

ABSTRACT
Human cytomegalovirus (HCMV) represents a major cause of clinical complications during pregnancy as well as immunosuppression, and the licensing of a protective HCMV vaccine remains an unmet global need. Here, we designed and validated novel Sendai virus (SeV) vectors delivering the T cell immunogens IE-1 and pp65. To enhance vector safety, we used a replication-deficient strain (rdSeV) that infects target cells in a nonproductive manner while retaining viral gene expression. In this study, we explored the impact that transduction with rdSeV has on human dendritic cells (DCs) by comparing it to the parental, replication-competent Sendai virus strain (rcSeV) as well as the poxvirus strain modified vaccinia Ankara (MVA). We found that wild-type SeV is capable of replicating to high titers in DCs while rdSeV infects cells abortively. Due to the higher degree of attenuation, IE-1 and pp65 protein levels mediated by rdSeV after infection of DCs were markedly reduced compared to those of the parental Sendai virus recombinants, but antigen-specific restimulation of T cell clones was not negatively affected by this. Importantly, rdSeV showed reduced cytotoxic effects compared to rcSeV and MVA and was capable of mediating DC maturation as well as secretion of alpha interferon and interleukin-6. Finally, in a challenge model with a murine cytomegalovirus (MCMV) strain carrying an HCMV pp65 peptide, we found that viral replication was restricted if mice were previously vaccinated with rdSeV-pp65. Taken together, these data demonstrate that rdSeV has great potential as a vector system for the delivery of HCMV immunogens.
IMPORTANCE HCMV is a highly prevalent betaherpesvirus that establishes lifelong latency after primary infection. Congenital HCMV infection is the most common viral complication in newborns, causing a number of late sequelae ranging from impaired hearing to mental retardation. At the same time, managing HCMV reactivation during immunosuppression remains a major hurdle in posttransplant care. Since options for the treatment of HCMV infection are still limited, the development of a vaccine to confine HCMV-related morbidities is urgently needed. We generated new vaccine candidates in which the main targets of T cell immunity during natural HCMV infection, IE-1 and pp65, are delivered by a replication-deficient, Sendai virus-based vector system. In addition to classical prophylactic vaccine concepts, these vectors could also be used for therapeutic applications, thereby expanding preexisting immunity in high-risk groups such as transplant recipients or for immunotherapy of glioblastomas expressing HCMV antigens.
KEYWORDS: cytomegalovirus, HCMV, MCMV, Sendai virus, vaccine, dendritic cells
INTRODUCTION
Human cytomegalovirus (HCMV) infects the majority of the human population and establishes lifelong latency with periodic reactivations. It represents one of the most common congenital infections worldwide, with an estimated prevalence of 0.64% at birth, and contributes significantly to morbidity and mortality in immunocompromised individuals (1, 2). Currently, no vaccine is available, and treatment options with antiviral agents such as ganciclovir are limited due to side effects and the emergence of drug-resistant virus strains (3–5). Of the numerous candidate vaccines that were developed over recent decades, only a few made it past the early stages of clinical trials (6). However, modest efficacies have previously been reported in phase II studies for both prophylactic as well as therapeutic approaches (7, 8). While these results are encouraging and imply that the development of an HCMV vaccine is feasible, they also underline the necessity to explore novel, alternative vaccine concepts.
Since clear immunological correlates of protection from HCMV infection have not been defined yet, vaccine candidates should ideally induce both humoral and cell-mediated immune responses (9). Several different glycoprotein complexes are expressed on the virion surface, all of which are targets for neutralizing humoral immunity. Until recently, subunit vaccines designed for the induction of protective antibody responses mostly focused on the fusion protein gB. The discovery of the pentameric complex (consisting of gH/gL and UL128/UL130/UL131a) as a crucial component for the infection of epithelial and endothelial cells revealed a new target for such vaccine concepts (10). Indeed, inclusion of the pentameric complex into subunit vaccines or attenuated virus led to promising results in animal models (11, 12).
At the same time, cell-mediated immunity plays a crucial role in controlling viral latency and limiting virus spread. This is well illustrated by the successful restoration of HCMV-specific cytotoxic T lymphocyte responses and concomitant virus control in immunocompromised patients through adoptive T cell transfer (13, 14). T cell responses are directed against a variety of viral epitopes, with the tegument protein pp65 and the transcriptional regulator IE-1 representing major targets (15, 16).
Viral vectors are a favored tool for the delivery of heterologous antigens, in part owing to their capability to efficiently prime T cell responses during vaccination (17). Several vectors, like the poxvirus strain modified vaccinia Ankara (MVA), are currently being evaluated as therapeutic vaccine candidates in clinical trials, although their efficacy has yet to be demonstrated (18, 19). However, repeated administration of an antigen by a given vector is impeded by the development of immunity to its backbone, which can be avoided by heterologous prime/boost immunizations (20). Hence, novel vectors should still be developed and assessed for their capacity to deliver HCMV immunogens. For such approaches, Sendai virus (SeV) might be a useful alternative. It is a nonsegmented, negative-strand RNA virus that belongs to the family Paramyxoviridae and causes respiratory infections in mice. A number of advantageous features have led to broad usage of SeV as a viral vector, including exclusive replication in the host cell cytoplasm, efficient transduction of both dividing and nondividing cells, broad target cell tropism, and replication to high titers in cell culture (21). Importantly, it is also considered to be nonpathogenic in humans (22, 23). Sendai virus is currently being tested as a Jennerian vaccine for human parainfluenza virus (with the first efforts on this concept dating back to the 1960s [24]) and as a viral vector for the delivery of human respiratory syncytial virus antigens (25–27). In appreciation of its many favorable characteristics, SeV is also emerging as a vector for the delivery of immunogens (e.g., Gag) from unrelated pathogens, such as HIV-1 (28, 29).
The aim of this study was to explore whether a highly attenuated, replication-defective Sendai virus strain might be a suitable vector for the delivery of HCMV antigens. SeV strains expressing IE-1 and pp65 were generated, as well as variants that were rendered replication deficient through partial deletion of the viral P gene, thus further contributing to vector safety (26, 30, 31). These new SeV strains were compared with recombinant MVA viruses expressing the same antigens in a series of ex vivo assays. The work is focused on the impact that transduction with these strains has on dendritic cells (DCs), partly because of their crucial role in initiating adaptive immune responses. In addition, DCs pulsed with HCMV antigens could be readily applied as a therapeutic vaccine, a strategy which is currently being employed with some success in clinical studies for the treatment of glioblastoma (32). Using monocyte-derived dendritic cells (moDCs), we found that Sendai virus vectors exhibit favorable features with regard to transduction rates, cytotoxicity, DC maturation, and antigen presentation. Importantly, immune responses elicited after vaccination of mice with replication-deficient SeV vectors are capable of limiting MCMV replication in vivo. Collectively, these data indicate that the pp65- and IE-1-expressing SeVs are promising candidates to be further assessed regarding their suitability for preventive and therapeutic vaccination purposes.
RESULTS
No case of symptomatic SeV infection in humans has been reported to date. Nevertheless, we wanted to achieve further vector attenuation, since next to HCMV-negative young women, possible target groups for an HCMV vaccine also include immunocompromised patients such as transplant recipients. We chose a previously described strain in which amino acids 2 to 77 of the viral P gene are deleted (31). Partial deletion of this gene prevents switching of the viral RNA-dependent RNA polymerase (vRdRp) from mRNA synthesis to genome replication, ultimately inhibiting the generation of progeny virus (33). This strain, here referred to as replication-deficient SeV (rdSeV), served as the backbone for the insertion of the HCMV antigens IE-1, pp65, or, as a control, green fluorescent protein (GFP) (Fig. 1A). For comparison, the same transgenes were inserted into the genome of the parental strain containing the full-length P gene, hence termed replication-competent SeV (rcSeV), and into the genome of the poxvirus strain MVA. Expression of all transgenes could be readily detected by Western blot analysis after successful transduction of Vero cells (Fig. 1B). To test whether partial deletion of the viral P gene in rdSeV is sufficient to prevent replication in human moDCs, we determined viral titers in the cell culture supernatant over a period of 48 h after infection. Whereas rcSeV was capable of replicating to high titers in moDCs, the amount of infectious particles was considerably below the baseline level after 24 h and was undetectable after 48 h for rdSeV, indicating that no virus was produced de novo (Fig. 1C). When a functional P gene was provided in trans by a Vero cell-based helper cell line (V3-10), rdSeV replication was restored and was similar to that of rcSeV in moDCs.
FIG 1.

