included in the calculation of the size of both the coumaryl-binding pocket and the cyclization site. Defining a cavity as an interior void not accessible to a solvent probe the CASTP methodology uses computational geometry to measure cavity volume. Using a variety of algorithms, including describing the molecular interior space through a collection of convex polygons, this protocol generates an analytically derived volume for a given protein domain. Volumes calculated for the cyclization sites and binding pockets for both STS (1Z1F) and CHS (1CGK) are shown in Fig 2C and 2D respectively.
3. Results and discussion
As identified in [11] polyketide elongation occurs through successive acetylations of the coumaryl ligand, driven by decarboxylation of the malonyl-CoA substrate and catalyzed by the residues of the active site. As such the growing polyketide is alternately attached and detached from the Cys164 residue by successive nucleophilic substitutions until ultimate formation of the bound tetraketide. Prior to the final attachment therefore the tetraketide, Fig 1A, is present as a free ligand. Geometry optimization with the semi-empirical AM1 Hamiltonian yields the extended conformation as the minimum energy structure for the CH3S-tetraketide. Single point energy calculations at the B3LYP/6-31G+(d,p) level for both enol and keto tautomers yielded the relative tautomeric stabilities reported in Table 1. For the C1 carbonyl the dual benefits of an intramolecular hydrogen bond and the extension of the conjugated π-system combine to overcome the more stable bond energies typical of the keto form. The QM results in Table 1 indicate that for the free tetraketide enolisation at C1 is favored by nearly 5 kcals mol−1.
The modeled structures for the bound tetraketide in STS and CHS are shown in Figs 2A and 2B respectively. While both models differ dramatically from the extended conformation predicted for the free structure, the 60° torsion identified in the x-ray structure of the RSVL template means that the carbonyls are predicted to be substantially more distorted from planarity when bound in STS than in CHS. Given that the conjugative benefit from enolization demands effective p-orbital overlap it is not surprising that QM/MM calculations predict that both structures favor the keto tautomer at C1, with the keto-enol difference substantially greater for the STS-bound tetraketide, Table 1. Frontier molecular Orbital (FMO) theory highlights the benefit of such conformations. FMO analysis of the C6 anion of the STS-bound tetraketide shows that in addition to bringing the C6 and C1 condensation sites into proximity, this conformation gives a highest occupied molecular orbital (HOMO) with the largest orbital coefficient at C6, Fig 3A, and a lowest unoccupied molecular orbital (LUMO) with the largest orbital coefficient at C1, Fig 3B. FMO analysis of the C2 anion of the CHS-bound tetraketide conformation gives a HOMO with the largest orbital coefficient at C2, Fig 3C, and a LUMO with the largest orbital coefficient at C7, Fig 3D. The HOMO-LUMO gap is quite similar for both, 28 kcal mol−1 for the aldol in STS versus 31 kcal mol−1 for the Claisen in CHS. Thus both the topology and energetics of the FMOs, as well as the proximity of key reaction sites, combine to favor an aldol condensation in STS and a Claisen condensation in CHS.
How these bound conformations correlate with the binding pocket and cyclization sites in STS and CHS can be seen in Figs 2C and 2D respectively. Since the aldol condensation, unlike the Claisen, spans the methylene group at C2 there is a mechanistic need for the cyclization site to be larger in STS than in CHS. Comparing the cavity volumes one sees that indeed the site is over twice as big in STS. Key to this enlargement is the proximate positioning of the Phe265 and Thr197 residues, thus at once enlarging the cyclization site by over 100%, while at the same time reducing the size of the binding pocket by about 20% relative to the size in CHS. This tradeoff is facilitated by the fact that in the STS-bound tetraketide the portion of the p-coumaryl fragment that is conjugated, and therefore planar, is substantially smaller than for the bound tetraketide in CHS. Whether the Phe265 conformational switch in STS favors the aldol condensation by enlarging the cyclization site, or whether the repositioning forces the dihedral twist found in the x-ray structure, is just a matter of perspective. A conformational tilt of Thr132 towards the Thr197 residue also serves to enlarge the cyclization site in STS at the expense of the binding pocket, and tracing the origin of this differential serves to connect the protein structure to the organic mechanism.