Sendai virus is capable of replicating in moDCs. (A) Schematic representation of viral genomes highlighting transgene insertion sites (not to scale; N, nucleoprotein; P, phosphoprotein; M, matrix protein; F, fusion protein; HN, hemagglutinin-neuraminidase; L, large protein). The modified SeV P gene is highlighted as Pmut. For MVA, letters refer to genome fragment sizes after HindIII digestion (66). (B) Western blot analysis of transgene expression 48 h postinfection (hpi) of Vero cells at an MOI of 1 with replication-competent (rcSeV) or replication-deficient (rdSeV) Sendai virus strains expressing the indicated genes, IE-1, pp65, or GFP. (C) Titration of cell culture supernatants at different time points after infection of human monocyte-derived dendritic cells (moDCs) with rcSeV-GFP or rdSeV-GFP at an MOI of 1. rdSeV was also used to infect the Vero cell line V3-10 (trans-complementing a full-length version of the viral P gene) at the same MOI. Three h postinfection, cells were washed once with medium and an aliquot was collected to determine baseline virus levels (residual virions that did not enter target cells and were not removed by washing). Viral titers are given as cell infectious units (CIU) per ml (bd, below detection limit).
We next wanted to assess whether expression of the heterologous antigens IE-1, pp65, and GFP is induced in dendritic cells upon infection with the different SeV strains. moDCs were generated by ex vivo differentiation of monocytes and infected at different multiplicities of infection (MOIs) with each vector. The percentage of antigen-positive cells was determined after 24 and 48 h by flow cytometry (Fig. 2). IE-1 and pp65 were stained intracellularly using labeled antibodies prior to the measurement (in contrast to GFP). In accordance with previously published data, we found that rcSeV is capable of efficiently infecting moDCs (34). For the GFP-carrying vectors, the percentage of antigen-positive cells was generally MOI dependent and at a similar level for rcSeV and MVA. Whereas median fluorescence intensity (MFI) levels were slightly higher for MVA at low MOIs, peak expression levels were highest with rcSeV at an MOI of 10 and 100, respectively. The percentage of GFP-positive cells as well as the monitored MFI were, however, clearly lowest when using rdSeV, showing that partial deletion of the P gene has a negative influence on overall transgene production. Nevertheless, the capacities to transduce dendritic cells and mediate transgene expression are preserved. Compared to GFP, intracellular detection of IE-1 and pp65 was altogether less sensitive for want of suitable flow cytometry antibodies. Consequently, expression of both IE-1 and pp65 was below the limit of detection for rdSeV, and signals were significantly lower than those obtained after infection with the corresponding rcSeV or MVA vector carrying GFP.
FIG 2.

SeV efficiently infects moDCs with rcSeV, eliciting higher transgene expression than rdSeV. MoDCs from 3 different HCMV seronegative blood donors were infected at the indicated MOIs, and the intracellular presence of the transgenes GFP, IE-1, and pp65 was quantified via flow cytometry after 24 (A and B) and 48 h (C and D) (nd, not determined). (A and C) Results are presented as the percentage of cells positive for a given antigen, with connected lines indicating values that were obtained using cells from an individual donor. (B and D) Median fluorescence intensity (MFI) values were normalized to the signals obtained from uninfected cells, with bars representing the means and standard deviations of values from all donors.
In addition to the vector-mediated expression of IE-1 and pp65 in dendritic cells, processing of the antigen and major histocompatibility complex (MHC) presentation are other critical prerequisites for the initiation or expansion of adaptive immune responses. As a surrogate marker for this process, we wanted to compare the tested vectors for their capacity to restimulate antigen-specific T cell clones after infection of dendritic cells. At 24 h after infection at various MOIs, we cocultivated HLA-matched moDCs with IE-1- or pp65-specific T cell clones (recognizing IE-188–96 on HLA-B8 and pp65417–426 on HLA-B7, respectively) and measured T cell reactivation by determining the intracellular presence of IFN-γ after another 6 h. All vectors efficiently restimulated T cells, with the maximum (approximately 80% of cells IFN-γ positive) reached at an MOI of 1 for MVA and 10 for both Sendai virus vectors (Fig. 3A). Whereas T cell responses were decreasing at higher MOIs when MVA was used, no such decrease in T cell restimulation was observed for the SeV vectors. Interestingly, rdSeV was equally as capable of eliciting moDC-driven T cell restimulation as rcSeV despite hardly detectable antigen levels in moDCs in flow cytometry (Fig. 2). Apart from minor differences, presumably reflecting differences in T cell receptor avidity of the clones used, the same trends were observable for IE-1 and pp65. To verify that the detected responses were antigen specific, moDCs were also infected with rcSeV-GFP, rdSeV-GFP, and MVA-GFP, followed by cocultivation with IE-1- or pp65-specific T cell clones. As expected, IFN-γ secretion was not above background levels at any MOI (data not shown).
FIG 3.
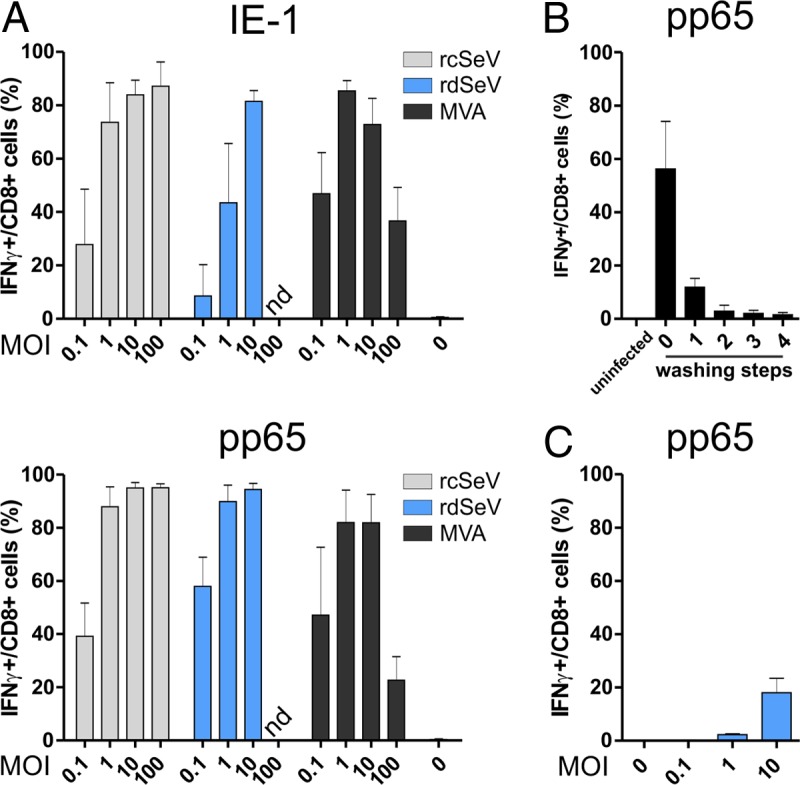
Infection with both Sendai vectors leads to efficient restimulation of T cells by infected moDCs. (A) Direct presentation assay. moDCs from 3 different HCMV seronegative blood donors were infected at the indicated MOIs with IE-1- or pp65-expressing vectors. At 24 hpi, antigen-specific T cell clones were added at an effector/target ratio of 1:1. After 6 h of cocultivation in the presence of brefeldin A (BFA), cells were stained for CD8 and intracellular IFN-γ and analyzed via flow cytometry. (B) Removal of extracellular virions before cross presentation: HeLa cells were transduced at an MOI of 10 with rdSeV-pp65. At 24 hpi, the supernatant from the overnight culture (lane 0) as well as 4 subsequent washing steps with cell culture medium (lanes 1 to 4) was collected and added to moDCs. Twenty-four h later, a pp65-specific, HLA-matched T cell clone was added to moDCs at an effector/target ratio of 1:1. After 6 h in the presence of BFA, CD8/IFN-γ staining and flow cytometry analysis were performed. (C) Cross-presentation assay. HeLa cells were transduced at the indicated MOIs with rdSeV-pp65. At 24 hpi, cells were washed 4 times and added to moDCs from 3 individual donors at a 1:1 ratio. After 24 h of cocultivation, an antigen-specific T cell clone was added for a HeLa/DC/T cell ratio of 1:1:1. T cell restimulation was measured after 6 h as described for panel A. Bars represent the means and standard deviations of values from all donors (A and C) or 3 independent experiments (B) (nd, not determined).
Despite efficient transduction of moDCs ex vivo, professional antigen-presenting cells will likely not be the main cell type producing antigenic proteins after in vivo administration of SeV vectors (e.g., via the intramuscular route). Thus, priming of CD8+ T cells requires secondary uptake and cross-presentation of antigens by DCs. We therefore wanted to test whether SeV-transduced cells or vesicles from those cells are internalized by moDCs and whether MHC-I presentation of antigens takes place. For this, an in vitro cross-presentation assay was performed. HeLa cells were infected at various MOIs with rdSeV-pp65, and 24 h postinfection (hpi) cells were added to moDCs, followed by cocultivation for 24 h. After this period, a pp65-specific T cell clone, HLA matched to the moDCs, was added, and after another 6 h, T cell restimulation was assessed by intracellular IFN-γ staining as described above. Direct presentation of pp65-derived peptides by HeLa cells can be ruled out in this experimental setup due to an HLA mismatch with the T cell clone used. rcSeV and MVA were not included in this experiment, since their replication in HeLa cells, and therefore also direct infection of moDCs during coculture, could not be excluded. Before addition of infected HeLa cells to moDCs, they were washed multiple times to remove residual extracellular rdSeV particles that might infect dendritic cells. The supernatants from those washing steps were separately incubated for 24 h with moDCs, followed by addition of T cell clones and assessment of IFN-γ production after 6 h to verify efficient removal of extracellular virions. Four washing steps proved sufficient to reduce the level of T cell reactivation to background levels (Fig. 3B). T cell restimulation as a result of cross-presentation did occur in this assay setup but was generally lower than that of direct infection of DCs (Fig. 3C, approximately 20% of T cells IFN-γ positive at an MOI of 10). However, the level of cross-presentation mediated by rdSeV at this MOI was still considerably higher than that of adenovirus 5 (Ad5), which was recently published in an identical experimental setting (35).
For efficient priming or expansion of T cell responses, SeV-infected DCs should exhibit a certain degree of longevity in vivo, which would otherwise hamper possible applications as a therapeutic vaccine. Sendai virus has long been known to cause strong cytopathic effects upon infection and was recently identified as a mediator of necroptotic cell death (36, 37). We hypothesized that the attenuation of rdSeV reduced undesired induction of cell death in dendritic cells. To test this, we infected moDCs at various MOIs with IE-1- or pp65-expressing virus strains and performed annexin V/7-aminoactinomycin D (7-AAD) staining after 24 and 48 h, respectively. All tested vectors caused cell death in an MOI-dependent manner and irrespective of the transgene, with MVA evidently being the most toxic variant (Fig. 4). Although IE-1 was previously described to inhibit apoptosis by activating the phosphatidylinositide 3′-OH kinase (PI3K) prosurvival pathway (38), we found that the amount of toxicity mediated by IE-1- and pp65-expressing vectors was comparable. At 24 h after infection, the percentage of annexin V/7-AAD-positive cells for a given MOI was comparable between both Sendai vectors. However, after 48 h, the proportion of healthy cells was further decreased when rcSeV was used, while it remained constant for rdSeV, confirming that this vector is clearly less toxic than the original strain or MVA.
FIG 4.

Attenuated rdSeV is less cytotoxic than rcSeV. moDCs from 3 different HCMV seronegative blood donors were infected at the indicated MOIs with IE-1- or pp65-expressing vectors. After 24 or 48 h, samples were costained with annexin V/7-AAD, and cells positive for one or both markers were quantified by flow cytometry. Bars represent the means and standard deviations (SD) of values from all donors (nd, not determined).
It is well established that priming of naive T cells by dendritic cells requires the latter to be in a fully mature state and that immature or only partially matured DCs are likely to induce tolerance or anergy in vivo (39). Phenotypically, maturation coincides with increased expression of surface molecules, such as CD80, CD83, CD86, and MHC-II. To assess whether any of these markers were upregulated in response to infection with SeV or MVA vectors, surface expression was measured via flow cytometry after 24 and 48 h (Fig. 5). We found that at an MOI of 0.1, MVA-IE-1 and MVA-pp65 induced upregulation of CD86 and HLA-DR, while CD80 upregulation was evident only after 48 h, and the expression of CD83 was largely unaltered at both time points. At higher MOIs, marker expression was generally lower than that of uninfected cells, with CD86 and HLA-DR being affected the most. In contrast, after infection with the SeV vectors, markers were upregulated in a time- and MOI-dependent manner, except for some downregulation of CD80 and CD83 when rcSeV-IE-1 was used. Especially at higher MOIs, SeV vectors were clearly superior to MVA in mediating maturation, with rdSeV inducing the highest overall upregulation. It is worth noting that SeV-induced maturation seemed less pronounced for IE-1-carrying vectors, which possibly reflects immunomodulatory properties of this protein. The observed tendencies were mostly more pronounced after 48 h than after 24 h.
FIG 5.

SeV induces maturation of dendritic cells. moDCs from 3 different HCMV seronegative blood donors were infected at the indicated MOIs with IE-1- or pp65-expressing vectors. After 24 or 48 h, samples were stained for CD80, CD83, CD86, or HLA-DR and analyzed via flow cytometry. Obtained median fluorescence intensity (MFI) values were normalized to those of uninfected cells. Log2 values which represent the means (± standard deviations) from all donors are displayed in a heatmap indicating upregulation (blue) or downregulation (red) of a given marker (nd, not determined).
Along with the upregulation of costimulatory molecules on the surface of dendritic cells, secretion of distinct cytokines is also critical for T cell priming and definition of the induced effector phenotype. To explore the cytokine release profile of moDCs in response to infection with SeV or MVA, we measured secretion of various cytokines in a cytometric bead multiplex assay. We found that all vectors induced production of interleukin-6 (IL-6), while the presence of the anti-inflammatory cytokine IL-10 was hardly detectable and did not surpass the level produced by uninfected cells (Fig. 6). IFN-α responses were above background for all vectors at an MOI of 1 and, at an MOI of 10, were only detectable when rdSeV was used. In contrast, tumor necrosis factor (TNF) secretion was lowest for rdSeV, and IL-18 release was only detectable after MVA infection. The observed trends were similar irrespective of the transgenes carried by the respective vectors, indicating that insertion of IE-1 or pp65 has no major influence on the cytokine profile of infected moDCs.
FIG 6.

SeV infection induces secretion of IL-6 and IFN-α. moDCs from 3 different HCMV seronegative blood donors were infected with either GFP-, IE-1-, or pp65-expressing vectors. At 48 h after infection, the presence of 13 different cytokines in the conditioned cell culture medium was assessed by a bead-based multiplex immunoassay. Uninfected cells that were untreated or stimulated with LPS (1 μg/ml) served as controls. Selected cytokine concentrations are depicted with bars representing the means and standard errors of the means (SEM) of 3 measurements from the same donor (performed in one experiment). Levels were not reproducibly above the limit of detection for IL-1β, IFN-γ, IL-12p70, IL-17A, IL-23, and IL-33, and no significant differences in secretion were detected for MCP-1 and IL-8 (data not shown).
We also tested whether other immune cells besides DCs can be infected by Sendai vectors for other potential gene delivery applications. Hence, we used rcSeV-GFP to infect human peripheral blood mononuclear cells (PBMCs) as a whole, and the amount of GFP-positive cells in different leukocyte populations was determined after 24 h to identify permissive cell types (Fig. 7). Except for B cells, all addressed subpopulations (NK cells, monocytes, and CD4+ and CD8+ T cells) exhibited GFP expression, although to varying degrees. Since partial deletion of the P gene likely has no influence on target cell tropism, rdSeV might be capable of transducing these cells as well. Monocytes were infected most efficiently, with approximately 80% of cells being GFP positive, followed by NK cells (50%) and T cells (20%), at an MOI of 10. Similar trends were observed at an MOI of 1, albeit with lower overall infection rates. These results suggest that Sendai virus could be a useful tool for transduction of a variety of immune cells.
FIG 7.

SeV is capable of infecting T cells, NK cells, and monocytes. (A) Gating strategy for discriminating different leukocyte populations. PBMCs from 3 different HCMV seronegative blood donors were infected at an MOI of 1 or 10 with rcSeV-GFP. (B) At 24 hpi, the amount of GFP-positive cells in the indicated populations was determined by flow cytometry. Bars represent the means and standard deviations (SD) of values from all donors.