The previously characterized “aldol switch” [1] involves a Ser338-stabilized crystallographic water connected through a hydrogen bonding network to Thr132 and Glu192. The position of this active water relative to Thr132 and Glu192 is shown in Fig 4C, and in relation to the cyclization site and binding pockets of STS in Fig 2C. Key to the thioesterase hydrolysis catalyzed by this water is a subtle displacement of the conserved Thr132 side-chain relative to that found in CHS, a displacement that facilitates the proton transfer mechanism necessary for hydrolysis. This displpacement is caused by a conformational change in the loop spanned by residues 131–137, a change which positions the Thr132 sidechain proximate to a Ser338-stabilized crystallographic water at an O-O distance of 2.88 Å [1]. By comparison in CHS the Thr132 sidechain oxygen is 4.27 Å from the Ser338-stabilized water, a distance similar to that found in PDB id 1C8U, an E. coli thioesterase II that produces over 95% chalcone but less than 3% of the stilbene [12]. And while this water is somewhat dynamic any movement to facilitate a hydrogen bond to the Thr132 in CHS would displace it from the active site and remove it as a nexus of the previously chatacterized hydrogen bonding network [11]. Analysis of this Thr132 displacement was undertaken by conformational sampling of the loop through a systematic search of the backbone dihedrals ϕ and ψ and optimization of the side chain conformations, using the LOOPER algorithm [13]. After energy minimization using CHARMm, including solvation energy calculated using the generalized Born approximation, scoring and ranking of the area 2 loops found in CHS (T131TSGVDH137) and STS (CHS (S131TTTPDL137) yield very different dynamic profiles. Whereas for CHS all 15 of the lowest energy conformers cluster around structures with O-O distances, from Ser338 stabilized water to Thr132O(H), of 6–7.5 Å, a majority of the STS low energy conformers adopt backbone conformations that permit hydrogen bonding to the Ser338 stabilized water. Four of these conformers cluster with a conformation nearly identical to the x-ray structure, another four adopt conformation with the Thr132 side-chain rotated by eighty degrees but still maintaining hydrogen-bonding contact with the Ser338 stabilized water through an O-O distance of 3.39 Å. As well as confirming that loop repositioning facilitates the “aldol switch” this result also reflects the findings that S131A and T132A mutants both exhibit increased chalcone production at the expense of stilbene production [1].
Fig 4.

(A) The mechanism of enol-keto conversion and Claisen condensation of the CHS-bound tetraketide intermediate, catalyzed by a Arg199-H2O- Thr197 proton transfer chain; (B) the mechanism for enol-keto conversion of the STS-bound tetraketide intermediate catalyzed by a Glu192-Thr132-H2O proton transfer chain that reconstitutes the active water; (C) hydrolysis of the Cys164-thioester, with proton abstraction by the Cys164 thiolate facilitating aldol condensation.
Structurally this displacement also contributes to the enlargement of the cyclization site in STS at the expense of reducing the volume of the binding pocket. However as can be seen in Fig 4B this water molecule is also proximate to the C1 in the STS-bound tetraketide, a tetraketide that prior to binding to Cys164 is predicted to favor enolization at C1, Fig 4B. And while the STS-bound keto tautomer is substantially more thermodynamically stable than the enol, the activation barrier for such conversions has been calculated to be as much as 60 kcals mol−1 in vacuo for simple linear β-diketones [14]. However theoretical calculations on β-cyclopentanedione showed that a chain of three proton transfers utilizing two active waters, in weakly acidic conditions, can reduce the barrier for tautomerization to just 7.9 kcal mol−1[15]. Based on the calculation results summarized in Table 1 we now have the possibility of a common mechanistic framework to explain cyclization in both STS and CHS, and just as importantly to provide an explicit mechanism to explain the “drive” for carbanion formation in CHS.
Given an energy difference of 4.6 kcal mol−1 in the free tetraketide, the STS-bound ligand will predominantly reflect a structure favoring an enol at C1. Given the proximate position of the active water and the necessary displacement of the Thr132 side-chain, a Glu192-Thr132-H2O proton transfer chain is predicted to catalyze the relatively facile conversion of the C1 enol, Fig 4B, to the C1 keto tautomer, Fig 4C. The isomerism reconstitutes the active water allowing it to subsequently catalyze hydrolysis of the Cys164-thioester, with proton abstraction by the newly liberated Cys164 thiolate facilitating attack from the C6 anion to the lately tautomerized C1 carbonyl, Fig 4C. Similar logic for CHS, namely that enol-keto conversion occurs prior or in concert with cyclization, would necessitate a similar proton transfer scheme. Comparing Figs 2C and 2D it can be seen that in addition to a displacement of Thr132 in STS, there is also an equally subtle displacement of the Thr197 at the base of the binding pocket in CHS. Inspection of the x-ray crystal structure reveals that the Thr197 side-chain displaces to a position intermediate between the C1 of the CHS-bound tetraketide and a crystallographic water. The water in turn is stabilized by a hydrogen bond to the side-chain of Arg199, creating a Arg199-H2O- Thr197 proton transfer chain. Analogous to the mechanism in STS this chain can catalyze tautomerization of the CHS-bound tetraketide from an enol at C1to a keto, Fig 4A. The lower exothermicity of this reaction, relative to that in STS, points to a later transition state (TS), one reflecting a greater buildup of charge at C2 compared to the TS predicted for STS. In the absence of a proton source interior to C2 this nascent carbanion is available to attack the C7 carbonyl via nucleophilic substitution i.e. a Claisen condensation.