Finally, we wanted to assess to what extent vaccination with rdSeV is capable of limiting cytomegalovirus infection in vivo. The strict species specificity of betaherpesviruses hampers preclinical testing of HCMV vaccine candidates in animals, so we turned to a novel, recently published mouse model to assess the efficacy of our rdSeV vectors in the context of an MCMV infection (40, 41). This model takes advantage of the strong T-cell immunogenicity of the HLA-A*0201 (HLA-A2.1)-restricted HCMV epitope pp65495–503 NLVPMVATV (here referred to as NLV). After vaccination of HLA-A2 transgenic mice with vaccine candidates containing HCMV pp65, challenge is performed with an MCMV strain (designated MCMV-NLV), in which the entire NLV peptide coding sequence had been inserted into the IE-2 gene along with its natural flanking amino acids. Murine T cell responses to the NLV peptide that were primed by preceding immunizations may then restrict the course of MCMV infection (40).
For proof of principle, A2Kb mice were immunized 3 times at weeks 0, 3, and 6 via the intranasal route with rdSeV-pp65. Eight weeks after the initial immunization, animals were challenged intravenously with MCMV-NLV, and after 4 more days, the infectious viral load was measured in homogenates of spleen, liver, and lungs by plaque assay and viral genome load was determined in parallel by quantitative PCR (qPCR). Compared to a nonimmunized control group, viral titers were markedly reduced in the liver and lungs after vaccination with rdSeV (Fig. 8). A similar, although not significant, trend was observed in the spleen. Likewise, viral genome numbers were reduced in the spleen and liver, but, interestingly, not in the lungs at the tested time point. Taken together, these data strongly indicate that immune responses primed by immunization with rdSeV-pp65 limit the course of systemic MCMV infection.
FIG 8.

Vaccination with rdSeV-pp65 limits viral replication after MCMV-NLV challenge in mice. HLA-A2 transgenic mice were separated into two groups of 5 animals each and immunized intranasally at weeks 0, 3, and 6 with rdSeV-pp65 (6 × 106 CIU per dose) or an equivalent volume of PBS as a negative control. Two weeks after the last immunization, mice were challenged intravenously with 5 × 105 PFU of MCMV-NLV. (A) Viral infectivity was quantified in the indicated organ homogenates 4 days after challenge infection by plaque assay. (B) Viral genome load was quantified in the same samples by qPCR. P values were calculated after log transformation by using unpaired, two-sided t test with Welch's correction.
DISCUSSION
In recent years, four different vaccine concepts utilizing viral vectors for the delivery of HCMV antigens have made it into clinical trials (reviewed in reference 18). Of those, a vector based on MVA containing pp65, as well as an exon of IE-1 and IE-2 each (HCMV-MVA triplex), is the only one currently being tested in phase II. The results from phase I, investigating safety and immunogenicity in a small cohort of healthy adults in a homologous prime/boost regimen, were recently published (19). While strong and durable T cell responses were induced upon priming, the booster immunization hardly showed an additional benefit. At the same time, anti-MVA immunity was detectable. In a different setting, however, it was previously reported that immune responses elicited by the closely related vaccinia strain NYVAC could be increased in a heterologous prime/boost regimen employing a DNA prime compared to the level for the NYVAC-alone group (42). Thus, it is likely that the immunogenicity of MVA could be improved upon by likewise combining it in a heterologous prime/boost regimen with secondary delivery modalities, such as the Sendai virus vectors described here.
Live attenuated virus vectors often pose residual health risks when administered as a vaccine, but usually they exhibit superior immunogenicity compared to inactivated virions. Although SeV has not been reported to cause disease in humans, clinical trials so far have only been conducted with healthy individuals. Thus, for immunocompromised individuals, which are the main target group for therapeutic HCMV vaccines, higher safety measures might have to be implemented. In order to increase the safety of the described Sendai virus-based HCMV vaccine candidates, we chose a replication-deficient SeV strain with a partial deletion of the P gene. This deletion interrupts the interaction between the viral P and N proteins and, as a consequence, renders the viral RNA-dependent RNA polymerase (vRdRp) incapable of switching from mRNA synthesis (using the negative-strand viral RNA genome as the template) to the de novo synthesis of minus-strand viral genomes from viral plus-strand mRNAs (33). Expression of SeV genes is mostly monocistronic. In this regard, however, the P locus is an exception, since it also encodes accessory proteins (C′, C, Y1, and Y2) that are translated through leaky ribosome scanning (43). Moreover, mRNA editing yields two additional viral proteins, termed V and W. Although all of these proteins seem to be dispensable for viral growth in cell culture, they were shown to promote pathogenicity in mice, for example, by counteracting interferon signaling (44). Partial deletion of the P gene in rdSeV presumably results in the production of truncated versions of V and W (whose translation is initiated from the same start codon as P) and C′ (which uses a non-AUG initiation site upstream of this locus) (45). At the same time, translation of C, Y1, and Y2 is abrogated entirely since the respective start codons are not present in rdSeV. The lack of these accessory proteins might contribute to the attenuated phenotype of rdSeV, thus further improving its safety profile without abrogating the capability to elicit protective immune responses (26). Further research will be required to determine to what extent the truncation or lack of these accessory proteins impacts the immunogenicity of rdSeV.
We demonstrated in this study that human dendritic cells are permissive for replication of SeV, which underlines the requirement for further attenuation to limit proliferation during a state of immunosuppression in vivo. In this regard, the N-terminal truncation of the phosphoprotein already proved to be sufficient to inhibit the generation of viral progeny upon infection of DCs. However, after entry of rdSeV into a cell, mRNA synthesis as well as protein translation still take place, thus preserving the advantages of live attenuated vectors to some extent. Although the deletion that was introduced into the genome of rdSeV mechanistically precludes the formation of progeny virus in any given cell type, it will be important to experimentally confirm this, for example, by using cells that were obtained from patients suffering from various types of innate or acquired immunodeficiency. Furthermore, vector safety could also be assessed more deeply in immunosuppressed animals such as NSG mice. While transgene expression is initiated in moDCs after SeV infection, we found that the overall amount of transgenic protein is markedly reduced when using rdSeV rather than the parental strain. Although reduced mRNA synthesis as a result of partial P deletion has been observed before, the underlying molecular mechanism has not been elucidated yet (30). This might be explained by the inability of the vRdRp complex from rdSeV to switch to genome replication. During infection with wild-type SeV strains, newly generated genome copies could serve as additional templates for subsequent rounds of mRNA synthesis, thereby amplifying overall gene expression. Importantly, despite decreased transgene levels, restimulation of antigen-specific T cells was hardly affected, which is in accordance with previously published data suggesting that the intracellular quantity of a given immunogen is not connected to the amount of MHC presentation on the cell surface (46).
All vectors that were compared in this study caused cell death in an MOI- and, with the exception of rdSeV, time-dependent manner. It is possible that cells which were negative for cell death markers after 24 h initially remained uninfected and that secondary infections by newly released virions, concomitant with induction of apoptosis, account for the increase in dying cells between 24 and 48 h. Because MVA was previously reported not to produce viral progeny in moDCs (47), such an effect is only conceivable for replication-competent SeV, which might explain why the proportion of dead cells remained constant over the observed time period for rdSeV. Information on the duration of paramyxovirus replication in cell culture is scarce (48), but we found that viral titers were above the baseline level already at 24 h after infection of moDCs with rcSeV (Fig. 1C), showing that the replication cycle can be completed in this time frame. Thus, one or more rounds of infection would be possible within 48 h. Nevertheless, an increase in dead cells over time was also evident for the replication-competent vectors at higher MOIs (10 and 100). Under these conditions, few cells should initially remain uninfected, which indicates that replication deficiency alone is an unlikely explanation for the reduced cytotoxicity of rdSeV. The discrepancy might also be explained by the different patterns of cytokines released after infection with the different vectors. For instance, secretion of TNF, a potent inducer of cell death (49), could be the cause of the toxicity observed here. In fact, the amounts of TNF produced upon infection mirror the degree of toxicity of the three vectors.
Priming of T cells in vivo requires a finely tuned combination of signals from dendritic cells, the main constituents of which are (i) processing and MHC presentation of a given peptide, (ii) presence of costimulatory molecules on the DC surface, and (iii) cytokine secretion (50). Already at low MOIs, CD8+ T cell clones were efficiently restimulated by moDCs after infection with all vectors tested, indicating no major viral interference with the antigen-processing and presentation machinery. However, the restimulatory capacity of MVA-infected DCs was diminished at higher MOIs, a phenomenon that was not observed for the SeV vectors and which may be linked to the higher cytotoxicity of the poxvirus strain. Reduced T cell stimulation by MVA may also be the result of a noticeable downregulation of costimulatory receptors from MOIs 1 to 100 as part of a previously described immune evasion mechanism (51). In contrast, maturation markers tended to be upregulated in an MOI-dependent manner by both SeV vectors with only a few exceptions, which is in accordance with findings by other groups describing Sendai virus as a potent inducer of DC maturation (52, 53). However, it was striking that upregulation of maturation markers after SeV infection was generally less pronounced for IE-1-expressing vectors than for their pp65-containing counterparts, with CD83 even being downregulated to some extent. During HCMV infection, IE-1 plays a central role in suppressing various innate immune response pathways (reviewed in reference 54) and may therefore also counteract DC maturation when expressed by heterologous vectors. The immediate-early protein IE-2 of HCMV was recently found to induce proteasomal degradation of CD83 (55), an immune evasion strategy that is similarly employed by other herpesviruses, such as herpes simplex virus 1 (56). Since IE-1 and IE-2 are alternative splicing products from the same gene locus with the first 85 amino acids being identical due to the shared usage of two exons, it is conceivable that IE-1 could likewise be capable of inducing CD83 degradation, but further research will be necessary to assess this. Such detrimental influences on DC maturation and function may be avoided by using a functionally inactivated IE-1 protein, as previously proposed by Tang and colleagues (57). Irrespective of a given transgene, SeV-infected DCs might indeed be capable of undergoing full maturation without the need for further components (like adjuvants) when taking into account that in addition to the upregulation of maturation markers, the Sendai vectors are also capable of inducing secretion of proinflammatory cytokines such as IL-6 and IFN-α.
Vaccination of HLA-A2 transgenic mice with rdSeV-pp65 limited MCMV-NLV replication compared to that of nonimmunized animals, as evidenced by reduced viral titers in the liver and lungs as well as lowered MCMV genome copy numbers in the spleen and liver. Reduced MCMV replication in organs other than the lung despite immunization via the intranasal route suggests that systemic immunity was elicited by the rdSeV vaccination. Given that HCMV is capable of spreading to a multitude of different organs, vaccine-mediated induction of immune responses that are not locally restricted is desirable, especially in therapeutic settings. Interestingly, although the amount of infectious virus was clearly reduced in the lungs of immunized animals, viral genome numbers were similar in both groups. This could be due to delayed clearance of viral genomes from the lung tissue even if the release of infectious virus has been limited already. Since the HCMV pp65-derived, HLA-A2-restricted NLV peptide represents the only antigenic overlap between the SeV vaccine strain and the MCMV challenge strain, NLV-specific CD8 T cells most likely are critical for the restricted viral replication that was observed in this experiment. As demonstrated by adoptive T cell transfer experiments, limitation of MCMV-NLV replication has clearly been attributed to NLV-specific T cells (40). Given that the chosen A2Kb mice still express endogenous H-2b molecules, it is possible that human and murine MHC molecules compete for the priming of pp65-specific T cell responses during vaccination with rdSeV-pp65, thereby impeding the priming of NLV-specific, HLA-A2-restricted T cells. Thus, usage of different animal models, such as humanized mice or nonhuman primates (reviewed by Crawford et al. [58] and Itell et al. [59]), might provide additional insights concerning the immunogenicity of rdSeV-pp65 and allow preclinical efficacy testing of IE-1-expressing vectors as well. It might also be promising to combine the rdSeV vectors introduced in this study with vaccine strategies that include B cell immunogens to induce both humoral and cell-mediated immunity.
Finally, we could demonstrate that SeV exhibits a broad target cell tropism. In addition to human moDCs, T cells, NK cells, and monocytes are efficiently infected by Sendai virus as well. This opens up a variety of possible gene delivery applications in basic research and gene therapy.
In conclusion, the favorable immunological characteristics of Sendai virus in combination with the enhanced safety profile of rdSeV emphasize the potential of this vector system. Thus, further testing of SeV as a vaccine platform in general and as an HCMV vaccine candidate in particular is warranted.
MATERIALS AND METHODS
Ethics statement.
The collection of blood donations from volunteers was approved by the ethics committee of the University of Regensburg (file reference 16-101-0347). Samples were taken from healthy adults who gave written informed consent beforehand. All animal experiments were approved by the ethics committee of the Landesuntersuchungsamt Rheinland-Pfalz, permit number 23177-07/G11-1-004.
Cells and viruses.
Baby hamster kidney (BHK-21; ATCC CCL-10) and Vero cells (ATCC CCL-81) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (all from PAN-Biotech), and 10% fetal calf serum (FCS). Peripheral blood mononuclear cells (PBMCs) were purified according to density using Ficoll-Paque (GE Healthcare) and subsequently cultured in RPMI 1640 supplemented with 2 mM l-glutamine (PAN-Biotech), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS. Primary monocytes were isolated via magnetic activated cell sorting (MACS) using anti-CD14 beads (Miltenyi Biotech) according to the manufacturer's instructions. Monocytes were differentiated to monocyte-derived dendritic cells by culturing them over a period of 5 to 6 days in RPMI medium containing 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 1% nonessential amino acids, 1% MEM vitamins, 1 mM sodium pyruvate, 10 μM β-mercaptoethanol (all from Gibco), 1,000 U/ml IL-4, and 1,000 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (both from Miltenyi Biotech). After 2 days, cells were given fresh RPMI medium containing all of the aforementioned supplements. Differentiation of monocytes to moDCs was verified via flow cytometry by checking the downregulation of CD14 and the upregulation of CD1a. The CD8+ T cell clones 4G6 (recognizing the pp65-derived peptide TPRVTGGGAM on HLA-B7) and 1C3 (recognizing the IE-1-derived peptide QIKVRVDMV on HLA-B8) were a kind gift from Dirk Busch (TU Munich). They were cultivated in RPMI 1640 with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS. The strain MVA-GFP was kindly provided by Mariano Esteban (CSIC Madrid), while all the SeV strains are based on the isolate D52 (ATCC VR-105). Primary mouse embryonic fibroblasts (MEF) were prepared from embryos of C57BL/6 mice (purchased from Janvier Labs) on day 14 of gestation by standard methods (60) and maintained in minimal essential medium (MEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS (all from Gibco). The chimeric bacterial artificial chromosome (BAC)-derived virus MCMV-NLV was described previously (40). In short, 69 nucleotides of HCMV open reading frame UL83 (n119,567 to n119,499; GenBank accession no. X17403) encoding 23 amino acids, including the pp65495–503 epitope and its natural flanking regions, were integrated into ORFm128 of the MCMV genome. Virus stocks of MCMV-NLV were prepared from infected C57BL/6 MEF by sucrose gradient ultracentrifugation as described previously (61).
Mice.
Mice were bred at the animal facility in the Central Laboratory Animal Facilities at the University Medical Center Mainz and maintained under specific-pathogen-free conditions. Transgenic A2Kb mice (a gift of Hakim Echchannaoui 3rd Medical Clinic, University Medical Center, Mainz, Germany) have been previously described (62) and express the human HLA-A2.1 molecule and mouse H-2Kb and H-2Dd molecules.
Generation of recombinant MVA vectors.
The recombinant poxvirus strains MVA-IE-1 and MVA-pp65 were generated by inserting the respective genes under the control of a synthetic early/late promoter (63) into the viral thymidine kinase (TK) locus J2R via homologous recombination, followed by plaque purification (64). Briefly, IE-1 (accession no. AAR31448.1) or pp65 (accession no. P06725.2) was cloned into the transfer vector pLZAW1 targeting the TK locus. BHK-21 cells were simultaneously transfected with the respective plasmid and infected with MVA-GFP. Isolation of recombinant MVA strains from BHK-21 cells as well as subsequent removal of the reporter gene lacZ were achieved over several rounds of plaque purification combined with blue-white screening. Complete separation of newly generated virus strains from the parental MVA strain and absence of the lacZ gene were verified by PCR screening. In addition, Western blot analysis as well as Sanger sequencing of the TK locus were performed to confirm correct integration of the transgenes. Virus stocks were produced by large-scale infection of BHK cells. Infected cells were lysed through 3 freeze/thaw cycles, and virus particles were purified from cell lysates through two ultracentrifugation rounds over a 30% sucrose cushion.
Generation, propagation, and titration of recombinant SeV strains.
All recombinant Sendai virus variants were generated from a cDNA template located on plasmid DNA. cDNA templates of rcSeV-GFP and rdSeV-GFP were generated previously (30). The new constructs rcSeV-IE-1, rcSeV-pp65, rdSeV-IE-1, and rdSeV-pp65 were cloned by exchanging the GFP transgene from the above-named GFP-expressing cDNA constructs against the respective transgenes (IE-1 and pp65) via NotI restriction digestion.