Both CHS and STS have recently been used as rational bioengineering platforms for the production of novel natural products. In one case a T197G STS mutant was used to synthesize a novel C17 resorcyclic acid. This was achieved through the addition of an extra acetyl group to the tetraketide intermediate, which was facilitated by the elongation of the binding pocket caused by the steric-reducing site mutation [16]. In another rational approach homology modeling using the CHS crystal structure was used to alter the substrate specificity of benzophenone synthase (BPS), with which it shares 59% sequence identity [17]. Here a T135L variant was produced resulting in the protrusion of the leucine side chain into the active site, thus opening a new binding pocket for the phenyl group of the intermediate. It is to be hoped that this elucidation of the mechanistic details of both hydroxystilbene and flavanone biosynthesis can aid in the bioengineering of even more novel natural products.
Supplementary Material
A unitary mechanism for the cyclization of tetraketides in stilbene and chalcone synthase
The identification of an active water that functions as a “Claisen switch” in chalcone synthase
QM-MM and FMO analysis of model tetraketide ligands highlighting common and disparate components of binding pockets and cyclization sites in stilbene and chalcone synthase
Acknowledgments
The authors wish to acknowledge the support of the National Institute of General Medical Services (1K12GM102745), as well as Welch Foundation (Grant# BH-0018) for its continuing support of the Chemistry Department at St. Edward’s University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Austin MB, Bowman ME, Ferrer J-L, Schröder J, Noel JP. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chemistry and biology. 2004;11:1179–1194. doi: 10.1016/j.chembiol.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Ferrer JL, Jez JM, Bowman ME, Dixon RA, Noel JP. Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nature structural biology. 1999;6(8) doi: 10.1038/11553. [DOI] [PubMed] [Google Scholar]
- 3.Shomura Y, Torayama I, Suh DY, Xiang T, Kita A, Sankawa U, Miki K. Crystal structure of stilbene synthase from Arachis hypogaea. Proteins: Structure, Function, and Bioinformatics. 2005;60:803–806. doi: 10.1002/prot.20584. [DOI] [PubMed] [Google Scholar]
- 4.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nature Structural & Molecular Biology. 2000;7:312. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 5.Buryanovskyy L, Fu Y, Boyd M, Ma Y, Hsieh TC, Wu JM, Zhang Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies DR, Mamat B, Magnusson OT, Christensen J, Haraldsson MH, Mishra R, … Kim H. Discovery of leukotriene A4 hydrolase inhibitors using metabolomics biased fragment crystallography. Journal of medicinal chemistry. 2009;52:4694–4715. doi: 10.1021/jm900259h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pineda-Sanabria SE, Robertson IM, Sykes BD. Structure of trans-resveratrol in complex with the cardiac regulatory protein troponin C. Biochemistry. 2011;50:1309–1320. doi: 10.1021/bi101985j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 9.Dewar MJS, Dewar Zoebisch EG, Healy EF, Stewart JJP. AM1: A New General Purpose Quantum Mechanical Molecular Model. J Am Chem Soc. 1985;107:3902. [Google Scholar]
- 10.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry. 2000;39(5):890–902. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Derewenda U, Dauter Z, Smith S, Derewenda ZS. Crystal structure of the Escherichia coli thioesterase II, a homolog of the human Nef binding enzyme. Nat Struct Biol. 2000;7:555–9. doi: 10.1038/76776. [DOI] [PubMed] [Google Scholar]
- 13.Spassov VZ, Flook PK, Yan L. LOOPER: a molecular mechanics-based algorithm for protein loop prediction. Protein Eng Des Sel. 2008 Jan 14;21:91–100. doi: 10.1093/protein/gzm083. [DOI] [PubMed] [Google Scholar]
- 14.Yamabe S, Tsuchida Na, Miyajima K. Reaction Paths of Keto-Enol Tautomerization of β-Diketones. J Phys Chem A. 2004;108:2750–2757. [Google Scholar]
- 15.D’Cunha C, Morozov AN, Chatfield DR. Theoretical Study of HOCl-Catalyzed Keto–Enol Tautomerization of β-Cyclopentanedione in an Explicit Water Environment. J Phys Chem A. 2013;117:8437–8448. doi: 10.1021/jp401409y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhan N, Li L, Cai C, Xu P, Linhardt RJ, Koffas MA. Enzymatic formation of a resorcylic acid by creating a structure-guided single-point mutation in stilbene synthase. Protein Science. 2015;24:167–173. doi: 10.1002/pro.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klundt T, Bocola M, Lütge M, Beuerle T, Liu B, Beerhues L. A single amino acid substitution converts benzophenone synthase into phenylpyrone synthase. Journal of Biological Chemistry. 2009;284:30957–30964. doi: 10.1074/jbc.M109.038927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.