Recombinant viruses were recovered through virus rescue experiments from transfected BSR-T7 cells as described before (33), with slight modifications. FuGENE6 (Roche) was used as the transfection reagent at 2.0 μl/μg DNA. Virus was harvested from the supernatant and amplified in Vero cells (replication-competent SeV) or in the helper cell line V3-10 (26) (replication-deficient SeV vector). Virus preparations were titrated as previously described (26), and titers are given as cell infectious units per milliliter (CIU/ml). The integrity of the various SeV vector genomes was confirmed by reverse transcription-PCR (RT-PCR) and sequencing.
Infection of monocyte-derived dendritic cells.
On day 5 or 6 after monocyte isolation, the culture medium of moDCs was replaced with RPMI 1640 devoid of any supplements. Virus suspensions were likewise diluted in RPMI 1640 and added to the cells. After 3 h, medium was removed and infected cells were cultivated in RPMI 1640 with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS for the desired amount of time.
Western blot analysis.
Cell extracts from Vero cells infected with various SeV strains were separated by SDS-PAGE and blotted onto nitrocellulose membranes. Proteins were detected using anti-IE-1 (clone IE1.G10; Abcam), anti-pp65 (ab49214; Abcam), or anti-GFP (sc-8334; Santa Cruz) antibodies and anti-mouse IgG-horseradish peroxidase (HRP) (Jackson ImmunoResearch) or anti-rabbit IgG-HRP (Dako) conjugates as secondary antibodies.
Intracellular antigen staining.
Twenty-four or 48 h after infection, moDCs were washed twice with phosphate-buffered saline (PBS) containing 1% FCS and 1 mg/ml NaN3 (wash buffer), treated for 30 min with PBS supplemented with 4% (wt/vol) paraformaldehyde (PFA) and 1% (wt/vol) Saponin (Cytofix/Cytoperm), and washed twice with PBS containing 0.1% (wt/vol) Saponin (Perm/Wash). Cells were stained for 30 min with the following primary antibodies (diluted 1:50 in Perm/Wash): anti-IE-1 (clone IE1.G10; Abcam) and anti-pp65 (ab53489; Abcam). Cells were then washed twice with Perm/Wash solution, stained for 30 min with phycoerythrin (PE)-goat anti-mouse IgG (poly4053; diluted 1:50 in Perm/Wash; BioLegend), and washed 2 more times afterwards. Samples infected with GFP-expressing vectors were only washed twice with wash buffer prior to measurement. Flow cytometry analysis was performed using a FACSCanto II device (BD Biosciences).
Restimulation of T cell clones and intracellular cytokine staining.
At 24 h after infection of moDCs, T cells were added at an effector/target cell ratio of 1:1 along with brefeldin A (BFA; 1 μg/ml). After 6 h of coincubation, cells were washed twice with wash buffer, treated for 30 min with Cytofix/Cytoperm solution, and washed two more times with Perm/Wash solution. Samples were then stained for 30 min with anti-CD8α-fluorescein isothiocyanate (clone RPA-T8; diluted 1:60 in Perm/Wash; BD Biosciences) and anti-IFN-γ-allophycocyanin (APC) (clone 4S.B3; diluted 1:60 in Perm/Wash; BioLegend) and finally washed twice with Perm/Wash. Data acquisition was performed on a FACSCanto II device (BD Biosciences). moDCs and T cells were first differentiated according to their forward scatter/side scatter (FSC/SSC) properties and further gated for CD8-positive cells. From those cells, a gate for IFN-γ-positive events was set that just excludes cells from an uninfected sample.
Cross presentation assay.
At 24 h after transduction of HeLa cells with rdSeV-pp65, they were washed 4 times with RPMI 1640 and detached from cell culture plates with trypsin-EDTA solution (PAN-Biotech). HeLa cells were added to moDCs at a 1:1 ratio, and after 24 h of coincubation, T cells were added along with BFA (1 μg/ml) at a moDC/T cell ratio of 1:1. After 6 h of coincubation, cells were stained for CD8 and IFN-γ and analyzed as described above.
Annexin V/7-AAD cytotoxicity assay.
To determine the amount of viable cells 24 or 48 h after infection, the APC Annexin V apoptosis detection kit with 7-AAD (BioLegend) was used according to the manufacturer's instructions. Flow cytometry analysis was performed using an Attune NxT flow cytometer (Life Technologies). Doublets were first excluded in an FSC-A/FSC-H plot, and cells debris was excluded according to FSC/SSC properties.
Analysis of DC maturation.
To assess the expression of maturation markers on the cell surface, moDCs were washed twice with wash buffer and incubated for 30 min with 4% (wt/vol) PFA in PBS. After two more washing steps with wash buffer, samples were stained for 30 min with anti-CD80-fluorescein isothiocyanate (clone L307.4; BD Biosciences), anti-CD83-PE-Cy7 (clone HB15e; BioLegend), anti-CD86-V450 (clone FUN-1; BD Biosciences), and anti-HLA-DR-APC (clone L243; all antibodies were diluted 1:60 in PBS; BioLegend). Finally, cells were washed twice with wash buffer and analyzed on a FACSCanto II device (BD Biosciences).
Cytokine profile.
At 48 h after infection, cell culture supernatants were collected and stored at −20°C until analysis. For cytokine quantification, the Legendplex human inflammation panel (13-plex kit; BioLegend) was used according to the manufacturer's instructions. Data acquisition was performed using an Attune NxT device (Life Technologies), and cytokine concentrations were determined using the software provided with the kit.
Leukocyte tropism.
PBMCs were infected with GFP-expressing vectors, and 24 hpi cells were washed twice with wash buffer and stained for 30 min with the following antibodies: anti-CD3-APC-Cy7 (clone SK7; BioLegend), anti-CD4-BV421 (clone RPA-T4; BioLegend), anti-CD8-AmCyan (clone SK1; BD Biosciences), anti-CD14-PE (clone 63D3; BioLegend), anti-CD19-APC (clone HIB19; BioLegend), and anti-CD56-peridinin chlorophyll protein (clone HCD56 [BioLegend]; all antibodies were diluted 1:60 in PBS). After staining, cells were washed twice with wash buffer and analyzed on a FACSCanto II device (BD Biosciences).
Immunization and MCMV challenge.
Ten- to 16-week-old anesthetized A2Kb mice were primed with 6 × 106 CIU of rdSeV-pp65 or PBS with 0.1% BSA in two 25-μl steps, each via the intranasal route as described previously (65). Boost immunizations were performed equally on day 21 and day 42 postpriming. On day 56 postpriming all groups were infected intravenously with 5 × 105 PFU MCMV-NLV. Four days postinfection, in vivo infectivity was determined from homogenates of spleen, lungs, and liver by plaque assay on MEF under conditions of centrifugal enhancement of infectivity (61). Viral genomes present in the respective organ lysates were quantitated by M55 (encoding gB)-specific qPCR normalized to cell number by pthrp-specific qPCR (61). Statistical analysis for group differences between two independent groups was performed after log transformation using Student's t test (unpaired, two sided; a P value of <0.05 was considered significant), with Welch's correction, with GraphPad Prism 6.04 (GraphPad Software, San Diego, CA, USA).
ACKNOWLEDGMENTS
This work was funded by the Bavarian Research Foundation (grant number AZ 1070-13, ForBIMed, to R.W.). N.AW.L. was funded by the Deutsche Forschungsgemeinschaft, SFB 1292, individual project TP11–viral evasion of innate and adaptive immune cells and “inbetweeners.”
We thank Dirk Busch and Magdalena Nauerth for the HCMV-specific T cell clones and Mariano Esteban for provision of MVA-GFP. M.A.W., C.K., and E.F. are paid employees of the company RSV Genius GmbH.
REFERENCES
- 1.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 3.Kimberlin DW, Lin C-Y, Sánchez PJ, Demmler GJ, Dankner W, Shelton M, Jacobs RF, Vaudry W, Pass RF, Kiell JM, Soong S, Whitley RJ, National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. 2003. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: a randomized, controlled trial. J Pediatr 143:16–25. doi: 10.1016/S0022-3476(03)00192-6. [DOI] [PubMed] [Google Scholar]
- 4.Drew WL, Miner RC, Busch DF, Follansbee SE, Gullett J, Mehalko SG, Gordon SM, Owen WF, Matthews TR, Buhles WC. 1991. Prevalence of resistance in patients receiving ganciclovir for serious cytomegalovirus infection. J Infect Dis 163:716–719. doi: 10.1093/infdis/163.4.716. [DOI] [PubMed] [Google Scholar]
- 5.Limaye AP. 2002. Ganciclovir-resistant cytomegalovirus in organ transplant recipients. Clin Infect Dis 35:866–872. doi: 10.1086/342385. [DOI] [PubMed] [Google Scholar]
- 6.Rieder F, Steininger C. 2014. Cytomegalovirus vaccine: phase II clinical trial results. Clin Microbiol Infect 20(Suppl 5):S95–S102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang M-L, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kharfan-Dabaja MA, Boeckh M, Wilck MB, Langston AA, Chu AH, Wloch MK, Guterwill DF, Smith LR, Rolland AP, Kenney RT. 2012. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis 12:290–299. doi: 10.1016/S1473-3099(11)70344-9. [DOI] [PubMed] [Google Scholar]
- 9.Wu SJ, Villarreal DO, Shedlock DJ, Weiner DB. 2015. Synthetic DNA approach to cytomegalovirus vaccine/immune therapy. Adv Exp Med Biol 848:131–148. doi: 10.1007/978-1-4939-2432-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wussow F, Chiuppesi F, Martinez J, Campo J, Johnson E, Flechsig C, Newell M, Tran E, Ortiz J, Rosa CL, Herrmann A, Longmate J, Chakraborty R, Barry PA, Diamond DJ. 2014. Human cytomegalovirus vaccine based on the envelope gH/gL pentamer complex. PLoS Pathog 10:e1004524. doi: 10.1371/journal.ppat.1004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu T-M, Wang D, Freed DC, Tang A, Li F, He X, Cole S, Dubey S, Finnefrock AC, ter Meulen J, Shiver JW, Casimiro DR. 2012. Restoration of viral epithelial tropism improves immunogenicity in rabbits and rhesus macaques for a whole virion vaccine of human cytomegalovirus. Vaccine 30:7469–7474. doi: 10.1016/j.vaccine.2012.10.053. [DOI] [PubMed] [Google Scholar]
- 13.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. 1992. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 14.Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Löffler J, Grigoleit U, Moris A, Rammensee H-G, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H. 2002. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99:3916–3922. doi: 10.1182/blood.V99.11.3916. [DOI] [PubMed] [Google Scholar]
- 15.Slezak SL, Bettinotti M, Selleri S, Adams S, Marincola FM, Stroncek DF. 2007. CMV pp65 and IE-1 T cell epitopes recognized by healthy subjects. J Transl Med 5:17. doi: 10.1186/1479-5876-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ura T, Okuda K, Shimada M. 2014. Developments in viral vector-based vaccines. Vaccines 2:624–641. doi: 10.3390/vaccines2030624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schleiss MR. 2016. Cytomegalovirus vaccines under clinical development. J Virus Erad 2:198–207. [PMC free article] [PubMed] [Google Scholar]
- 19.La Rosa C, Longmate J, Martinez J, Zhou Q, Kaltcheva TI, Tsai W, Drake J, Carroll M, Wussow F, Chiuppesi F, Hardwick N, Dadwal S, Aldoss I, Nakamura R, Zaia JA, Diamond DJ. 2017. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 129:114–125. doi: 10.1182/blood-2016-07-729756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivière C, Danos O, Douar AM. 2006. Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther 13:1300–1308. doi: 10.1038/sj.gt.3302766. [DOI] [PubMed] [Google Scholar]
- 21.Iida A, Inoue M. 2013. Concept and technology underlying sendai virus (SeV) vector development, p 69–89. In Nagai Y. (ed), Sendai virus vector. Springer, Tokyo, Japan. [Google Scholar]
- 22.Nagai Y. 1999. Paramyxovirus replication and pathogenesis. Reverse genetics transforms understanding. Rev Med Virol 9:83–99. [DOI] [PubMed] [Google Scholar]
- 23.Slobod KS, Shenep JL, Luján-Zilbermann J, Allison K, Brown B, Scroggs RA, Portner A, Coleclough C, Hurwitz JL. 2004. Safety and immunogenicity of intranasal murine parainfluenza virus type 1 (Sendai virus) in healthy human adults. Vaccine 22:3182–3186. doi: 10.1016/j.vaccine.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 24.Stark JE, Heath RB. 1967. The development of antibodies against Sendai virus in childhood. Arch Gesamte Virusforsch 20:438–444. doi: 10.1007/BF01275224. [DOI] [PubMed] [Google Scholar]
- 25.Adderson E, Branum K, Sealy RE, Jones BG, Surman SL, Penkert R, Freiden P, Slobod KS, Gaur AH, Hayden RT, Allison K, Howlett N, Utech J, Allay J, Knight J, Sleep S, Meagher MM, Russell CJ, Portner A, Hurwitz JL. 2015. Safety and immunogenicity of an intranasal Sendai virus-based human parainfluenza virus type 1 vaccine in 3- to 6-year-old children. Clin Vaccine Immunol 22:298–303. doi: 10.1128/CVI.00618-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiegand MA, Gori-Savellini G, Gandolfo C, Papa G, Kaufmann C, Felder E, Ginori A, Disanto MG, Spina D, Cusi MG. 2017. A respiratory syncytial virus vaccine vectored by a stable chimeric and replication-deficient Sendai virus protects mice without inducing enhanced disease. J Virol 91:e02298-. doi: 10.1128/JVI.02298-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones BG, Sealy RE, Rudraraju R, Traina-Dorge VL, Finneyfrock B, Cook A, Takimoto T, Portner A, Hurwitz JL. 2012. Sendai virus-based RSV vaccine protects African green monkeys from RSV infection. Vaccine 30:959–968. doi: 10.1016/j.vaccine.2011.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii H, Matano T. 2015. Development of an AIDS vaccine using Sendai virus vectors. Vaccine 33:6061–6065. doi: 10.1016/j.vaccine.2015.06.114. [DOI] [PubMed] [Google Scholar]
- 29.Nyombayire J, Anzala O, Gazzard B, Karita E, Bergin P, Hayes P, Kopycinski J, Omosa-Manyonyi G, Jackson A, Bizimana J, Farah B, Sayeed E, Parks CL, Inoue M, Hironaka T, Hara H, Shu T, Matano T, Dally L, Barin B, Park H, Gilmour J, Lombardo A, Excler J-L, Fast P, Laufer DS, Cox JH, Ingabire SSR, Ouattara G, Steele A, Gumbe A, Chinyenze K, Welsh S, Verlinde C, King D, Bishop C, Chetty P, Clark L, Booley M, Zachariah D, Syvertsen K, Anas K, Naarding M, Cormier E, Ackland J, Hasegawa M, S001 Study Team. 2017. First-in-human evaluation of the safety and immunogenicity of an intranasally administered replication-competent Sendai virus-vectored HIV type 1 Gag vaccine: induction of potent T-cell or antibody responses in prime-boost regimens. J Infect Dis 215:95–104. doi: 10.1093/infdis/jiw500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bossow S, Schlecht S, Schubbert R, Pfeiffer M, Neubert WJ, Wiegand M. 2012. Evaluation of nucleocapsid and phosphoprotein P functionality as critical factors during the early phase of paramyxoviral infection. Open Virol J 6:73–81. doi: 10.2174/1874357901206010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiegand M, Gori-Savellini G, Martorelli B, Bossow S, Neubert WJ, Cusi MG. 2013. Evaluation of a novel immunogenic vaccine platform based on a genome replication-deficient Sendai vector. Vaccine 31:3888–3893. doi: 10.1016/j.vaccine.2013.06.053. [DOI] [PubMed] [Google Scholar]
- 32.Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, Norberg P, Xie W, Herndon JE, Healy P, McLendon RE, Friedman AH, Friedman HS, Bigner D, Vlahovic G, Mitchell DA, Sampson JH. 2017. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res 23:1898–1909. doi: 10.1158/1078-0432.CCR-16-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiegand MA, Bossow S, Schlecht S, Neubert WJ. 2007. De novo synthesis of N and P proteins as a key step in Sendai virus gene expression. J Virol 81:13835–13844. doi: 10.1128/JVI.00914-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armeanu S, Bitzer M, Smirnow I, Bossow S, Appel S, Ungerechts G, Bernloehr C, Neubert WJ, Lauer UM, Brossart P. 2005. Severe impairment of dendritic cell allostimulatory activity by Sendai virus vectors is overcome by matrix protein gene deletion. J Immunol 175:4971–4980. [DOI] [PubMed] [Google Scholar]
- 35.Kiener R, Fleischmann M, Schwegler C, Ruzsics Z, Thirion C, Schrödel S, Asbach B, Wagner R. 2018. Vaccine vectors based on adenovirus 19a/64 exhibit broad cellular tropism and potently restimulate HCMV-specific T cell responses ex vivo. Sci Rep 8:1474. doi: 10.1038/s41598-018-19874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bitzer M, Prinz F, Bauer M, Spiegel M, Neubert WJ, Gregor M, Schulze-Osthoff K, Lauer U. 1999. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32). J Virol 73:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, Coscoy L, Winoto A. 2017. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 24:615–625. doi: 10.1038/cdd.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y, Alwine JC. 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J Virol 76:3731–3738. doi: 10.1128/JVI.76.8.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudek AM, Martin S, Garg AD, Agostinis P. 2013. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol 4:438. doi: 10.3389/fimmu.2013.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas S, Klobuch S, Podlech J, Plachter B, Hoffmann P, Renzaho A, Theobald M, Reddehase MJ, Herr W, Lemmermann NAW. 2015. Evaluating human T-cell therapy of cytomegalovirus organ disease in HLA-transgenic mice. PLoS Pathog 11:e1005049. doi: 10.1371/journal.ppat.1005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemmermann NAW, Reddehase MJ. 2016. Refining human T-cell immunotherapy of cytomegalovirus disease: a mouse model with “humanized” antigen presentation as a new preclinical study tool. Med Microbiol Immunol (Berlin) 205:549–561. doi: 10.1007/s00430-016-0471-0. [DOI] [PubMed] [Google Scholar]
- 42.McCormack S, Stöhr W, Barber T, Bart P-A, Harari A, Moog C, Ciuffreda D, Cellerai C, Cowen M, Gamboni R, Burnet S, Legg K, Brodnicki E, Wolf H, Wagner R, Heeney J, Frachette M-J, Tartaglia J, Babiker A, Pantaleo G, Weber J. 2008. EV02: a phase I trial to compare the safety and immunogenicity of HIV DNA-C prime-NYVAC-C boost to NYVAC-C alone. Vaccine 26:3162–3174. doi: 10.1016/j.vaccine.2008.02.072. [DOI] [PubMed] [Google Scholar]
- 43.Russell CJ, Hurwitz JL. 2016. Sendai virus as a backbone for vaccines against RSV and other human paramyxoviruses. Expert Rev Vaccines 15:189–200. doi: 10.1586/14760584.2016.1114418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato A, Ohnishi Y, Hishiyama M, Kohase M, Saito S, Tashiro M, Nagai Y. 2002. The amino-terminal half of Sendai virus C protein is not responsible for either counteracting the antiviral action of interferons or down-regulating viral RNA synthesis. J Virol 76:7114–7124. doi: 10.1128/JVI.76.14.7114-7124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato A, Ohnishi Y, Kohase M, Saito S, Tashiro M, Nagai Y. 2001. Y2, the smallest of the Sendai virus C proteins, is fully capable of both counteracting the antiviral action of interferons and inhibiting viral RNA synthesis. J Virol 75:3802–3810. doi: 10.1128/JVI.75.8.3802-3810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croft NP, Smith SA, Wong YC, Tan CT, Dudek NL, Flesch IEA, Lin LCW, Tscharke DC, Purcell AW. 2013. Kinetics of antigen expression and epitope presentation during virus infection. PLoS Pathog 9:e1003129. doi: 10.1371/journal.ppat.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chahroudi A, Garber DA, Reeves P, Liu L, Kalman D, Feinberg MB. 2006. Differences and similarities in viral life cycle progression and host cell physiology after infection of human dendritic cells with modified vaccinia virus Ankara and vaccinia virus. J Virol 80:8469–8481. doi: 10.1128/JVI.02749-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mocarski ES, Shenk T, Griffiths P, Pass RF. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 49.Wajant H, Pfizenmaier K, Scheurich P. 2003. Tumor necrosis factor signaling. Cell Death Differ 10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 50.Reis e Sousa C. 2006. Dendritic cells in a mature age. Nat Rev Immunol 6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 51.Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Steinman RM, Bhardwaj N. 1999. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol 163:6762–6768. [PubMed] [Google Scholar]
- 52.Hosoya N, Miura T, Kawana-Tachikawa A, Koibuchi T, Shioda T, Odawara T, Nakamura T, Kitamura Y, Kano M, Kato A, Hasegawa M, Nagai Y, Iwamoto A. 2008. Comparison between Sendai virus and adenovirus vectors to transduce HIV-1 genes into human dendritic cells. J Med Virol 80:373–382. doi: 10.1002/jmv.21052. [DOI] [PubMed] [Google Scholar]
- 53.López CB, Moltedo B, Alexopoulou L, Bonifaz L, Flavell RA, Moran TM. 2004. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J Immunol 173:6882–6889. [DOI] [PubMed] [Google Scholar]
- 54.Paulus C, Nevels M. 2009. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses 1:760–779. doi: 10.3390/v1030760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heilingloh CS, Grosche L, Kummer M, Mühl-Zürbes P, Kamm L, Scherer M, Latzko M, Stamminger T, Steinkasserer A. 2017. The major immediate-early protein IE2 of human cytomegalovirus is sufficient to induce proteasomal degradation of CD83 on mature dendritic cells. Front Microbiol 8:119. doi: 10.3389/fmicb.2017.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heilingloh CS, Mühl-Zürbes P, Steinkasserer A, Kummer M. 2014. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J Gen Virol 95:1366–1375. doi: 10.1099/vir.0.062810-0. [DOI] [PubMed] [Google Scholar]
- 57.Tang A, Freed DC, Li F, Meschino S, Prokop M, Bett A, Casimiro D, Wang D, Fu T-M. 2017. Functionally inactivated dominant viral antigens of human cytomegalovirus delivered in replication incompetent adenovirus type 6 vectors as vaccine candidates. Hum Vaccin Immunother 13:2763–2771. doi: 10.1080/21645515.2017.1308988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crawford LB, Streblow DN, Hakki M, Nelson JA, Caposio P. 2015. Humanized mouse models of human cytomegalovirus infection. Curr Opin Virol 13:86–92. doi: 10.1016/j.coviro.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Itell HL, Kaur A, Deere JD, Barry PA, Permar SR. 2017. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr Opin Virol 25:126–133. doi: 10.1016/j.coviro.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Podlech J, Holtappels R, Grzimek NKA, Reddehase MJ. 2002. Animal models: murine cytomegalovirus, p 493–525. In Kaufmann SHE, Kabelitz D (ed), Methods in microbiology: immunology of infection, vol. 32 Academic Press, Oxford, United Kingdom. [Google Scholar]
- 61.Lemmermann NAW, Podlech J, Seckert CK, Kropp KA, Grzimek NKA, Reddehase MJ, Holtappels R. 2010. CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model, p 369–429. In Kabelitz D, Kaufmann SHE (ed), Methods in microbiology: immunology of infection, 2nd ed, vol 37 Academic Press, Oxford, United Kingdom. [Google Scholar]
- 62.Vitiello A, Marchesini D, Furze J, Sherman LA, Chesnut RW. 1991. Analysis of the HLA-restricted influenza-specific cytotoxic T lymphocyte response in transgenic mice carrying a chimeric human-mouse class I major histocompatibility complex. J Exp Med 173:1007–1015. doi: 10.1084/jem.173.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chakrabarti S, Sisler JR, Moss B. 1997. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 23:1094–1097. [DOI] [PubMed] [Google Scholar]
- 64.Perdiguero B, Gómez CE, Cepeda V, Sánchez-Sampedro L, García-Arriaza J, Mejías-Pérez E, Jiménez V, Sánchez C, Sorzano CÓ Oliveros SJC, Delaloye J, Roger T, Calandra T, Asbach B, Wagner R, Kibler KV, Jacobs BL, Pantaleo G, Esteban M. 2015. Virological and immunological characterization of novel NYVAC-based HIV/AIDS vaccine candidates expressing clade C trimeric soluble gp140(ZM96) and Gag(ZM96)-Pol-Nef(CN54) as virus-like particles. J Virol 89:970–988. doi: 10.1128/JVI.02469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oduro JD, Redeker A, Lemmermann NAW, Ebermann L, Marandu TF, Dekhtiarenko I, Holzki JK, Busch DH, Arens R, Čičin-Šain L. 2016. Murine cytomegalovirus (CMV) infection via the intranasal route offers a robust model of immunity upon mucosal CMV infection. J Gen Virol 97:185–195. doi: 10.1099/jgv.0.000339. [DOI] [PubMed] [Google Scholar]
- 66.DeFilippes FM. 1982. Restriction enzyme mapping of vaccinia virus DNA. J Virol 43:136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]